
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA T-shaped Ni[κ2-(CF2)4–] NHC complex: unusual Csp3–F and M–CF bond functionalization reactions†
Nicholas O.
Andrella
,
Alexandre J.
Sicard
,
Serge I.
Gorelsky
,
Ilia
Korobkov
and
R. Tom
Baker
*
Department of Chemistry and Centre for Catalysis Research and Innovation(CCRI), University of Ottawa, 30 Marie Curie, Ottawa, ON K1N 6N5 Canada
First published on 6th August 2015
Abstract
A T-shaped octafluoronickelacyclopentane–NHC complex is prepared and characterized. While the solid-state structure includes a weak isopropyl-CH3 agostic interaction, the reactivity of this complex with Lewis- and Brønsted acids is clearly enhanced by its low coordination number. Reaction with Me3SiOTf, for example, yielded a rare metal–heptafluorocyclobutyl complex whereas carboxylic acids gave substitution at the α-carbon and/or Ni–CF bond protonolysis to afford thermally robust 4H-octafluorobutyl Ni complexes.
Introduction
Fluorocarbons and their derivatives are valuable as refrigerants, agrochemicals, unique solvents/surfactants and fluoropharmaceuticals, with annual sales of the latter alone in the billions of dollars.1 As the demand for fluorinated chemicals has increased, so too have synthetic methods for introducing fluorine and fluorocarbon groups.2–4 Despite recent advances, transition metal-mediated/-catalyzed routes are rare in comparison to the well-developed organometallic chemistry of hydrocarbons.5 The challenge rests in the stability of metal–perfluoroalkyl (M–RF) bonds, relative to metal–alkyl bonds.6 M–CF bonds are typically inert to processes such as insertion/alkyl migration reactions, vital to metal-mediated catalytic cycles.7 Moreover, C–F bonds are stronger than C–H bonds,1a,b posing another obstacle to metal-based approaches.We are investigating perfluoronickelacyclopentane complexes (PNCPs) as platforms for functionalized fluorocarbons with an initial focus on fundamental stoichiometric reactions. PNCPs have been synthesized previously by reaction of tetrafluoroethylene (TFE, CF2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CF2) with Ni0 complexes. The displacement of P ligands by bidentate ligands has also been reported (Scheme 1).8
CF2) with Ni0 complexes. The displacement of P ligands by bidentate ligands has also been reported (Scheme 1).8
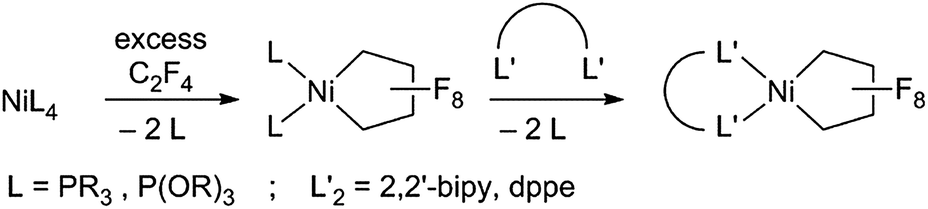 | ||
| Scheme 1 Synthesis of perfluoronickelacyclopentanes. | ||
To date, reports concerning the reactivity of PNCPs are sparse: Burch and co-workers found that Lewis acidic BF3 effects fluoride abstraction from Cα and phosphine migration to the activated carbon (Scheme 2a).9 Extending this reaction to the unsymmetrical P^S chelate, we showed that treatment with excess isonitrile effected cleavage of the Ni–CF bond (Scheme 2b).10 With phosphite co-ligands, a remarkable hydrogenolysis reaction enables the selective catalytic hydrodimerization of TFE (Scheme 2c).11 As far as we know, this reaction is the only example of a perfluoro-metallacyclopentane participating in a catalytic cycle.12
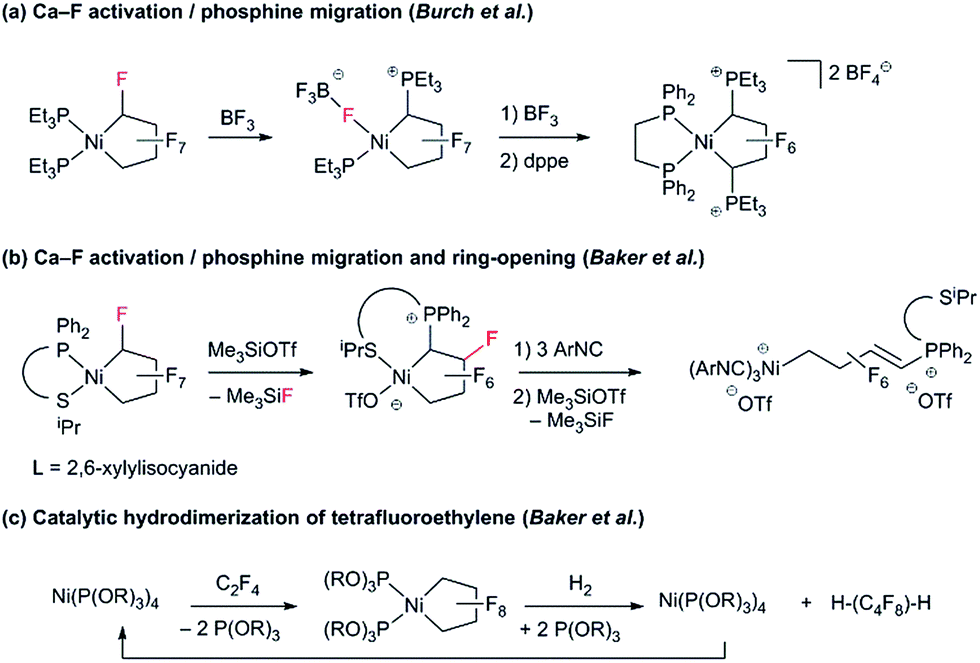 | ||
