
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
RhI/RhIII catalyst-controlled divergent aryl/heteroaryl C–H bond functionalization of picolinamides with alkynes†
Ángel Manu
Martínez
,
Javier
Echavarren
,
Inés
Alonso
 ,
Nuria
Rodríguez
*,
Ramón
Gómez Arrayás
* and
Juan C.
Carretero
*
,
Nuria
Rodríguez
*,
Ramón
Gómez Arrayás
* and
Juan C.
Carretero
*
Universidad Autónoma de Madrid (UAM), Cantoblanco 28049, Madrid, Spain. E-mail: n.rodriguez@uam.es; ramon.gomez@uam.es; juancarlos.carretero@uam.es
First published on 29th June 2015
Abstract
The ability to establish switchable site-selectivity through catalyst control in the direct functionalization of molecules that contain distinct C–H bonds remains a demanding challenge that would enable the construction of diverse scaffolds from the same starting materials. Herein we describe the realization of this goal, namely a divergent heteroaryl/aryl C–H functionalization of aromatic picolinamide derivatives, targeting two distinct C–H sites, either at the pyridine ring or at the arene unit, to afford isoquinoline or ortho-olefinated benzylamine (or phenethylamine) derivatives. This complementary reactivity has been achieved on the basis of a RhIII/RhI switch in the catalyst, resulting in different mechanistic outcomes. Notably, a series of experimental and DFT mechanistic studies revealed important insights about the mechanism of the reaction and reasons behind the divergent regiochemical outcome.
Introduction
The great potential of metal-catalyzed C–H bond functionalization to streamline synthetic schemes has been illustrated with many elegant methods featuring exquisite and predictable site-selectivity in the presence of multiple reactive C–H bonds.1 However, despite the fast-paced development of this field, the discovery of procedures capable of divergent functionalization at distinct C–H sites through catalyst control is relatively uncommon,2 yet highly appealing. In particular, achieving distinctive positional reactivities by simply varying the ligand environment and oxidation state of the catalytically active metal species could provide a unique opportunity for the construction of diverse scaffolds from the same starting materials.Rh-catalyzed coupling reactions of alkynes involving C–H cyclometalation/annulation of (hetero)arenes provide an atom- and step-economical route to heterocycles, ubiquitous structural elements in nature, medicinal chemistry and material science.3–7 Also, the use of alkynes as coupling partners allows access to aromatic compounds with a pendant ortho-vinyl group,6 that could serve as a versatile synthetic handle. In both contexts, rhodium(III)-catalysts, most often introduced as Cp*RhIIILn precursors in combination with the classical CuI/CuII redox couple, have proven to be particularly useful.3–5 However, in contrast to the tremendous strides made with functionalized arenes,3–6 there are few methods for the RhIII-catalyzed C–H activation of electron-deficient aza-heterocycles containing a basic nitrogen such as pyridine.7 This deficiency is somewhat surprising given that nitrogen-containing heterocyclic compounds are privileged structures in medicinal chemistry.
We envisaged that N-benzyl-2-picolinamides would provide an opportunity for developing a divergent C–H functionalization procedure targeting selectively either the pyridyl unit or the benzyl moiety. Our plan is outlined in Fig. 1. There are several challenges behind the choice of this substrate. Firstly, the aminocarbonyl group at C2 might strengthen the interaction between the pyridinic nitrogen and the metal through a bidentate coordination, thereby preventing the catalyst from interacting with the target pyridinic C–H bond.8 In fact, the picolinamide (COPy) has been extensively used as a directing group in a variety of C(sp2)– and C(sp3)–H functionalization reactions.9 In contrast, there have only been isolated examples of successful derivatization at the pyridine ring,10 thus highlighting the challenging nature of this task. Recently, the groups of Shi11 and our own8 managed to overcome this difficulty and reported the RhIII-catalyzed ortho-olefination/annulation of picolinamides with electron-deficient olefins. Secondly, the benzylamine unit embedded in the substrate is prone to dehydrogenation at the benzylic position under the oxidative Rh/CuII system, potentially leading to imine-type intermediates.12a,b The scarcity of precedents for the functionalization of benzylamine derivatives12 compared to the variety of methods available for benzoic acid derivatives3 points toward a challenging transformation.
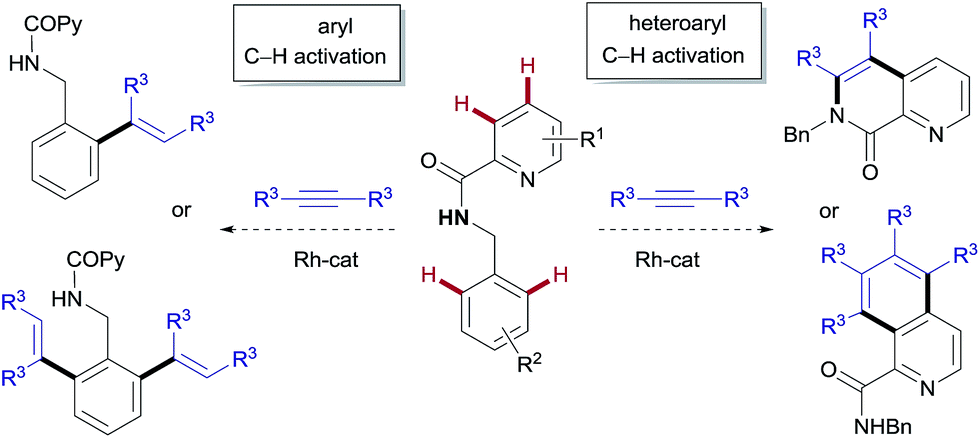 | ||
| Fig. 1 Proposed catalyst-controlled divergent aryl/heteroaryl C–H functionalization. | ||
Herein we describe the catalyst-controlled divergent heteroaryl/aryl functionalization of picolinamide derivatives that provides selective straightforward access to either isoquinoline-1-carboxamide or ortho-olefinated benzylamine (or phenethylamine) derivatives. This complementary reactivity has been achieved by simply choosing between either a RhIII or a RhI catalyst.2 To our knowledge, RhI/RhIII divergent control in C–H activation on the same substrate remains undocumented.
