
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe first chiral diene-based metal–organic frameworks for highly enantioselective carbon–carbon bond formation reactions†
Takahiro
Sawano
,
Pengfei
Ji
,
Alexandra R.
McIsaac
,
Zekai
Lin
,
Carter W.
Abney
and
Wenbin
Lin
*
Department of Chemistry, University of Chicago, 929 E 57th St, Chicago, Illinois 60637, USA. E-mail: wenbinlin@uchicago.edu
First published on 14th September 2015
Abstract
We have designed the first chiral diene-based metal–organic framework (MOF), E2-MOF, and postsynthetically metalated E2-MOF with Rh(I) complexes to afford highly active and enantioselective single-site solid catalysts for C–C bond formation reactions. Treatment of E2-MOF with [RhCl(C2H4)2]2 led to a highly enantioselective catalyst for 1,4-additions of arylboronic acids to α,β-unsaturated ketones, whereas treatment of E2-MOF with Rh(acac)(C2H4)2 afforded a highly efficient catalyst for the asymmetric 1,2-additions of arylboronic acids to aldimines. Interestingly, E2-MOF·Rh(acac) showed higher activity and enantioselectivity than the homogeneous control catalyst, likely due to the formation of a true single-site catalyst in the MOF. E2-MOF·Rh(acac) was also successfully recycled and reused at least seven times without loss of yield and enantioselectivity.
Introduction
In the past 15 years, metal–organic frameworks (MOFs) have emerged as a novel class of highly porous molecular materials with great potential for many applications, including gas storage,1 chemical sensing,2 biomedical imaging,2a,3 drug delivery,4 nonlinear optics,5 and catalysis.6 Although a number of excellent MOF-based catalytic systems have been developed recently,7 examples of highly enantioselective asymmetric reactions catalyzed by MOFs are still limited despite their potential utility in the synthesis of high-value fine chemicals. Asymmetric MOF catalysts not only enable the recycling and reuse of expensive chiral ligands and precious metals, but also prevent the leaching of toxic metals into organic products which can be a significant issue for the pharmaceutical industry.Since the first report of a MOF-based asymmetric catalyst with modest enantioselectivity in 2000,8 a number of highly enantioselective MOF catalysts with Lewis acid reactivity have been designed, including Ti(IV)-BINOL-based MOFs9 and Mn(III)- and Co(III)-salen-based MOFs.10 To expand the scope of MOF-catalyzed asymmetric reactions, Lin and coworkers recently developed BINAP-based MOFs for a number of important asymmetric catalytic reactions.7h However, the bulky BINAP and derivatives reduce the channel/cavity sizes of BINAP-MOFs and hinder their applications in asymmetric reactions involving sterically demanding transition states/intermediates. Herein we report the design and synthesis of the first chiral diene-based MOFs and their use in asymmetric C–C bond formation reactions (Fig. 1). With less steric demand than their BINAP predecessors, the diene-MOFs metalated with Rh(I) complexes are excellent catalysts with high activities and enantioselectivities for 1,4-additions of arylboronic acids to α,β-unsaturated ketones and 1,2-additions of arylboronic acids to aldimines.11
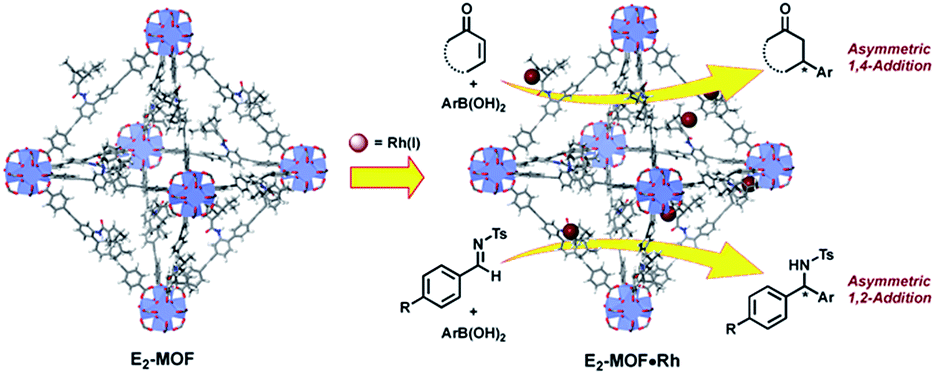 | ||
| Fig. 1 Postsynthetically Rh-metalated E2-MOF catalysts for asymmetric reactions. | ||
Chiral dienes have been applied to a broad range of asymmetric reactions since being reported independently by Hayashi12 and Carreira.13,14 In particular, asymmetric Rh-diene complexes provide powerful methods to construct chiral centers in C–C bond formations. For example, 1,4-additions of electron-deficient olefins and 1,2-addition of imines with arylboronic acids in the presence of rhodium and a chiral diene provide highly desirable synthetic methods to obtain the addition products with high yields and enantioselectivities.15,16 In these reactions, Rh complexes of chiral dienes typically afford higher yields and enantiomeric excesses (ee's) than the corresponding Rh-BINAP complexes.