| Scheme 2 Previously reported reactivity of perfluoronickelacyclopentanes. | ||
Our approach to metallacycle functionalization hinges on the reactivity of metal-activated Cα–F bonds13 wherein we replace a C–F bond by C–Nu vs. the current paradigm C–L (Nu = nucleophile, L = ligand). Using N-heterocyclic carbenes (NHCs),14 we aimed to access low-coordinate PNCPs wherein the strong M–CNHC bond may also prevent ligand migration to Cα. There is considerable precedent for such an approach to low-coordinate metal complexes.15 Hillhouse and coworkers prepared a two-coordinate nickel–imido complex bearing the exceptionally bulky IPr* ligand (analog of IPr with 2,6-bis(diphenyl-methyl)phenyl groups instead of 2,6-diisopropylphenyl).16 Similarly, Miyazaki and coworkers synthesized a T-shaped three-coordinate nickel(I) chloride species [Ni(IPr)2Cl] by treatment of two-coordinate [Ni(IPr)2] with aryl chlorides.17 Also, Hartwig et al. synthesized a low-valent, three-coordinate palladium(II) norbornyl species [Pd(SIPr)(NHAr)(Nor)], which underwent facile C–N bond reductive elimination when heated.18
In this report we show that low-coordinate NHC Ni perfluorometallacycles undergo facile Csp3–F and M–CF bond cleavage as well as Cα-functionalization.9 We also demonstrate the first migration of a fluoroalkyl to a reactive carbon center. This is significantly different from the reactivity previously observed for phosphine Ni perfluorometallacycle complexes.9
Results and discussion
Starting from bis(phosphite) PNCPs10 (1a and b) we were able to cleanly synthesize coordinatively-saturated or -unsaturated nickel perfluorometallacycles. Thus, 1a reacts smoothly with 1 equiv. of ItBu (ItBu = 1,3-di-tert-butylimidazol-2-ylidene) to afford the NHC/phosphite product 2 (Scheme 3, top; X-ray structural characterization presented in ESI†).19 Significantly, the reaction between the larger SIPr ligand [SIPr = 1,3-bis(2,6-diisopropylphenyl)imidazolin-2-ylidene] and a nickel metallacycle with sterically-demanding co-ligands (1b) results in displacement of both phosphite ligands, yielding the pseudo-three-coordinate/14e-Ni(II) metallacycle 3 (Scheme 3, bottom).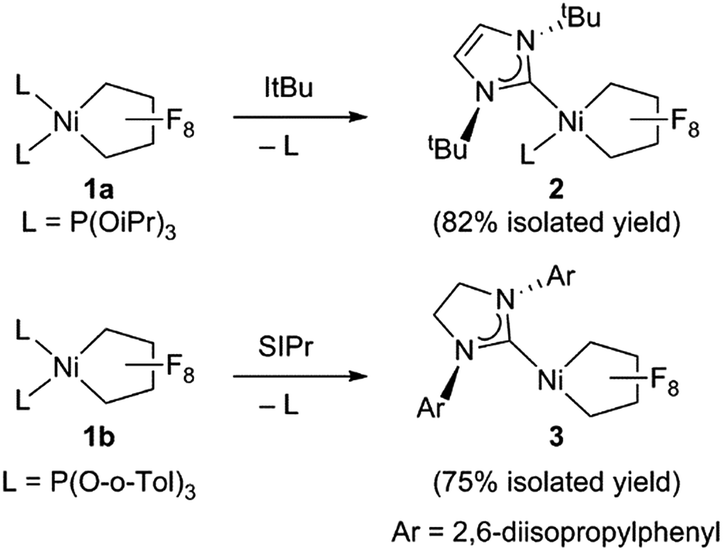 | ||
| Scheme 3 Synthesis of NHC perfluoronickelacyclopentane complexes. | ||
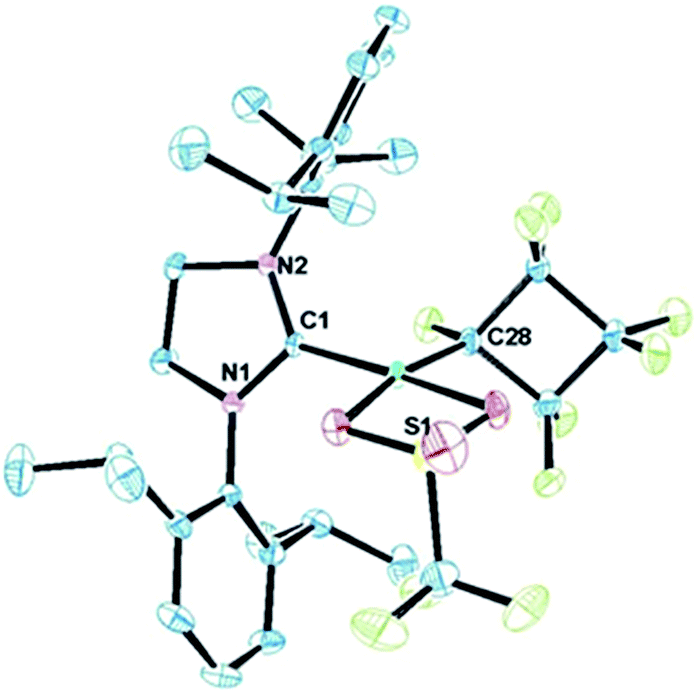
The molecular structure of complex 3, as determined by single crystal X-ray diffraction, exhibits a T-shaped coordination about the Ni and features a weak agostic interaction with the isopropyl methyl group (Ni–C = 2.757(1) Å; compare Ni–CF bond distance trans to the NHC (1.934(1) Å) with that trans to the agostic interaction (1.875(1) Å) (Fig. 1a). The 19F NMR spectrum of 3 in C6D6 is consistent with ideal C2v symmetry at room temperature, with only two distinct singlet resonances at −101.9 (Fα) and −138.6 ppm (Fβ). While these resonances both broaden significantly at 213 K, it is apparent that the T-flip interconversion encounters only a small energy barrier.20 To confirm this, we carried out DFT calculations (at the B3LYP/TZVP level with and without the empirical dispersion correction of Grimme).21 Intriguingly, the