Results and discussion
Optimization studies
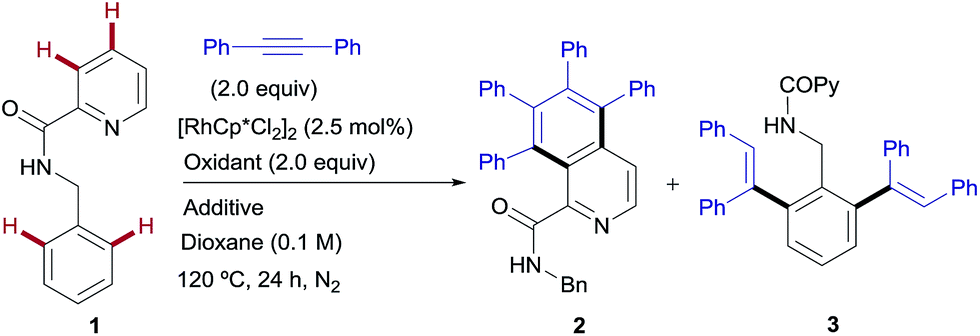
The model reaction between N-benzylpicolinamide (1) and diphenylacetylene was chosen for the optimization studies (Table 1). A low but promising outcome was obtained with [RhCp*Cl2]2 (2.5 mol%) in conjunction with Cu(OAc)2 (2 equiv.), providing a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.7 mixture of the isoquinoline-1-carboxamide derivative 2 (ref. 13) and the di-olefinated benzylamine derivative 3,13 both resulting from two C–H activations and two alkyne insertions at either the pyridyl or the benzene unit (entry 1). The replacement of Cu(OAc)2 with Cu(TFA)2 led to suppression of the catalytic activity, likely due to the lower basicity of trifluoroacetate compared to acetate (entry 2). In fact, the addition of 4 equiv. of NaOAc to the Rh/Cu(TFA)2 system restored the catalytic activity (entry 3), suggesting that the oxidant is a source of acetate, necessary for the reaction to proceed. Further investigation (see the ESI†), led us to find that the addition of AgSbF6 (10 mol%) to sequester the chloride ligands remarkably improved both the reactivity and the site selectivity, allowing a clean and complete conversion of 1 into isoquinoline 2 as the single coupling product (entry 4). Control experiments determined that the product formation is completely inhibited in the absence of the Rh catalyst (entry 5) or with the omission of the copper salt, even when using O2 as an external co-oxidant (entry 6). Interestingly, however, the reactivity was partially restored but lead selectively to the di-olefinated product 3, albeit with moderate yield, without the CuII salt but in the presence of NaOAc (entry 7). These optimization studies are evidence for the critical role played by both the Cu(OAc)2, as both the oxidant and carboxylate source, and AgSbF6, responsible for promoting the ligand exchange at Rh, in determining the catalyst activity and selectivity towards the formation of isoquinoline 2.
:3.7 mixture of the isoquinoline-1-carboxamide derivative 2 (ref. 13) and the di-olefinated benzylamine derivative 3,13 both resulting from two C–H activations and two alkyne insertions at either the pyridyl or the benzene unit (entry 1). The replacement of Cu(OAc)2 with Cu(TFA)2 led to suppression of the catalytic activity, likely due to the lower basicity of trifluoroacetate compared to acetate (entry 2). In fact, the addition of 4 equiv. of NaOAc to the Rh/Cu(TFA)2 system restored the catalytic activity (entry 3), suggesting that the oxidant is a source of acetate, necessary for the reaction to proceed. Further investigation (see the ESI†), led us to find that the addition of AgSbF6 (10 mol%) to sequester the chloride ligands remarkably improved both the reactivity and the site selectivity, allowing a clean and complete conversion of 1 into isoquinoline 2 as the single coupling product (entry 4). Control experiments determined that the product formation is completely inhibited in the absence of the Rh catalyst (entry 5) or with the omission of the copper salt, even when using O2 as an external co-oxidant (entry 6). Interestingly, however, the reactivity was partially restored but lead selectively to the di-olefinated product 3, albeit with moderate yield, without the CuII salt but in the presence of NaOAc (entry 7). These optimization studies are evidence for the critical role played by both the Cu(OAc)2, as both the oxidant and carboxylate source, and AgSbF6, responsible for promoting the ligand exchange at Rh, in determining the catalyst activity and selectivity towards the formation of isoquinoline 2.
|
|
||||
|---|---|---|---|---|
| Entry | Oxidant | Additive | Conversionb (%) | 2/3c (%) |
| a 1 (0.15 mmol), alkyne (0.30 mmol), [RhIII]-cat. (5 mol%), oxidant (2.0 equiv.), dioxane (0.1 M), 120 °C, 24 h. b Determined by 1H NMR from the crude mixture. c [RhIII]-cat. (10 mol%). d 4.0 equiv. e 10 mol%. f Without Rh-catalyst. g Under O2. | ||||
| 1 | Cu(OAc)2 | — | 47 | 21/79 |
| 2 | Cu(TFA)2 | — | — | —/— |