Results and discussion
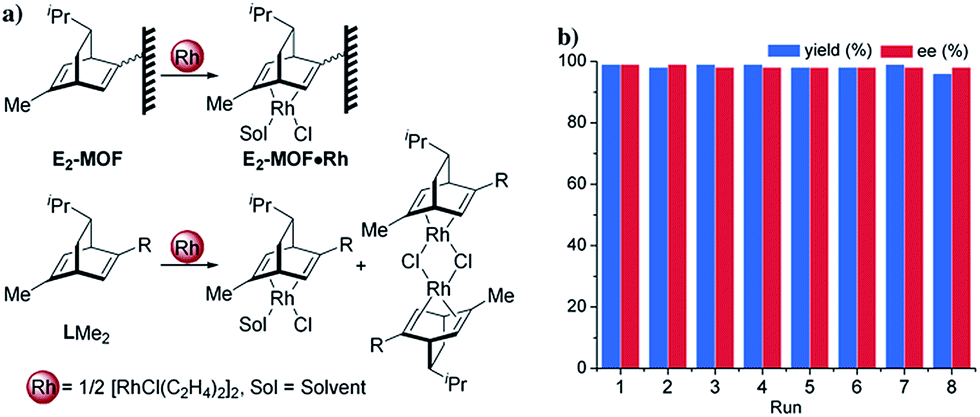
We targeted the synthesis of the chiral diene-based MOF, E2-MOF, based on the linear dicarboxylate ligand containing an orthogonal chiral diene group and the Zr6(μ3-O)4(μ3-OH)4 secondary building unit (SBU). Zr MOFs of the UiO structure not only provide a highly tunable platform for designing functional materials but also are stable under a broad range of reaction conditions.17 The diene ligand (LH2) was synthesized from a known chiral diene-carboxylic acid compound (Scheme 1).18 Upon treatment with oxalyl chloride, the corresponding acid chloride was reacted with the dicarboxylic ester possessing an orthogonal amino group to afford the methyl ester of the chiral diene (LMe2) in 57% yield. Subsequent saponification of LMe2 afforded enantiopure LH2 in 77% yield. E2-MOF was synthesized as colorless crystals in 42% yield by treating ZrCl4 with 1 equiv. of LH2 and a small amount of trifluoroacetic acid (TFA) in dimethylformamide (DMF) at 70 °C for 5 d.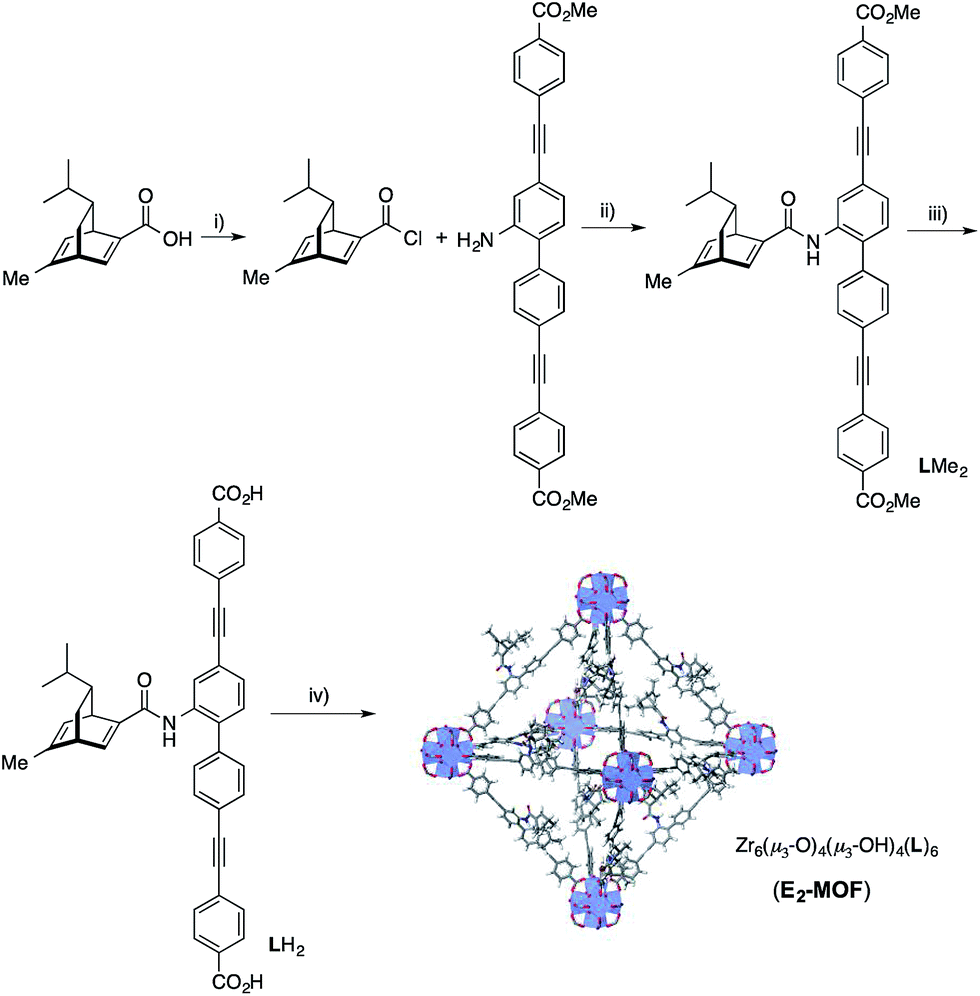 | ||
| Scheme 1 Synthesis of LH2. (i) oxalyl chloride, CH2Cl2; (ii) TEA, THF, 57% yield over 2 steps; (iii) NaOH, THF, EtOH, 77% yield; (iv) ZrCl4, TFA, DMF, 70 °C, 5 d, 42% yield. | ||
Single crystal X-ray diffraction revealed that E2-MOF crystallizes in the Fm![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m space group and adopts the UiO structure. The chiral diene moieties are randomly distributed in the framework and could not be located in the electron density map. The 1H NMR spectra of digested E2-MOF confirms that the chiral diene groups remain intact during the MOF crystal growth (Fig. S1 and S3, ESI†) and E2-MOF has a formula of Zr6(μ3-O)4(μ3-OH)4(L)6·143DMF·109H2O. Thermogravimetric analysis indicated that E2-MOF contains 73% solvent (Fig. S2, ESI†), suggesting a highly porous framework structure. However, nitrogen sorption measurements afforded negligible surface areas, presumably due to framework distortion upon solvent removal (Fig. S4 and S5, ESI†).9c,19 The pore accessibility of E2-MOF, E2-MOF·RhCl, and E2-MOF·Rh(acac) was demonstrated by dye absorption measurements which shows the uptake of 5.32 (112 wt%), 2.53 (98 wt%), and 4.47 (107 wt%) of Brilliant Blue R-250 per unit cell by E2-MOF, E2-MOF·RhCl, and E2-MOF·Rh(acac), respectively (Fig. S7, ESI†).9c
m space group and adopts the UiO structure. The chiral diene moieties are randomly distributed in the framework and could not be located in the electron density map. The 1H NMR spectra of digested E2-MOF confirms that the chiral diene groups remain intact during the MOF crystal growth (Fig. S1 and S3, ESI†) and E2-MOF has a formula of Zr6(μ3-O)4(μ3-OH)4(L)6·143DMF·109H2O. Thermogravimetric analysis indicated that E2-MOF contains 73% solvent (Fig. S2, ESI†), suggesting a highly porous framework structure. However, nitrogen sorption measurements afforded negligible surface areas, presumably due to framework distortion upon solvent removal (Fig. S4 and S5, ESI†).9c,19 The pore accessibility of E2-MOF, E2-MOF·RhCl, and E2-MOF·Rh(acac) was demonstrated by dye absorption measurements which shows the uptake of 5.32 (112 wt%), 2.53 (98 wt%), and 4.47 (107 wt%) of Brilliant Blue R-250 per unit cell by E2-MOF, E2-MOF·RhCl, and E2-MOF·Rh(acac), respectively (Fig. S7, ESI†).9c