calculations reveal two spin-singlet structures with a very small energy difference (ΔG298 K = 0.0–1.5 kcal mol−1). The first computed structure coincides well with the observed solid-state structure of 3; the 3-center bond order index between the Ni and the corresponding C–H bond of 0.05 is much less than 8/27 (∼0.3), the maximum possible value for a 3-center 2-electron bond. As a result, the Mayer valence index for Ni in structure 3 is only 3.09. The second structure (3′) features a weak η3 interaction between the aryl group of the NHC ligand at the 4th coordination site of the Ni atom (Fig. 1b). Mayer bond orders for the corresponding three Ni–C interactions are in the 0.02–0.05 range, with a total bond order of 0.09. This suggests a semi-bidentate binding mode for the class of NHC ligands possessing pendant aryl groups. From calculations with the dispersion correction, structure 3′ has the same Gibbs free energy as 3. Without the dispersion correction, structure 3′ is actually 1.5 kcal mol−1 lower in energy than 3. The 3-coordinate structure with trigonal coordination around Ni and symmetric binding of the C4F8 ligand is a transition state with a low energy (ΔGǂ298 K = 2.1 kcal mol−1 relative to 3). Thus, it is clear that cleavage of the weak agostic and/or η3-aryl bond is facile and allows for rapid reorientation of the ligands around the Ni center. Attempts to obtain evidence for structure 3′ by low temperature NMR were frustrated by dynamic processes associated with the T-flip and hindered rotations about the M–C and perhaps N–C bonds.
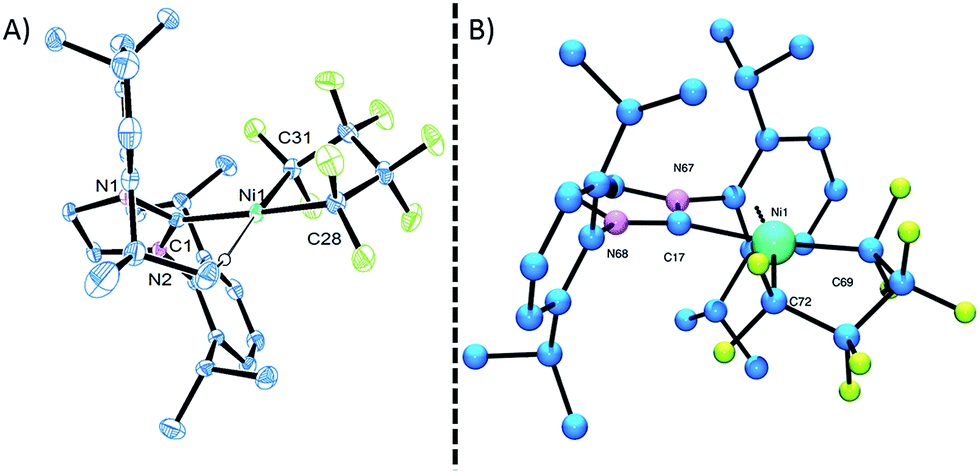 | ||
| Fig. 1 (A) ORTEP representation of the molecular structure of 3. Thermal-ellipsoid probabilities are set to 35% with hydrogen atoms omitted for clarity. The Ni–C(1) distance is 1.854(2) Å. (B) Optimized structure of low energy Ni–aryl isomer 3′; Ni–Caryl distances are = 2.818, 3.329, 3.379, 4.166, 4.204, 4.543 Å. The Ni–C(17) distance is 1.989 Å. | ||
The HOMO of 3 (ε = −6.01 eV; Fig. 2, left) is localized on the Ni (87%), primarily from a dz2 orbital contribution (71%). Lower-lying orbitals display interactions between metal dxz, dyz orbitals and the π-system of the aryl group.22 The LUMO (ε = −1.96 eV; Fig. 3, right) is an anti-bonding combination of the metal dx2−y2 orbital (total Ni character of 45%) with the π-donor orbitals of the NHC and C4F8 ligands. Thus, reactivity of the M–C bond is likely under orbital control and arises from an interaction with the HOMO of 3. In contrast, C–F bond activation is likely a combination of orbital and charge control with a slant towards the latter as the hardness of the Lewis acid increases.23
 | ||
| Fig. 2 The HOMO (left) and LUMO (right) of 3. Isosurface values of 0.04 au are used. | ||
 | ||
| Fig. 3 ORTEP representation of the molecular structure of 4a with thermal ellipsoid probabilities set to 30% and hydrogen atoms omitted for clarity. The Ni–C(1) distance is 1.854(2) Å. | ||