| 3 | Cu(TFA)2 | NaOAcd | 66 | 44/56 |
| 4 | Cu(OAc)2 | AgSbF6e | >98 | >98/<2 |
| 5f | Cu(OAc)2 | AgSbF6e | — | —/— |
| 6g | — | AgSbF6e | — | —/— |
| 7 | — | NaOAcd/AgSbF6e | 46 | <2/>98 |
RhIII-catalyzed pyridyl C–H functionalization: synthesis of isoquinoline derivatives
The scope of this aromatic homologation method allows for the construction of variously substituted polyarylated isoquinoline derivatives (Scheme 1). It is important to note that the isoquinoline moiety forms the core of many biologically active molecules.14 In this study microwave heating was generally applied since it dramatically reduced reaction times (from 24 h to just 1 h) while preserving the high site-selectivity, as exemplified in the isolation of 2 in 90% yield. Both electron-donating and electron-withdrawing substituents at either the pyridine (14–15, 54 and 62%) or the diarylacetylene15 (11–12, 61% and 63%) coupling partners were well tolerated. It is also remarkable that the Cl substituent survived the reaction conditions (15, 62%). The use of substituents on the amide nitrogen other than a benzyl group was also tolerated, as demonstrated by the good reactivity displayed by the substrate bearing an ethyl substituent (R = Et), which provided the corresponding isoquinoline derivative 13 in 85% yield.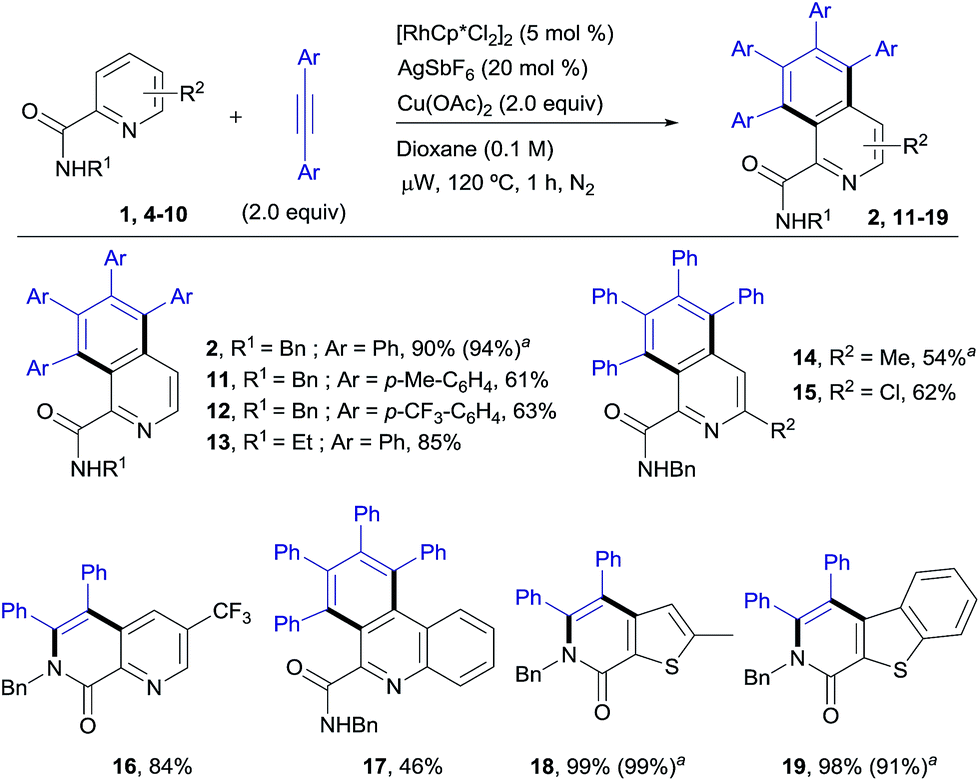 | ||
| Scheme 1 RhIII-catalyzed pyridyl C–H functionalization, leading to the isoquinoline derivatives. Conditions: benzylamine derivative (0.15 mmol), alkyne (0.30 mmol), [RhCp*Cl2]2 (5 mol%), Cu(OAc)2 (2.0 equiv.), dioxane (0.1 M), μW, 120 °C, 1 h. aUnder conventional heating at 120 °C for 24 h. | ||
Interestingly, the presence of a CF3 group at the C5 of the pyridine ring, residing in close proximity to one of the reactive C–H bonds, interrupted the aromatic homologation and led exclusively to the 1,7-naphthyridin-8(7H)-one derivative 16 (ref. 13) (84% yield), resulting from a double C–H/N–H activation2c and only one alkyne insertion. Although factors affecting these reactivity differences remain to be elucidated, this result suggests that the second alkyne insertion/C–H activation is sensitive to steric effects, so that the presence of a substituent in the aryl ortho-position to the reactive C–H site may impart a significant steric demand, thereby bypassing the normal reaction outcome and favouring the competitive trapping of the plausible alkenyl rhodium intermediate by the amidic N–H. Extension of this reaction to heteroaryl 2-carboxamides with quinoline, thiophene or benzo[b]thiophene skeletons proved successful, but whereas the first substrate evolved to give the expected aromatic homologation product in moderate yield (17, 46%), in the other two cases the reaction proceeded through the “interrupted” pathway, leading to the C–H/N–H cyclization products 18 (ref. 13) and 19 in excellent yields (98–99%).
RhI-catalyzed C–H ortho-olefination at the benzylamine unit
Our desired goal of developing a divergent C–H functionalization protocol guided us to revisit the low-yielding but encouragingly selective formation of the di-olefinated benzylamine 3, observed in the reaction of 1 with diphenylacetylene in the absence of Cu(OAc)2 but using NaOAc as an acetate ion source (see Table 1, entry 7, 46% NMR conversion). We reasoned that in the absence of the CuII-terminal oxidant, RhI species,16 rather than RhIII, could be a competent catalyst leading to the di-ortho-olefination product through a distinct mechanistic pathway. To our delight, this was indeed the case and the desired product 3 was obtained in 88% yield when using [Rh(cod)Cl]2 (ref. 17) (2.5 mol%) under conditions very similar to those in entry 7 of Table 1 (Scheme 2).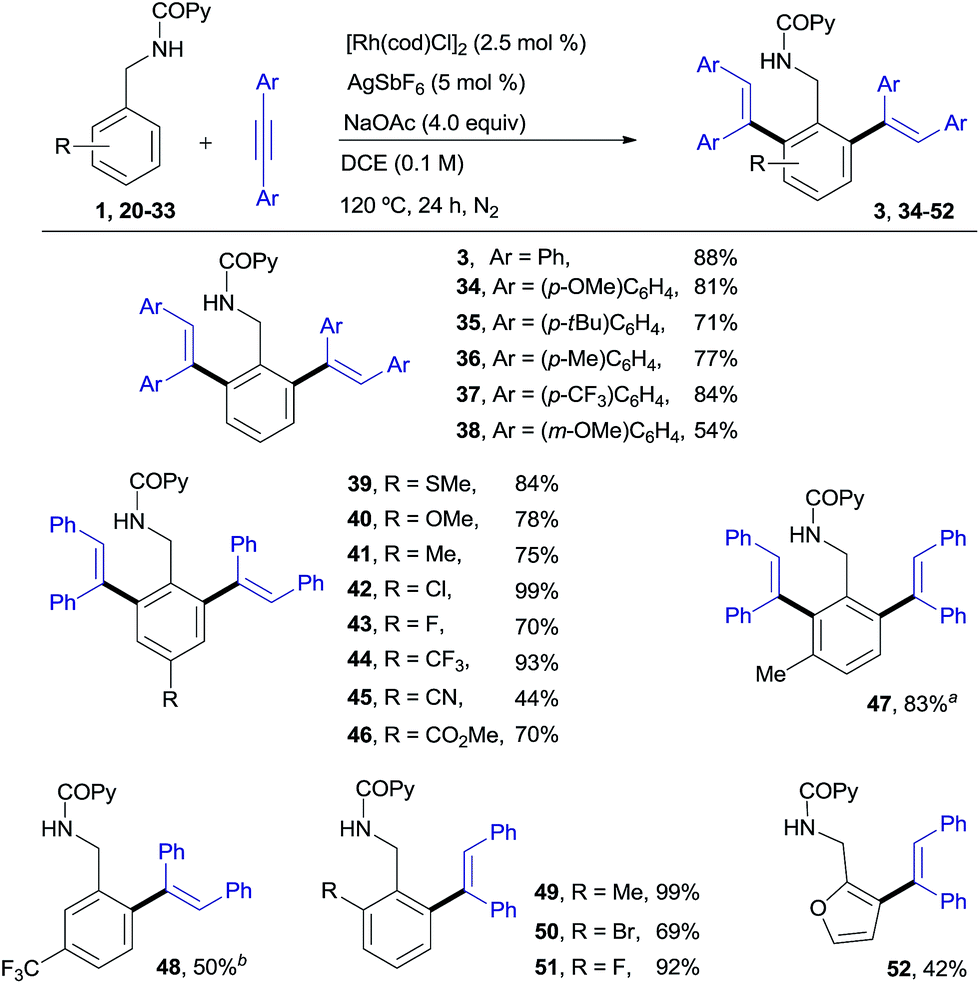 | ||
| Scheme 2 RhI-catalyzed ortho-olefination of benzylamine derivatives. aThe mono-ortho-olefinated product was also isolated in 8% yield. bThe di-olefinated product was also isolated in 6% yield. | ||