Postsynthetic metalation of E2-MOF was carried out by treatment with 1 equiv. of [RhCl(C2H4)2]2 or 1 equiv. of Rh(acac)(C2H4)2, (based on the Rh equivalent with respect to the L equivalents in E2-MOF, ESI†; acac is acetylacetonate). Powder X-ray diffraction (PXRD) studies indicated that E2-MOF·RhCl and E2-MOF·Rh(acac) remained crystalline and adopted the same structure as the original E2-MOF (Fig. 2a). Inductively coupled plasma mass spectrometry (ICP-MS) was used to determine the extent of metalation in E2-MOF based on the Rh to Zr ratios. E2-MOF·RhCl achieved 66% metalation whereas E2-MOF·Rh(acac) only had 13% of the L ligands metalated.
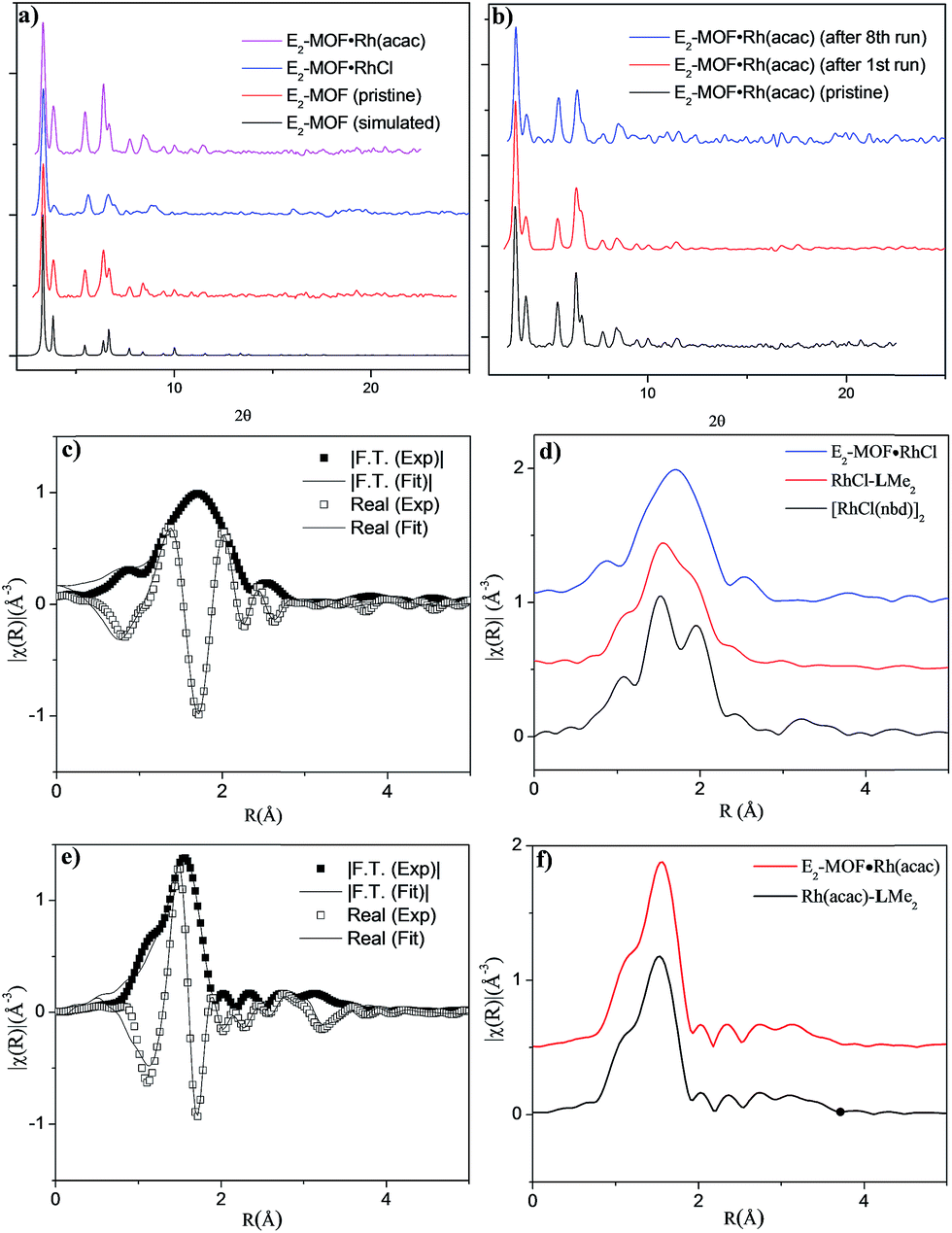 | ||
| Fig. 2 (a) PXRD patterns of pristine E2-MOF (simulated from the CIF file, black; experimental, red), and freshly prepared E2-MOF·RhCl (blue) and E2-MOF·Rh(acac) (pink). (b) PXRD patterns of pristine E2-MOF·Rh(acac) (black) and E2-MOF·Rh(acac) recovered from 1,2-addition reactions (after 1st run (red) and 8th run (blue)). (c) EXAFS data (squares) and best fits(lines) for E2-MOF·RhCl. Data are displayed in R-space containing both magnitude of Fourier transform and real components. An R-factor of 0.01 was obtained for the fit. (d) A comparison of EXAFS data for E2-MOF·RhCl, RhCl(LMe2), and the [RhCl(nbd)]2 dimer. (e) EXAFS data (squares) and best fits (lines) for E2-MOF·Rh(acac). An R-factor of 0.016 was obtained for the fit. (f) A comparison of EXAFS data for E2-MOF·Rh(acac) and Rh(acac)-LMe2. | ||
Due to the positional disorder and incomplete metalation of the diene moiety, the Rh coordination environments could not be determined by traditional crystallographic techniques. X-ray absorption fine structure (XAFS) spectroscopy at the Rh K-edge was used to investigate the local coordination environment of Rh in E2-MOF·RhCl, E2-MOF·Rh(acac), Rh-metalated LMe2, and the dimeric [RhCl(nbd)]2 standard. Data were processed and analyzed using the Athena and Artemis programs of the IFEFFIT package based on FEFF 6. E2-MOF·RhCl was fitted with a monomeric model where the Rh coordination sphere is occupied by norbornadiene, chloride, and a THF molecule (Fig. 2c). The spectra for [RhCl(nbd)]2 was fitted by the corresponding crystal structure (Fig. S13†). Compared to the spectra for E2-MOF·RhCl, a significant peak was observed in R-space at ∼2 Å which is largely attributable to a second Rh–Cl direct scattering path; amplitude from Rh–Rh direct scattering paths can also be observed at ∼3.2 