Initial studies on the C–F bond activation reactions of 3 are promising in the context of synthesizing functionalized fluoro-carbons by metal-mediated approaches. Firstly, when 3 is treated with the Lewis acid TMSOTf (TMS = Me3Si, OTf = SO3CF3), α-fluoride-abstraction, accompanied by Ni–CF bond cleavage and CF–CF bond formation, furnishes a rare perfluorocyclobutyl complex 4a (Scheme 4, 75% isolated yield).24 The driving force behind this transformation is likely related to the triflate leaving group ability and the formation of a strong C–C bond.25 Importantly, the NHC remains bound to the nickel atom (i.e., does not migrate to Cα), potentially opening new pathways to functionalized fluorocarbon derivatives. Upon heating complex 4a (80 °C in C6D6, 24 h), perfluorocyclobutene is produced, presumably via a β-fluoride elimination mechanism, although the metal-containing co-product(s) have not yet been identified.26 Interestingly, a single OTf containing product can be discerned by 19F NMR (−93.37 ppm) but a Ni–F signal could not be located. The 1H NMR shows that the NHC remains intact. Upon addition of PPh3 to the reaction mixture, PPh3F2 was identified as a major product, suggesting formation of a Ni–F thermolysis co-product.
 | ||
| Scheme 4 Synthesis and decomposition of 4a. | ||
The distorted square planar structure of complex 4a (Fig. 3) features a bidentate triflate ligand which can also likely access the κ1-mode in solution as evidenced by the simple 19F NMR spectrum27 and observed tendency to eliminate. The perfluorocyclobutyl ring is nearly planar, the Ni–C bond is short [1.890(2) Å] due to the weak σ-trans influence ligand, and the Cα–F bond distance (1.384(3) Å) is considerably longer than the other C–F bonds (average of 1.33 Å).
The reactivity enhancement offered by low-coordinate 3 is evidenced by the sluggish reaction of 4-coordinate complex 2 with TMSOTf to give a mixture of unidentified products. Indeed, monitoring the reaction of 3 and TMSOTf at −25 °C allowed for the identification of a Ni–C4F7 intermediate 5a apparently containing a Cα–OTf linkage (triflate CF319F NMR resonance is coupled to Cα–F: 5JFF = 11 Hz). This is in contrast to previous suggestions of a metal fluorocarbene intermediate (Scheme 5).9
 | ||
| Scheme 5 Possible intermediates in the reaction of 3 with TMSOTf. | ||
Having established that the NHC ligand remains bound to the metal upon fluoride-abstraction from 3, we shifted our focus to C–F bond functionalization using Brønsted acids. Treatment of 3 with trifluoroacetic acid [TFA; pKa = 3.4 (DMSO)]28a gives HF and the more stable (vs.5a) trifluoroacetate-substituted metallacycle 5b that could be characterized spectroscopically at room temperature (Scheme 6). Nonetheless, accompanying formation of perfluoro-cyclobutene, presumably formed via an analogous structure to 4a, led us to move to weaker Brønsted acids. Remarkably, reaction of 3 with acetic acid [pKa = 12 (DMSO)]28b (Scheme 6, bottom) yielded the stable ester metallacycle 5c (30% isolated yield) as well as the Ni–CF bond cleavage product 6a in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio. At a similar acidity [pKa = 11 (DMSO)]28c but increased steric bulk, 2,4,6-trimethyl-benzoic acid gave a 10:1 mixture favouring the ring cleavage product, 6b.