As shown in Scheme 2, N-benzylpicolinamide 1 smoothly reacted with a variety of diarylacetylenes15 equipped with both electron-rich and electron-poor para-substituted aryl groups, to give the corresponding di-olefinated benzylamine derivatives in good yield (34–37, 71–84%). meta-Substitution at the diaryl acetylene is also possible, albeit with lower efficiency (38, 54%), while no reaction was observed with the more sterically hindered ortho-substituted diaryl acetylenes (not shown).18 A broad range of para-, ortho- and meta-substituents at the benzylamine unit with very different electronic properties proved to be suitable substrates (39–51, 44–99% yield). The functional-group compatibility is remarkable, including coordinating functionalities (CN or SMe), and halogens (Cl and, especially, the challenging Br). A meta-Me substituent led to the di-olefinated product in good yield (47, 83% yield), while a meta-CF3 resulted mainly in mono-olefination at the sterically less hindered ortho-position (48, 50% yield). ortho-Substitution, which often results in reduced reactivity for steric reasons, was well tolerated (49–51, 69–99%). Likewise, the successful use of a heteroaromatic substrate turned out to be viable, albeit in a lower yield (furanyl derivative 52, 42%).
Exploration of unsymmetrical alkyl-substituted internal alkynes
We next explored unsymmetrical alkynes, for which regiocontrol in the insertion step becomes an issue of concern. Unsymmetrical aliphatic-substituted internal alkynes are a more challenging type of substrate due to their diminished reactivity and poor regioselectivity in the 1,2-migratory insertion often observed in the context of rhodium-catalyzed C–H functionalization.4f Interestingly, it was found that β-alkyl acetylenic esters did participate in the pyridyl/phenyl divergent C–H functionalization with excellent selectivity, albeit with a different reaction outcome than the diarylalkynes (Scheme 3). For instance, the reaction of 1 with ethyl pent-2-ynoate under the RhIII-catalyzed conditions (i.e., the isoquinoline formation conditions) led to an ortho-functionalization at the pyridine ring but it did not yield the corresponding isoquinoline. Instead, the 5,5-fused bicyclic ester 53, with a valuable 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine architecture holding a quaternary carbon center, was obtained as the sole reaction product in good yield (90%). This compound seems to arise from a competitive evolution of the alkyne insertion complex that prevents the second alkyne insertion/C–H activation. On the other hand, this result demonstrates that the reaction outcome can be significantly influenced by changes in the alkyne substitution. However, when the same two reacting partners (1 + ethyl 2-pentynoate) were submitted to the RhI-catalyzed conditions, a clean formation of the di-olefinated benzylamine derivative 54 was observed, yet in modest yield (40%). In the latter case, the reaction was found to be accelerated under aerobic conditions (air or a balloon of O2). Remarkably, both of the RhIII and RhI C–H functionalization processes led to products with complete regioselectivity regarding the alkyne insertion (in both cases at the β-position of the ethyl 2-pentynoate).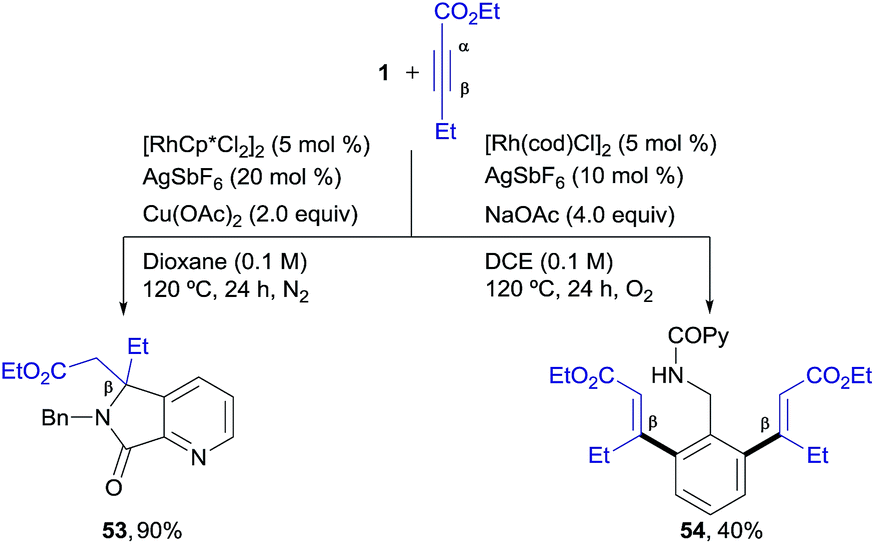 | ||
| Scheme 3 Rhodium-controlled divergent aryl/heteroaryl C–H functionalization in the reaction of 1 with ethyl pent-2-ynoate. | ||
The use of enynes as another type of non-aromatic alkyne coupling partner with an electronic bias for highly regioselective insertion, elegantly introduced by Huestis and co-workers in the context of C–H functionalization,4q led us to easily prepare di-ortho-dienyl benzylamine derivatives in good yields (products 55–57, 72–93% yield, Scheme 4). This reaction revealed the tolerance of this catalyst system towards a sensitive alkyl chloride substituent (57, 72% yield). As occurred in the case of the acetylenic esters, higher reaction rates were observed under aerobic conditions and in all cases studied the conjugated moiety attached to the alkyne ended up at the vinylic position away from the phenyl ring with complete regiocontrol.