Å (Fig. 2d). The RhCl-LMe2 system was best fitted with a combination of monomeric (∼85%) and dimeric (∼15%) models (Fig. S15, ESI†), which was confirmed by 1H NMR spectroscopy (Fig. S18, ESI†). E2-MOF·Rh(acac) and Rh(acac)-LMe2 were fitted with a reported crystal structure where the Rh coordination sphere is occupied by a diene and an acac ligand.18a There is little difference between E2-MOF·Rh(acac) and Rh(acac)-LMe2 in their EXAF spectra (Fig. 2f) presumably due to the similarity of one chelating diene and two bridging diene on each Rd center in EXAFS, but 1H NMR of Rh(acac)-LMe2 indicated that the Rh(acac)-LMe2 contained a complex mixture including oligomeric/polymeric species in the homogeneous control (Fig. S19, ESI†). These results indicate that E2-MOF·RhCl is a true single-site catalyst by prohibiting any such dimer formation owing to site isolation.7a–j,20
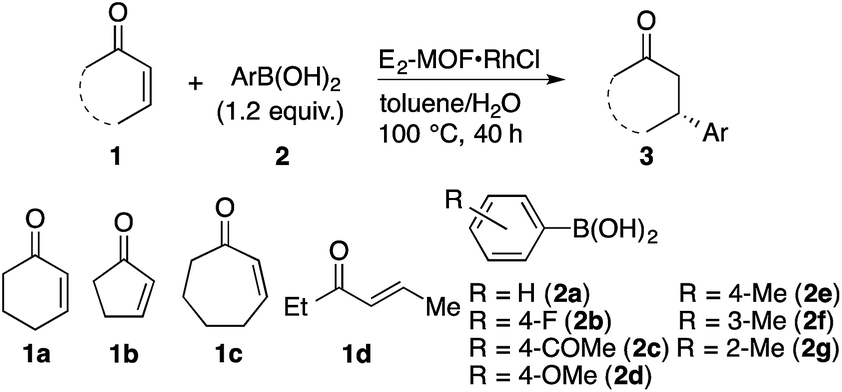
E2-MOF·RhCl is a highly effective catalyst for 1,4-additions of arylboronic acids to α,β-unsaturated ketones. The reaction of 2-cyclohexenone (1a) with phenylboronic acid (2a) in the presence of 0.01 mol% E2-MOF·RhCl gave the addition product in 97% yield and 95% ee (Table 1, entry 1). At 0.005 mol% Rh loading, the reaction proceeded to give the addition product in 67% yield and 94% ee, leading to a high turnover number (TON) of 13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 400 (entry 2). In comparison, the 1,4-addition reaction with 0.005 mol% Rh of E2-MOF·Rh(acac) gave the addition product in 46% yield with 93% ee. These results are comparable with those of the homogeneous control catalyst (Table 1, entry 3). As expected, the catalytic activity of E2-MOF·RhCl is much higher than BINAP-MOF·RhCl (Table 1, entries 2 vs. 4). E2-MOF·RhCl catalyzed 1,4-addition reactions have a broad substrate scope for both arylboronic acids and α,β-unsaturated ketones. Both electron donating groups and electron withdrawing groups can be installed to the aromatic ring of arylboronic acids, giving the addition products in high yields and high ee's (Table 1, entries 5–8). The addition of arylboronic acids having a substituent at the meta and ortho position also proceeded (Table 1, entries 9 and 10). For α,β-unsaturated ketones, the reactions proceeded with five-membered ring and seven-membered ring substrates (Table 1, entries 11 and 12) as well as with a linear ketone (Table 1, entry 13). Heterogeneity of the 1,4-addition reaction was confirmed by ICP-MS, which indicates the leaching of only small amounts of Rh (1.3%) and Zr (<0.01%) into the solution. However, the recovered E2-MOF·RhCl showed reduced catalytic activity (Scheme S1, ESI†), which might be due to the gradual loss of MOF crystallinity during the course of the reaction (Fig. S19, ESI†). Consistent with this, E2-MOF soaked in 1 M HCl, water, or 1 M NaOH for 40 h lost crystallinity as judged by PXRD (Fig. S6, ESI†).