:1 ratio. At a similar acidity [pKa = 11 (DMSO)]28c but increased steric bulk, 2,4,6-trimethyl-benzoic acid gave a 10:1 mixture favouring the ring cleavage product, 6b.
The molecular structure of 5c features similar Ni–C bond distances (1.895(7) vs. 1.896(6) Å) and a distorted square planar coordination (Fig. 4). The functionalized heptafluoro-metallacyclopentane ring is puckered with the smallest C–C bond distance being C(32)α–C(39)β [1.49(1) Å]. The carbonyl oxygen completes the nickel coordination sphere (Ni–O1 = 1.969(4) Å).
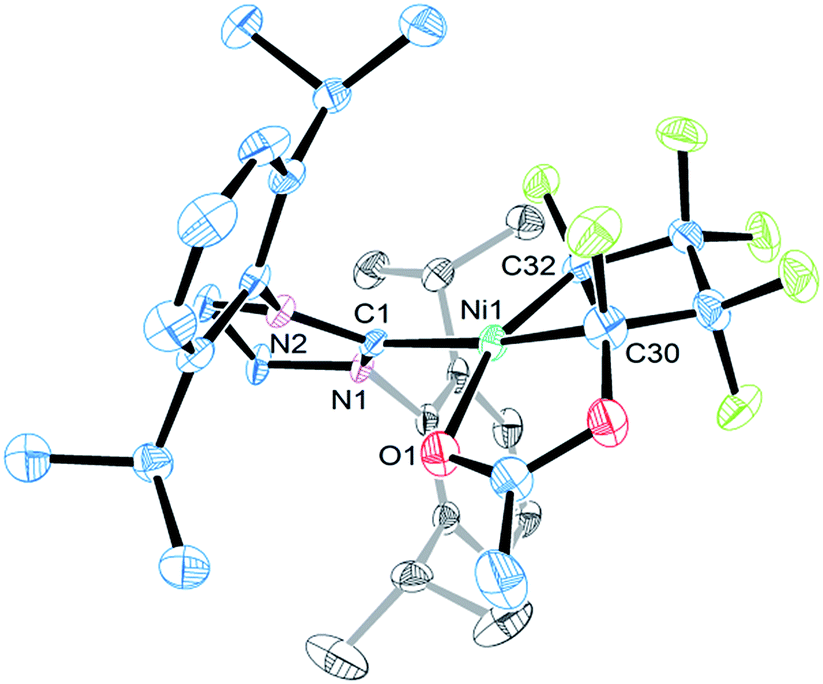 | ||
| Fig. 4 ORTEP representation of the molecular structure of 5c with thermal ellipsoid probabilities set to 30% and hydrogen atoms omitted for clarity. The Ni–C(1) distance is 1.928(2) Å. | ||
The 19F NMR spectra of 5a–d are very similar and support our original proposal for the low temperature intermediate 5a in the reaction of 3 with TMSOTf. The Cα–F 19F chemical shifts of the functionalized carbon, (−117.2 and −119.2 ppm) can be compared with those of the phosphonium analogs (−115.6 and −117.8 ppm) shown in Scheme 2.
As expected, the ring-opened products 6a and b display nearly identical 19F NMR chemical shift patterns with the Cγ– and Cδ–F resonances distinguished by F–H coupling of 6 and 52 Hz, respectively. These unique complexes have been identified as their potassium cation adducts using ESI-MS (747.2 g mol−1 and 851.4 g mol−1 respectively) and are surprisingly inert to thermolysis at 80 °C in C6D6 for 20 h.
Considering the importance of esters as synthons in organic transformations29 this C–O bond-forming reaction 3 → 5 is very appealing from the standpoint of synthesizing functionalized fluorocarbons. As such, understanding competing pathways for M–C vs. C–F bond cleavage would be valuable.30 Viable reaction pathways can be considered as proceeding via either 5- or 6-membered transition states (Scheme 7). The selective HF elimination observed for TFA, is eroded as Ni–C bond protonolysis (orbital control) competes using acids of intermediate acidity (e.g. pKa ∼11).31 With the bulkier trimethylbenzoic acid, kinetic acidity factors in the tighter 5-membered ring transition state could severely limit HF elimination.32
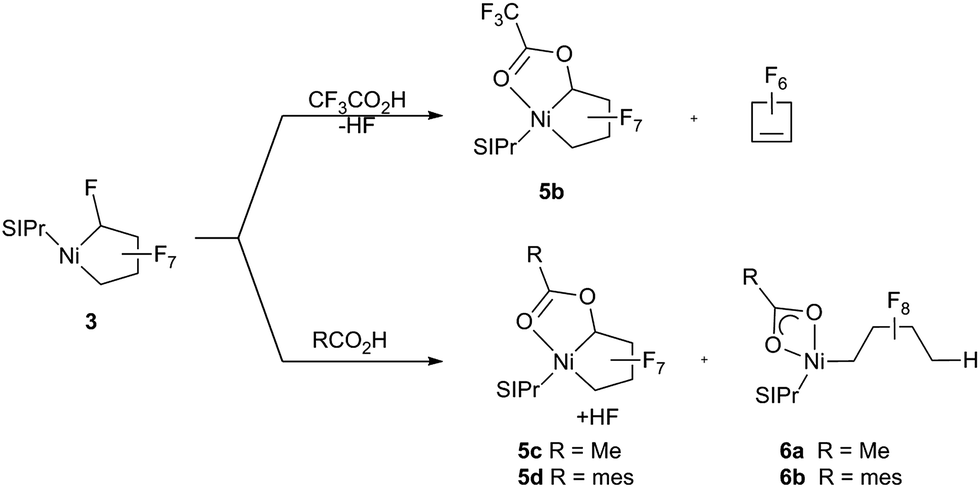 | ||
| Scheme 6 Reaction of 3 with trifluoroacetic, acetic and 2,4,6-trimethylbenzoic acids. | ||
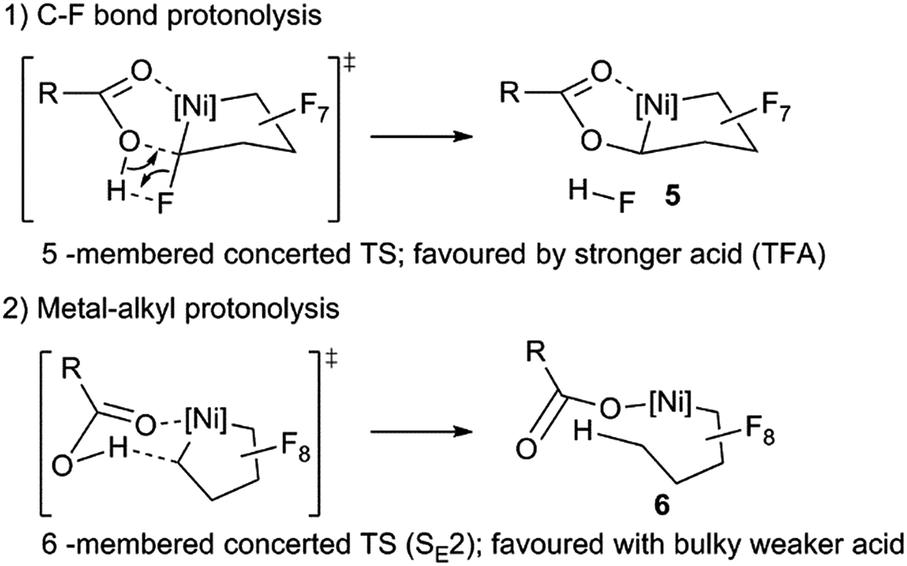 | ||
| Scheme 7 Proposed reaction pathways for C–F activation vs. Ni–RF protonolysis. | ||
Conclusions