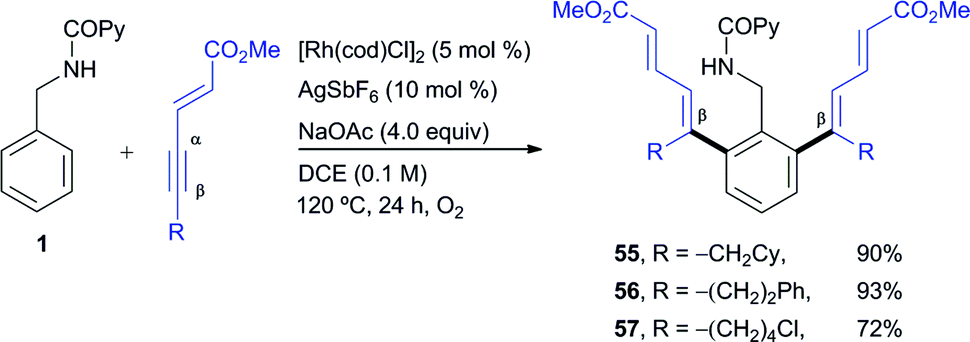 | ||
| Scheme 4 RhI-catalyzed ortho-olefination of 1 with 1,3-enynes. | ||
Finally, as shown in Scheme 5, some unsymmetrical alkyl–aryl–alkynes, such as cyclohexyl–aryl–acetylenes, also participated in the RhI-catalyzed cross-coupling reaction, affording the desired di-olefinated products as single regioisomers and stereoisomers (products 58–60, 76–88%) showing that, as in the previous examples, there is complete regiocontrol in favour of functionalization at the β-position of the starting conjugated alkyne. In contrast, very poor conversion was observed with oct-1-yn-1-ylbenzene while internal dialkyl-alkynes such as 2-butyne resulted in a total lack of reactivity (not shown).
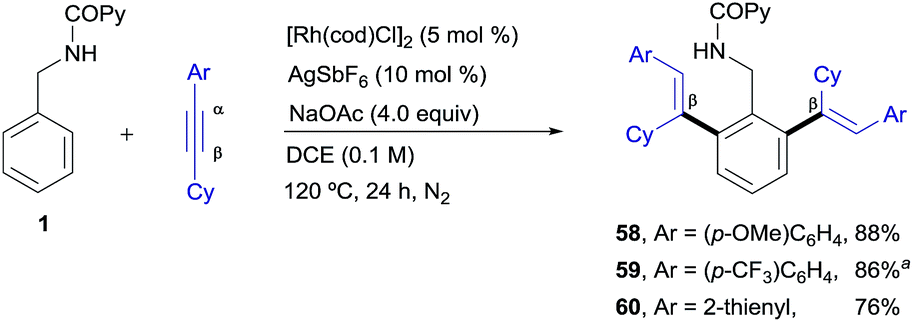 | ||
| Scheme 5 RhI-catalyzed ortho-olefination of 1 with aryl-cyclohexyl-acetylenes. aUsing 5 mol% [Rh(cod)Cl]2 and 10 mol% of AgSbF6. | ||
Extension of the reactions to phenethylamine derivatives
Pleasingly, this method could be extended to phenethylamine derivatives, which have a tether that is one carbon longer with regard to the directing group. N(COPy)-phenethylamine (61)19 reacted smoothly with diphenylacetylene under the optimized conditions to give the di-olefinated product 73 (ref. 13) in 76% yield (Scheme 6). In terms of scope, the results parallel those found with the benzylamine derivatives, with the applicability to naphthalene (86, 42%) and heteroaromatic (87, 97%) compounds being of particular relevance. This structural flexibility is noteworthy, since very often the precise tether length of the directing group is found to be crucial for reactivity in C–H functionalizations.20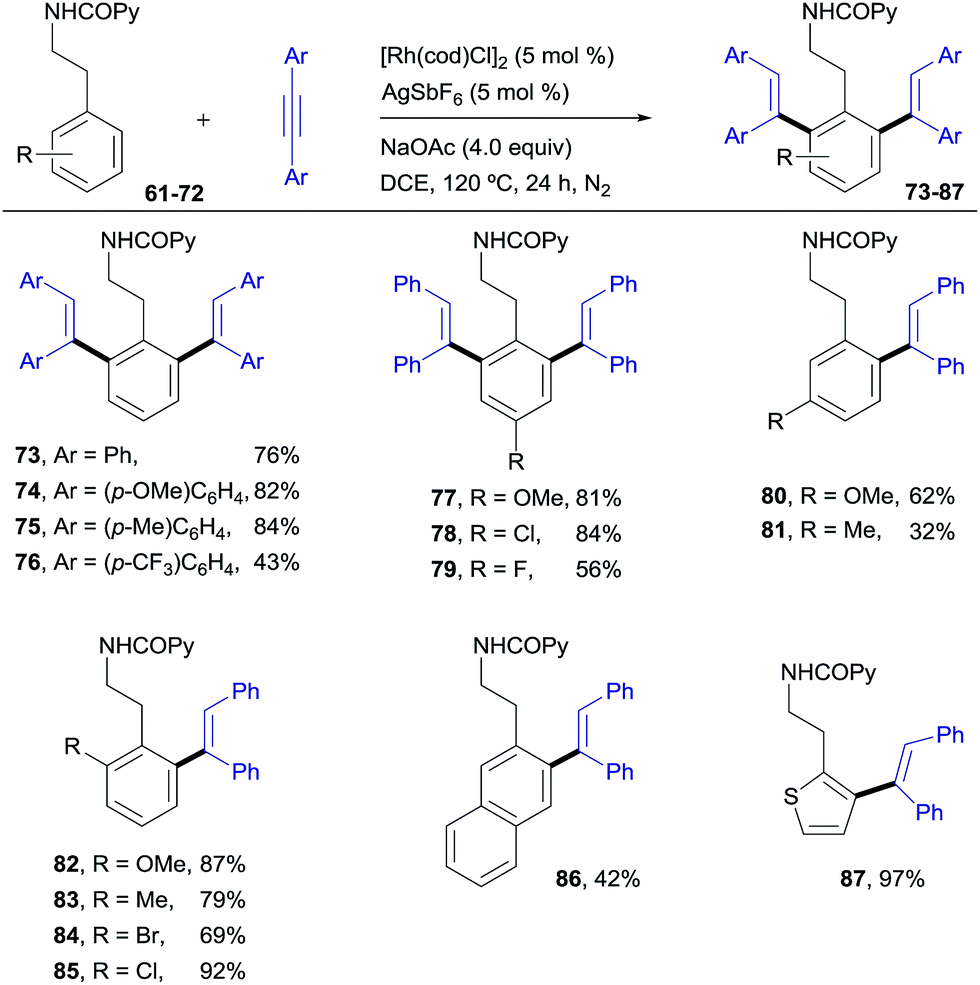 | ||
| Scheme 6 RhI-catalyzed ortho-olefination of phenethylamine derivatives. | ||
The complementary reactivity of the RhIII-catalyzed oxidative alkenylation/annulation was also briefly explored with N-(2-picolinamide)-protected phenethylamine substrates (Scheme 7). As in the model reaction, when the parent substrate 61 was submitted to the standard optimized reaction conditions, the 1,7-naphthyridin-8(7H)-one 88 was produced as a single product in 90% yield. This product results from a double C–H/N–H activation and only one alkyne insertion (referred to as the “interrupted” pathway) rather than the aromatic homologation via the two-fold C–H activation previously observed for the reaction of the analogous benzylamine derivative under identical reaction conditions (product 2, 94% yield). This result adds additional weight to the noticed sensitivity of this catalyst system to steric hindrance, which appears to strongly influence the reaction outcome.