400 (entry 2). In comparison, the 1,4-addition reaction with 0.005 mol% Rh of E2-MOF·Rh(acac) gave the addition product in 46% yield with 93% ee. These results are comparable with those of the homogeneous control catalyst (Table 1, entry 3). As expected, the catalytic activity of E2-MOF·RhCl is much higher than BINAP-MOF·RhCl (Table 1, entries 2 vs. 4). E2-MOF·RhCl catalyzed 1,4-addition reactions have a broad substrate scope for both arylboronic acids and α,β-unsaturated ketones. Both electron donating groups and electron withdrawing groups can be installed to the aromatic ring of arylboronic acids, giving the addition products in high yields and high ee's (Table 1, entries 5–8). The addition of arylboronic acids having a substituent at the meta and ortho position also proceeded (Table 1, entries 9 and 10). For α,β-unsaturated ketones, the reactions proceeded with five-membered ring and seven-membered ring substrates (Table 1, entries 11 and 12) as well as with a linear ketone (Table 1, entry 13). Heterogeneity of the 1,4-addition reaction was confirmed by ICP-MS, which indicates the leaching of only small amounts of Rh (1.3%) and Zr (<0.01%) into the solution. However, the recovered E2-MOF·RhCl showed reduced catalytic activity (Scheme S1, ESI†), which might be due to the gradual loss of MOF crystallinity during the course of the reaction (Fig. S19, ESI†). Consistent with this, E2-MOF soaked in 1 M HCl, water, or 1 M NaOH for 40 h lost crystallinity as judged by PXRD (Fig. S6, ESI†).
|
|
||||||
|---|---|---|---|---|---|---|
| Entry | Enone | Aryl boronic acid | Catalyst loading (mol% Rh) | Yieldb (%) | eec (%) | TON |
| a Reaction conditions: 1 (1 equiv.), 2 (1.2 equiv.), toluene, H2O at 100 °C for 40 h. b Isolated yield. c Determined by chiral HPLC. d [RhCl(C2H4)2]2 and LMe2 were used as catalyst. e BINAP-MOF·RhCl was used as catalyst. f Not determined. g 2.0 equiv. of PhB(OH)2. | ||||||
| 1 | 1a | 2a | 0.01 | 97 | 95 | 9700 |
| 2 | 1a | 2a | 0.005 | 67 | 94 | 13400 |
| 3d | 1a | 2a | 0.005 | 70 | 91 | 14000 |
| 4e | 1a | 2a | 0.005 | 3 | —f | 600 |
| 5 | 1a | 2b | 0.025 | 90 | 94 | 3600 |
| 6 | 1a | 2c | 0.05 | 80 | 91 | 1600 |
| 7 | 1a | 2d | 0.01 | 84 | 96 | 8400 |
| 8 | 1a | 2e | 0.05 | 87 | 95 | 1740 |
| 9 | 1a | 2f | 0.05 | 84 | 94 | 1680 |
| 10 | 1a | 2g | 0.05 | 82 | 74 | 1640 |
| 11 | 1b | 2a | 0.1 | 82 | 90 | 820 |
| 12g | 1c | 2a | 0.1 | 93 | 70 | 930 |
| 13g | 1d | 2a | 0.25 | 84 | 90 | 336 |
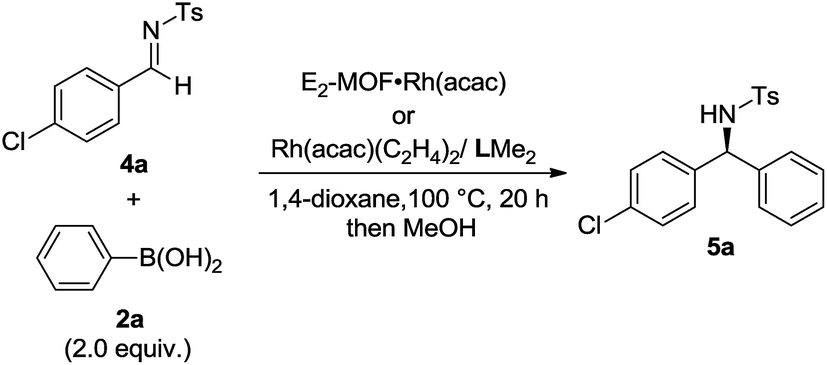
Asymmetric 1,2-addition of arylboronic acids to aldimines proceeded in the presence of E2-MOF·Rh(acac).21 At 0.2 mol% Rh loading, the reaction gave the addition product in 55% yield and 98% ee (Table 2, entry 1). Quantitative yield of the addition product was obtained at 3 mol% Rh loading (Table 2, entry 3). Interestingly, E2-MOF·Rh(acac) performed better than the homogeneous control catalyst both in terms of yields and ee's. For the homogeneous control, the product yield can be increased by increasing the catalyst loading but at the expense of the obtained ee's. The ee's of the E2-MOF·Rh(acac)-catalyzed reactions remained constant at different catalyst loadings. This striking difference can be attributed to the desirable site isolation provided by the MOF, which exclusively affords the desired monomeric Rh species; in contrast, the homogeneous control can form a dimeric/oligomeric species which might be less enantioselective. This monomer/dimer equilibrium was proved by EXAFS and 1H NMR studies for the RhCl(LMe2) system (Fig. 2d and 3a).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Catalyst loading | Yieldb (%) | eec (%) | TON |
| a 4a (1.0 equiv.), 2a (2.0 equiv.), catalyst, 1,4-dioxane, 100 °C, 20 h. b NMR yield based on internal standard. c Determined by chiral HPLC analysis. Ts = p-toluenesulfonyl. | |||||
| 1 | E2-MOF·Rh(acac) | 0.2 mol% | 55 | 98 | 275 |