In summary, we have prepared the first NHC–perfluorometallacyclopentane complexes and exploited the bulky SIPr ligand to stabilize a pseudo-three-coordinate nickelacycle, 3. Importantly, 3 undergoes Cα–F abstraction reactions without migration of the NHC ligand. Instead, we see an unprecedented migration of the fluoroalkyl to the reactive carbon center, giving rise to the novel perfluorocyclobutyl complex via Ni–CF bond cleavage. More importantly, the low-coordinate nature of 3 allows for ring functionalization. Strong acids favour selective Cα functionalization, but the resulting products are unstable with respect to competing metallacycle ring contraction and elimination of perfluorocyclobutene. With less acidic reagents stable ring-functionalized products are formed but a competing Ni–CF bond cleavage pathway comes into play and dominates for bulkier carboxylic acids. These are the first examples of selective functionalization of a PNCP and synthesis of thermally stable Ni–C4F8H complexes. These results are encouraging in the context of developing metallacycle-based routes to functionalized fluorocarbons.Ongoing work is focused on (a) expanding the scope of ring functionalization substrates suitable for reactions with 3 and (b) reductive (see Scheme 2, above) and oxidative approaches for removing the functionalized fluorocarbon fragments from the metal. Preliminary results of the hydrogenolysis of compound 3 indicate enhanced reactivity towards H2 (i.e., at 7 psig and 25 °C) vs. reported 4-coordinate phosphite variants.11a However, loss of selectivity33 is observed with the synthesis of two distinct products. Full details of these results will be published in due time.
Acknowledgements
We thank the NSERC and the Canada Research Chairs program for generous financial support and the University of Ottawa, Foundation for Innovation and Ontario Ministry of Economic Development and Innovation for essential infrastructure. N. O. A. gratefully acknowledges support from the province of Ontario, NSERC and the University of Ottawa (OGS and CGS-M/D).Notes and references
- (a) R. D. Chambers, Fluorine in Organic Chemistry, Blackwell, Oxford, 2004 Search PubMed; (b) P. Kirsch, Modern Fluoroorganic Chemistry: Synthesis, Reactivity, Applications, Wiley-VCH, Weinheim, 2 edn, 2013 Search PubMed; (c) S. Purser, P. R. Moore, S. Swallow and V. Gouverneur, Chem. Soc. Rev., 2008, 37, 320 RSC; (d) I. Ojima, Fluorine in Medicinal Chemistry and Chemical Biology, John Wiley & Sons, Ltd, Chichester, 2009 Search PubMed.