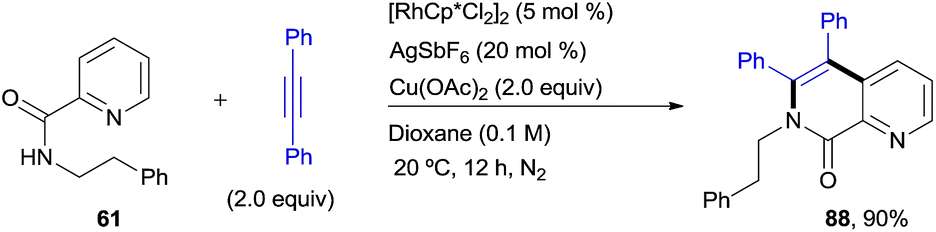 | ||
| Scheme 7 RhIII-catalyzed pyridyl C–H functionalization leading to a 1,7-naphthyridin-8(7H)-one derivative. | ||
Chemoselective deprotection and removal of the auxiliary COPy group
Scheme 8 illustrates the chemoselective N-deprotection of 2 to give the isoquinoline-2-carboxamide derivative 89 (82%), as well as the facile removal of the auxiliary picolinamide directing group in both the benzyl- and phenethylamine di-olefinated products (90 and 91, 86% and 89%, respectively).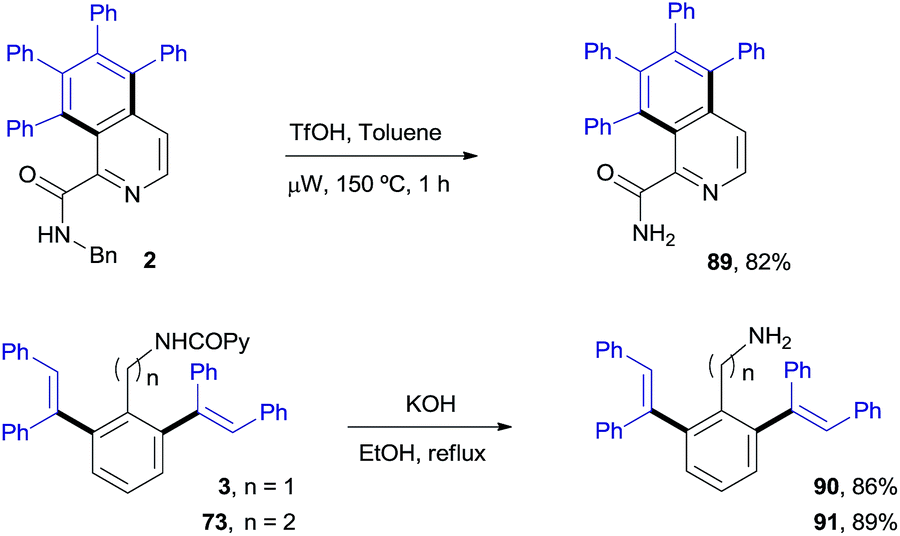 | ||
| Scheme 8 Deprotection of the N-benzyl group and removal of the COPy directing group. | ||
Mechanistic insights
:30 mixture of the isoquinoline derivative 2 and the di-olefinated product 3, in the presence of NaOAc at 120 °C in only 4 h (Scheme 9). It is worth remarking that no reactivity is observed in the absence of NaOAc.
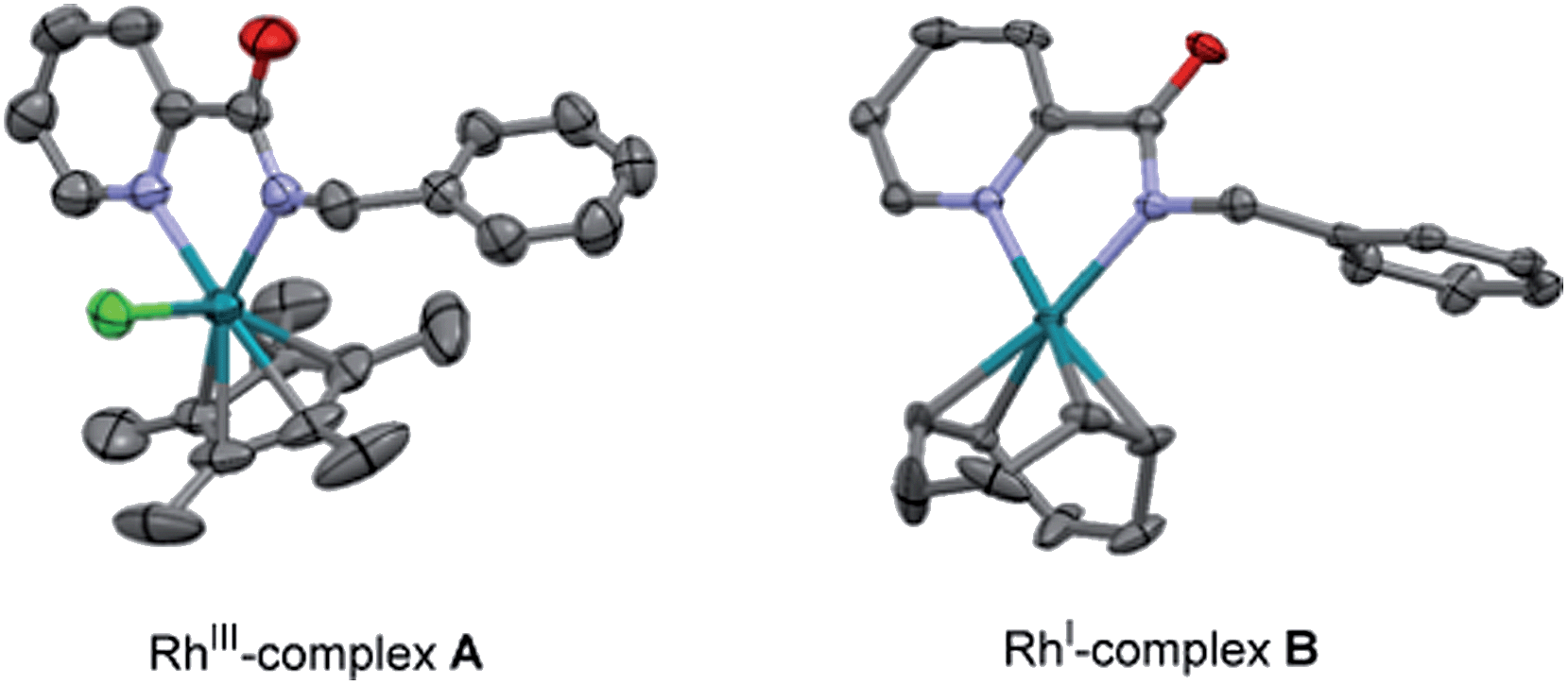 | ||
| Fig. 2 ORTEP view of RhIII-complex A and RhI-complex B. The hydrogen atoms have been removed for simplicity. | ||
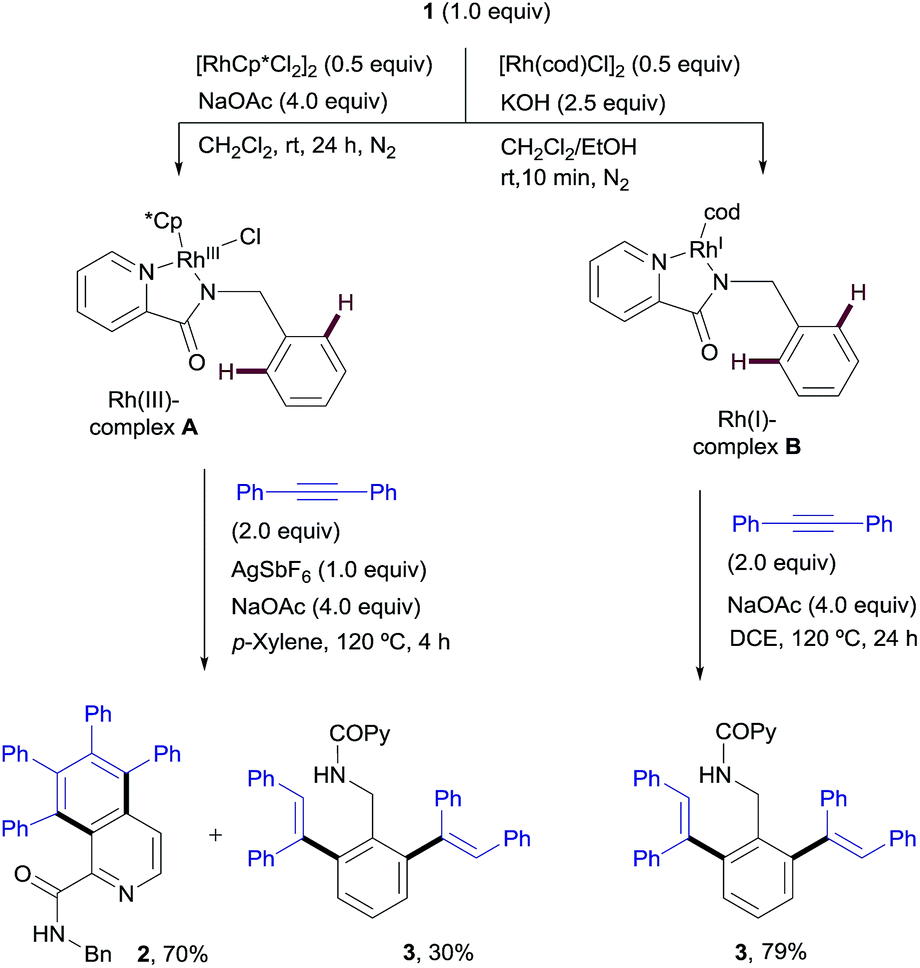 | ||
| Scheme 9 Stoichiometric studies with isolated RhIII- and RhI-picolinamide complexes. | ||