| 2 | E2-MOF·Rh(acac) | 0.6 mol% | 71 | 98 | 118 |
| 3 | E2-MOF·Rh(acac) | 3 mol% | 99 | 98 | 33 |
| 4 | Rh(acac)/LMe2 | 0.2 mol% | 11 | 94 | 55 |
| 5 | Rh(acac)/LMe2 | 0.6 mol% | 55 | 89 | 92 |
| 6 | Rh(acac)/LMe2 | 3 mol% | 87 | 83 | 29 |
 | ||
| Fig. 3 (a) Structures of rhodium-coordinated diene complexes in MOF catalyst and the homogeneous control catalyst. (b) Plot of yield (%) and ee (%) of 1,2-addition product at various runs in the recycle and reuse of E2-MOF·Rh(acac) (6 mol% Rh) for 1,2-addition of aldimine 4a with phenylboronic acid (2a). | ||
E2-MOF·Rh(acac)-catalyzed 1,2-addition reactions have a broad substrate scope for both arylboronic acids and aldimines to give addition products with excellent ee's (ranging from 97% to >99%). The reaction works with aldimines and arylboronic acids having electron donating groups or electron withdrawing groups (Table 3, entries 1–6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | R1 | R2 | Yieldb (%) | eec (%) | TON |
| a 4 (1.0 equiv.), 2 (2.0 equiv.), E2-MOF·Rh(acac) (3 mol% Rh), 1,4-dioxane, 100 °C, 20 h. b NMR yield based on internal standard. c Determined by chiral HPLC analysis. | |||||
| 1 | Cl (4a) | H (2a) | 99 | 98 | 33 |
| 2 | Cl (4a) | F (2b) | 95 | 99 | 32 |
| 3 | Cl (4a) | OMe (2d) | 80 | 97 | 27 |
| 4 | H (4b) | F (2b) | 97 | 99 | 32 |
| 5 | H (4b) | OMe (2d) | 96 | 97 | 32 |
| 6 | OMe (4c) | H (2a) | 98 | 99 | 33 |
| 7 | OMe (4c) | F (2b) | 99 | >99 | 33 |
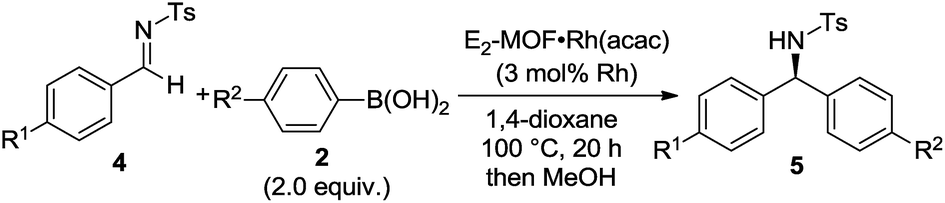
Several experiments proved that E2-MOF·Rh(acac) is a true heterogeneous and reusable catalyst. First, the MOF catalyst (6 mol% Rh) could be recycled and reused for at least 7 times without loss of yield and ee (Fig. 3b). Second, the crystallinity of the MOF catalyst recovered from the 1st and 8th runs was still maintained as the PXRD of the recovered catalyst remained the same as the freshly prepared E2-MOF·Rh(acac) (Fig. 2b). Third, ICP-MS analysis showed negligible leaching of Rh (0.49%) and Zr (0.07%) during the reaction. Fourth, the progress of the reaction was stopped by removing the MOF catalyst from the reaction mixture, indicating that the supernatant is inactive in catalyzing the 1,2-addition reactions (Scheme S3, ESI†).
Conclusions
We have developed catalytically active chiral Rh-diene-based MOFs for asymmetric C–C formation reactions. The metalated MOFs catalyzed 1,4-addition of arylboronic acids to α,β-unsaturated ketones with a TON of 13400 and 1,2-addition of arylboronic acids to aldimines with excellent enantioselectivity (up to >99% ee). E2-MOF·Rh(acac) showed higher activity and enantioselectivity than the homogeneous control catalyst owing to the formation of a single-site catalyst in the MOF, and was reused for at least 7 times without loss of yield and ee. Our work thus establishes metalated diene-MOFs as highly active and enantioselective single-site solid catalysts for the construction of carbon–carbon bonds.
Acknowledgements
This work was supported by NSF (CHE-1464941). We thank C. Poon for help with ICP-MS analyses. Single crystal diffraction studies were performed at ChemMatCARS (Sector 15), APS, Argonne National Laboratory. ChemMatCARS is principally supported by the Divisions of Chemistry (CHE) and Materials Research (DMR), National Science Foundation, under grant number NSF/CHE-1346572. XAFS studies were performed at beamlines 9BM-B and 20BM-B at the APS. Use of the APS, an Office of Science User Facility operated for the U.S. DOE Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357.Notes and references
- (a) M. P. Suh, H. J. Park, T. K. Prasad and D.-W. Lim, Chem. Rev., 2012, 112, 782–835 CrossRef CAS PubMed; (b) M. Dincă and J. R. Long, Angew. Chem., Int. Ed., 2008, 47, 6766–6779 CrossRef PubMed; (c) J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670–4679 CrossRef CAS PubMed.