- Stoichiometric [Cu]–CF3 reagents for trifluoromethylation of organic substrates, particularly aryl halides: (a) H. Morimoto, T. Tsubogo, N. D. Litvinas and J. F. Hartwig, Angew. Chem., Int. Ed., 2011, 50, 3793 CrossRef CAS PubMed; (b) G. G. Dubinina, H. Furutachi and D. A. Vicic, J. Am. Chem. Soc., 2008, 130, 8600 CrossRef CAS PubMed; (c) D. M. Wiemers and D. J. Burton, J. Am. Chem. Soc., 1986, 108, 832 CrossRef CAS.
- Metal-catalyzed C–F and C–CF3 bond formation: (a) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470 CrossRef CAS PubMed; (b) E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1678 Search PubMed; (c) V. V. Grushin, Acc. Chem. Res., 2010, 43, 160 CrossRef CAS PubMed; (d) J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2009, 48, 8610 CrossRef CAS PubMed; (e) K. L. Hull, W. Q. Anani and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 7134 CrossRef CAS PubMed.
- Non-metal based approaches to introducing fluorocarbon fragments to organic substrates: (a) F. Wang, T. Luo, J.-B. Hu, Y. Wang, H. S. Krishnan, P. V. Jog, S. K. Ganesh, G. K. S. Prakash and G. A. Olah, Angew. Chem., Int. Ed., 2011, 50, 7153 CrossRef CAS PubMed (CF2 addition to alkenes); (b) R. P. Singh, G. Cao, R. L. Kirchmeier and J. M. Shreeve, J. Org. Chem., 1999, 64, 2873 CrossRef CAS PubMed . (Me3Si–CF3 addition to carbonyl groups).
- (a) H.-J. Arpe, Industrial Organic Chemistry, Wiley-VCH, Weinheim, 5th edn, 2010 Search PubMed; (b) Applied Homogeneous Catalysis with Organometallic Compounds, ed. B. Cornils and W. A. Herrmann, Wiley-VCH, New York, 1996 Search PubMed.
- (a) F. L. Taw, A. E. Clark, A. H. Mueller, M. T. Janicke, T. Cantat, B. L. Scott, P. J. Hay, R. P. Hughes and J. L. Kiplinger, Organometallics, 2012, 31, 1484 CrossRef CAS; (b) R. P. Hughes, Adv. Organomet. Chem., 1990, 31, 183 CrossRef CAS; (c) J. A. Morrison, Advances in Organometallic Chemistry: Trifluoromethyl-Containing Transition Metal Complexes, Academic Press, 1993, vol. 35 Search PubMed; (d) P. J. Brothers and W. R. Roper, Chem. Rev., 1988, 88, 1293 CrossRef CAS.
- [Mn]–CF3 inert to CO migratory-insertion: S. K. Shin and J. L. Beauchamps, J. Am. Chem. Soc., 1990, 112, 2057 CrossRef CAS.
- (a) M. Ohashi, M. Shibata, H. Saijo, T. Kambara and S. Ogoshi, Organometallics, 2013, 32, 3631 CrossRef CAS; (b) W. Gasafi-Martin, G. Oberendfellner and K. von Werner, Can. J. Chem., 1996, 74, 1922 CrossRef CAS; (c) M. Green, A. Laguna, J. L. Spencer and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1977, 1010 RSC; (d) J. Browning, H. D. Empsall, M. Green and F. G. A. Stone, J. Chem. Soc., Dalton Trans., 1973, 381 RSC; (e) C. S. Cundy, M. Green and F. G. A. Stone, J. Chem. Soc. A, 1970, 1647 RSC; (f) M. Green, R. B. L. Osborn, A. J. Rest and F. G. A Stone, J. Chem. Soc. A, 1968, 2525 RSC; (g) P. M. Treichel and F. G. A. Stone, Adv. Organomet. Chem., 1964, 1, 143 CrossRef CAS.
- R. R. Burch, J. C. Calabrese and S. D. Ittel, Organometallics, 1988, 7, 1642 CrossRef CAS.
- K. A. Giffin, D. J. Harrison, I. Korobkov and R. T. Baker, Organometallics, 2013, 32, 7424 CrossRef CAS.
- (a) R. T. Baker, R. P. Beatty, W. B. Farnham and R. L. Wallace Jr, US Pat., 5670679, 1997; (b) Generation of perfluorocyclobutene or perfluorocyclobutane from an iron perfluorometallacyclopentane complex, under thermal or thermal/oxidizing(Br2) conditions, respectively, T. A. Manuel, S. L. Stafford and F. G. A. Stone, J. Am. Chem. Soc., 1961, 83, 249 CrossRef CAS.
- M. Ohashi and S. Ogoshi, Catalytic Transformations of Fluorinated Olefins. in Topics in Organometallic Chemistry, Springer, Switzerland, 2014 Search PubMed.