On the other hand, the stoichiometric reaction of 1 with [Rh(cod)Cl]2, under similar conditions to those employed in the formation of complex A, provided the RhI-complex B, whose X-ray structure showed a similar N,N-bidentate metal coordination (Fig. 2, see the ESI† for details).13 Remarkably, the reaction of complex B with diphenylacetylene afforded the di-olefinated product 3 as the only product (Scheme 9). Control experiments confirmed again that NaOAc is crucial for the reaction to proceed.
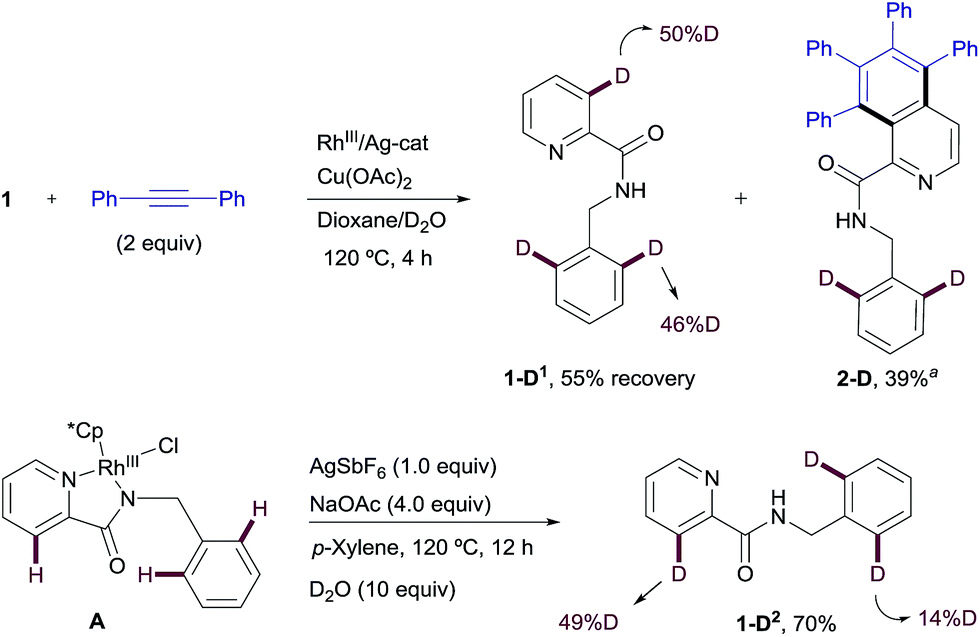 | ||
| Scheme 10 H/D exchange experiments in the RhIII-promoted C–H functionalization process. aThe H/D exchange was detected using mass spectrometry in this case (the exact deuterium content could not be determined by NMR). | ||
Likewise, when RhIII-complex A was dissolved in a p-xylene/D2O mixture and heated at 120 °C in the presence of NaOAc and AgSbF6 for 12 h but in the absence of an alkyne, 1-D2 was recovered in 70% yield showing 49% of deuterium incorporation at the C3–Py and 14% of H/D scrambling at the ortho-positions of the benzyl moiety (Scheme 10). This result seems to indicate that with stoichiometric amounts of Rh, C–H insertion at both the aryl and heteroaryl sites also become reversible in the absence of the alkyne.