- (a) D. Liu, K. Lu, C. Poon and W. Lin, Inorg. Chem., 2014, 53, 1916–1924 CrossRef CAS PubMed; (b) L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed; (c) Y. Cui, Y. Yue, G. Qian and B. Chen, Chem. Rev., 2012, 112, 1126–1162 CrossRef CAS PubMed; (d) M. D. Allendorf, C. A. Bauer, R. K. Bhakta and R. J. T. Houk, Chem. Soc. Rev., 2009, 38, 1330–1352 RSC.
- W. Lin, W. J. Rieter and K. M. L. Taylor, Angew. Chem., Int. Ed., 2009, 48, 650–658 CrossRef CAS PubMed.
- (a) P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed; (b) J. D. Rocca, D. Liu and W. Lin, Acc. Chem. Res., 2011, 44, 957–968 CrossRef PubMed; (c) R. C. Huxford, J. D. Rocca and W. Lin, Curr. Opin. Chem. Biol., 2010, 14, 262–268 CrossRef CAS PubMed.
- (a) C. Wang, T. Zhang and W. Lin, Chem. Rev., 2012, 112, 1084–1104 CrossRef CAS PubMed; (b) O. R. Evans and W. Lin, Acc. Chem. Res., 2002, 35, 511–522 CrossRef CAS PubMed.
- (a) J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS; (b) M. Zhao, S. Ou and C.-D. Wu, Acc. Chem. Res., 2014, 47, 1199–1207 CrossRef CAS PubMed; (c) M. Yoon, R. Srirambalaji and K. Kim, Chem. Rev., 2012, 112, 1196–1231 CrossRef CAS PubMed; (d) L. Ma, C. Abney and W. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC; (e) J. Lee, O. K. Farha, J. Roberts, K. A. Scheidt, S. T. Nguyen and J. T. Hupp, Chem. Soc. Rev., 2009, 38, 1450–1459 RSC.
- (a) K. Manna, T. Zhang, F. X. Greene and W. Lin, J. Am. Chem. Soc., 2015, 137, 2665–2673 CrossRef CAS PubMed; (b) C. M. McGuirk, M. J. Katz, C. L. Stern, A. A. Sarjeant, J. T. Hupp, O. K. Farha and C. A. Mirkin, J. Am. Chem. Soc., 2015, 137, 919–925 CrossRef CAS PubMed; (c) H. Fei and S. M. Cohen, J. Am. Chem. Soc., 2015, 137, 2191–2194 CrossRef CAS PubMed; (d) M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed; (e) K. Manna, T. Zhang, M. Carboni, C. W. Abney and W. Lin, J. Am. Chem. Soc., 2014, 136, 13182–13185 CrossRef CAS PubMed; (f) K. Manna, T. Zhang and W. Lin, J. Am. Chem. Soc., 2014, 136, 6566–6569 CrossRef CAS PubMed; (g) H. Fei, J. Shin, Y. S. Meng, M. Adelhardt, J. Sutter, K. Meyer and S. M. Cohen, J. Am. Chem. Soc., 2014, 136, 4965–4973 CrossRef CAS PubMed; (h) J. M. Falkowski, T. Sawano, T. Zhang, G. Tsun, Y. Chen, J. V. Lockard and W. Lin, J. Am. Chem. Soc., 2014, 136, 5213–5216 CrossRef CAS PubMed; (i) K. Mo, Y. Yang and Y. Cui, J. Am. Chem. Soc., 2014, 136, 1746–1749 CrossRef CAS PubMed; (j) J. Canivet, S. Aguado, Y. Schuurman and D. Farrusseng, J. Am. Chem. Soc., 2013, 135, 4195 CrossRef CAS PubMed; (k) D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed; (l) M. H. Beyzavi, R. C. Klet, S. Tussupbayev, J. Borycz, N. A. Vermeulen, C. J. Cramer, J. F. Stoddart, J. T. Hupp and O. K. Farha, J. Am. Chem. Soc., 2014, 136, 15861–15864 CrossRef CAS PubMed; (m) D. T. Genna, A. G. Wong-Foy, A. J. Matzger and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 10586–10589 CrossRef CAS PubMed; (n) Y. Liu, S. Liu, S. Liu, D. Liang, S. Li, Q. Tang, X. Wang, J. Miao, Z. Shi and Z. Zheng, ChemCatChem, 2013, 5, 3086–3091 CrossRef CAS PubMed.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS PubMed.
- (a) C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS PubMed; (b) C.-D. Wu and W. Lin, Angew. Chem., Int. Ed., 2007, 46, 1075–1078 CrossRef CAS PubMed; (c) L. Ma, J. M. Falkowski, C. Abney and W. Lin, Nat. Chem., 2010, 2, 838–846 CrossRef CAS PubMed; (d) S. Regati, Y. He, M. Thimmaiah, P. Li, S. Xiang, B. Chen and J. C.-G. Zhao, Chem. Commun., 2013, 49, 9836–9838 RSC.