- Activation of C–F bonds in [M]–RF complexes: (a) R. P. Hughes, J. Fluorine Chem., 2010, 131, 1059 CrossRef CAS PubMed; (b) J. Goodman, V. V. Grushin, R. B. Larichev, S. A. Macgregor, W. J. Marshall and D. C. Roe, J. Am. Chem. Soc., 2009, 131, 4236 CrossRef CAS PubMed; (c) S. A. Garratt, R. P. Hughes, I. Kovacik, A. J. Ward, S. Willemsen and D. Zhang, J. Am. Chem. Soc., 2005, 127, 15585 CrossRef CAS PubMed; (d) H. Torrens, Coord. Chem. Rev., 2005, 249, 1957 CrossRef CAS PubMed; (e) D. Huang, P. R. Koren, K. Folting, E. R. Davidson and K. G. Caulton, J. Am. Chem. Soc., 2000, 122, 8916 CrossRef CAS; (f) T. G. Richmond and D. F. Shriver, Organometallics, 1984, 3, 314 CrossRef CAS; (g) T. G. Richmond and D. F. Shriver, Organometallics, 1983, 2, 1061 CrossRef CAS , see also ref. 8a.
- (a) R. H. Crabtree, J. Organomet. Chem., 2005, 690, 5451 CrossRef CAS PubMed; (b) D. J. D. Wilson, S. A. Couchman and J. L. Dutton, Inorg. Chem., 2012, 51, 7657 CrossRef CAS PubMed.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC.
- C. A. Laskowsky, A. J. Miller, G. L. Hillhouse and T. R. Cundari, J. Am. Chem. Soc., 2011, 133, 771 CrossRef PubMed.
- S. Miyazaki, Y. Koga, T. Matsumoto and K. Matsubara, Chem. Commun., 2010, 46, 1932 RSC.
- P. S. Hanley, S. L. Marquard, T. R. Cundari and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 15281 CrossRef CAS PubMed.
- Ni–RF bond lengths for structure 2 (see ESI†): 1.956(1) Å (trans to P); 1.945(1) Å (trans to C). Ni–C(NHC) bond length for 2: 1.945(1) Å.
- See ESI page S14 for low temperature NMR spectra.†.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787 CrossRef CAS PubMed.
- See ESI page S29.†.
- N. Islam and D. C. Gosh, Theoretical and Computational Research in the 21st century, ed. N. Islam, CRC Press, Boca Raton, 2014, ch. 1, pp. 1–58 Search PubMed.
- To the best of our knowledge, the only previously reported metal perfluorocyclobutyl complexes are: (a) B. L. Dyatkin, B. I. Martynov, L. G. Martynova, N. G. Kizim, S. R. Sterlin, Z. A. Stumbrevichute and L. A. Federov, J. Organomet. Chem., 1973, 57, 423 CrossRef CAS; (b) B. L. Dyatkin, S. R. Sterlin, B. I. Martynov, E. I. Mysov and I. L. Knunyants, Tetrahedron, 1971, 27, 2843 CrossRef CAS.
- (a) The leaving group ability of OTf, R. K. Crossland, W. E. Wells and V. J. Shiner, J. Am. Chem. Soc., 1971, 93, 4217 CrossRef CAS.
- (a) T. Miura, Y. Ito and M. Murakami, Chem. Lett., 2008, 37, 1006 CrossRef CAS; (b) C. J. Bourgeois, R. P. Hughes, J. Yuan, A. G. DiPasquale and A. L. Rheingold, Organometallics, 2006, 25, 2908 CrossRef CAS.
- For NMR spectra of compound 4a see pages S21–S22.†.
- (a) M. H. Haindl, M. B. Schmid, K. Zeitler and R. M. Gschwind, RSC Adv., 2012, 2, 5941 RSC; (b) T. Cohen, D. A. Bennett and A. J. Mura Jr, J. Org. Chem., 1976, 41, 2507 CrossRef; (c) J. Jover, R. Bosque and J. Sales, QSAR Comb. Sci., 2008, 27, 563 CrossRef CAS PubMed.
- W. Riemenschneider and H. M. Bolt, Esters, Organic, in Ullman's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2005 Search PubMed.
- Previous work showed protonation of C–F bond instead of Ir–CH3 using a strong acid: R. P. Hughes, D. Zhang, L. N. Zakharov and A. L. Rheingold, Organometallics, 2002, 21, 4902 CrossRef CAS.
- E. W. Kalberer, J. F. Houlis and D. M. Roddick, Organometallics, 2004, 23, 4112–4115 CrossRef CAS.
- Similar reactivity was previously observed for 3-membered perfluorinated metallacycles: W. Xu, H. Sun, Z. Xiong and X. Li, Organometallics, 2013, 32, 7122 CrossRef CAS.
- R. P. Hughes and J. M. Smith, J. Am. Chem. Soc., 1999, 121, 6084 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, NMR spectra and X-ray crystallographic information. CCDC files 968465 (2), 968466 (3), 968467 (4a), 1028645 (5c) and 1412522 (3·H2O); For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01886b |
| This journal is © The Royal Society of Chemistry 2015 |