- (a) S.-H. Cho, B. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563–2565 RSC; (b) F. Song, C. Want, J. M. Falkowski, L. Ma and W. Lin, J. Am. Chem. Soc., 2010, 132, 15390–15398 CrossRef CAS PubMed; (c) F. Song, C. Wang and W. Lin, Chem. Commun., 2011, 47, 8256–8258 RSC; (d) A. M. Shultz, A. A. Sarjeant, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2011, 133, 13252–13255 CrossRef CAS PubMed; (e) J. M. Falkowski, C. Wang, S. Liu and W. Lin, Angew. Chem., Int. Ed., 2011, 50, 8674–8678 CrossRef CAS PubMed; (f) A. M. Shultz, O. K. Farha, D. Adhikari, A. A. Sarjeant, J. T. Hupp and S. T. Nguyen, Inorg. Chem., 2011, 50, 3174–3176 CrossRef CAS PubMed; (g) C. Zhu, G. Yuan, X. Chen, Z. Yang and Y. Cui, J. Am. Chem. Soc., 2012, 134, 8058–8061 CrossRef CAS PubMed.
- Chiral diene-based covalent organic framework was recently reported and used for rhodium-catalyzed 1,4-addition reaction at modest TONs and ee’s. J. Dong, Y. Liu and Y. Cui, Chem. Commun., 2014, 50, 14949–14952 RSC.
- T. Hayashi, K. Ueyama, N. Tokunaga and K. Yoshida, J. Am. Chem. Soc., 2003, 125, 11508–11509 CrossRef CAS PubMed.
- C. Fischer, C. Defieber, T. Suzuki and E. M. Carreira, J. Am. Chem. Soc., 2004, 126, 1628–1629 CrossRef CAS PubMed.
- C. Defieber, H. Grützmacher and E. M. Carreira, Angew. Chem., Int. Ed., 2008, 47, 4482–4502 CrossRef CAS PubMed.
- (a) R. Shintani, Y. Tsutsumi, M. Nagaosa, T. Nishimura and T. Hayashi, J. Am. Chem. Soc., 2009, 131, 13588–13589 CrossRef CAS PubMed; (b) T. Nishimura, J. Wang, M. Nagaosa, K. Okamoto, R. Shintani, F. Kwong, W. Yu, A. S. C. Chan and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 464–465 CrossRef CAS PubMed; (c) R. Shintani, M. Takeda, T. Nishimura and T. Hayashi, Angew. Chem., Int. Ed., 2010, 49, 3969–3971 CrossRef CAS PubMed; (d) T. Nishimura, Y. Takiguchi and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 9086–9089 CrossRef CAS PubMed.
- (a) N. Tokunaga, Y. Otomaru, K. Okamoto, K. Ueyama, R. Shintani and T. Hayashi, J. Am. Chem. Soc., 2004, 126, 13584–13585 CrossRef CAS PubMed; (b) R. Shintani, M. Takeda, T. Tsuji and T. Hayashi, J. Am. Chem. Soc., 2010, 132, 13168–13169 CrossRef CAS PubMed; (c) Z. Cui, H.-J. Yu, R.-F. Yang, W.-Y. Gao, C.-G. Feng and G.-Q. Lin, J. Am. Chem. Soc., 2011, 133, 12394–12397 CrossRef CAS PubMed; (d) T. Nishimura, A. Noishiki, G. C. Tsui and T. Hayashi, J. Am. Chem. Soc., 2012, 134, 5056–5059 CrossRef CAS PubMed; (e) Y. Luo, H. B. Hepburn, N. Chotsaeng and H. W. Lam, Angew. Chem., Int. Ed., 2012, 51, 8309–8313 CrossRef CAS PubMed; (f) T. Nishimura, A. Noishiki, Y. Ebe and T. Hayashi, Angew. Chem., Int. Ed., 2013, 52, 1777–1780 CrossRef CAS PubMed.
- (a) J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed; (b) M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632–6640 CrossRef CAS.
- (a) K. Okamoto, T. Hayashi and V. H. Rawal, Chem. Commun., 2009, 4815–4817 RSC; (b) G. Pattison, G. Piraux and H. W. Lam, J. Am. Chem. Soc., 2010, 132, 14373–14375 CrossRef CAS PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 217–225 CrossRef PubMed.
- (a) P. V. Dau and S. M. Cohen, Inorg. Chem., 2015, 54, 3134–3138 CrossRef CAS PubMed; (b) Y.-R. Lee, Y.-M. Chung and W.-S. Ahn, RSC Adv., 2014, 4, 23064–23067 RSC; (c) Y. Yang, H.-F. Yao, F.-G. Xi and E.-Q. Gao, J. Mol. Catal. A: Chem., 2014, 390, 198–205 CrossRef CAS PubMed; (d) T. Toyao, M. Fujiwaki, Y. Horiuchi and M. Matsuoka, RSC Adv., 2013, 3, 21582–21587 RSC; (e) B. Li, Y. Zhang, D. Ma, L. Li, G. Li, G. Li, Z. Shi and S. Feng, Chem. Commun., 2012, 48, 6151–6153 RSC; (f) P. Wu, J. Wang, Y. Li, C. He, Z. Xie and C. Duan, Adv. Funct. Mater., 2011, 21, 2788–2794 CrossRef CAS PubMed; (g) R. Srirambalaji, S. Hong, R. Natarajan, M. Yoon, R. Hota, Y. Kim, Y. Ho Ko and K. Kim, Chem. Commun., 2012, 48, 11650–11652 RSC; (h) F. Vermoortele, R. Ameloot, A. Vimont, C. Serre and D. de Vos, Chem. Commun., 2011, 47, 1521–1523 RSC.
- The 1,2-addition reaction did not proceed with E2-MOF·RhCl.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental section; synthesis and characterization of ligand LH2 and E2-MOF; XAFS experiments and analyses; MOF-catalyzed asymmetric additions of arylboronic acids to α,β-unsaturated ketones and aldimines. CCDC 1064133. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc02100f |
| This journal is © The Royal Society of Chemistry 2015 |