Similar deuterium labeling studies were performed in the RhI-promoted C–H functionalization process (Scheme 11). When substrate 1 was allowed to react with diphenylacetylene in a DCE/D2O mixture at 120 °C for 12 h under otherwise standard RhI-catalyzed conditions {[Rh(cod)Cl]2 (2.5 mol%)/AgSbF6 (5 mol%) and NaOAc (4 equiv.)}, unreacted 1 was recovered (in 8% yield) with significant deuterium incorporation at the ortho-position of the benzylamine moiety (57%D) but no H/D exchange detected at the pyridine ring. The main component of the reaction mixture was the di-olefinated product 3-D1 (73% isolated yield), which showed high levels of deuterium incorporation at the vinylic position (85%D, Scheme 11a). This result suggests a reversible metalation/deutero(proto) demetalation at the reactive C–H sites, whereas activation at the pyridine ring appears to be less favorable. The high degree of deuteration at the vinylic positions of product 3-D1 is compatible with a mechanism of arene activation via oxidative insertion (which should retain the H/D incorporation from the starting material) in which the hydride/deuterium ligand exchange with D2O in the RhIII-complex, resulting from the oxidative addition of RhI into the ortho-C–H bond of 1, readily occurs prior to reductive elimination.22
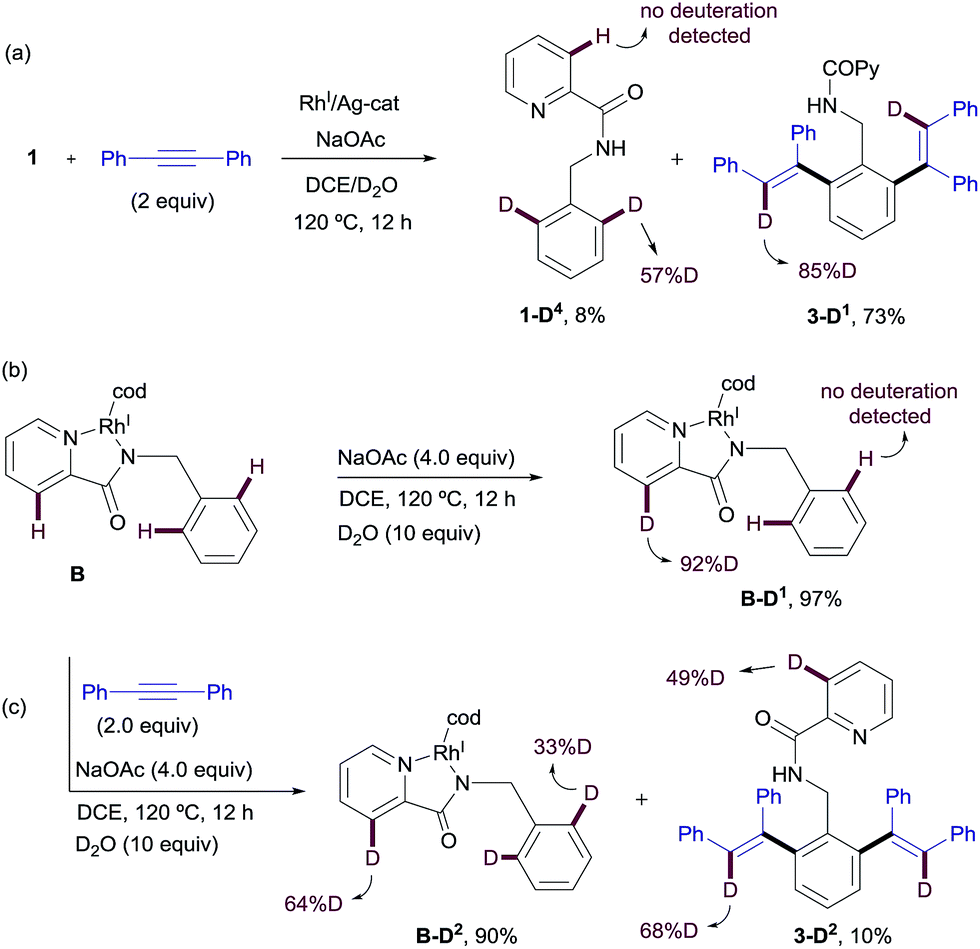 | ||
| Scheme 11 H/D exchange experiments in the RhI-promoted C–H functionalization process. | ||
The evaluation of the potential of RhI-complex B for metalation/deutero(proto) demetalation in the absence of an alkyne using a hydrogen/deuterium exchange process led to almost complete deuteration of the C3–Py position in RhI-complex B (B-D1, 92%D), with no deuteration being observed at the benzylamine part (Scheme 11b). This result was in contrast to the high selectivity towards the benzylamine moiety observed under catalytic RhI in the presence of an alkyne, where no deuteration was observed at the pyridine ring. Product B-D1 may arise from dissociation of the pyridinic nitrogen ligand from Rh (e.g., through displacement by the acetate ion), followed by metalation/deutero-demetalation at the ortho 2-picolinamide moiety. Finally, when RhI-complex B was mixed with the diphenylacetylene in a DCE/D2O mixture at 120 °C (Scheme 11c), a very low conversion to the dialkenylation product 3-D2 was observed (10% isolated yield after 12 h), which showed significant deuterium incorporation at both 3-pyridyl (49%D) and vinylic (68%D) positions. The unreacted complex was recovered in 90% isolated yield with 64% H/D scrambling at the C3–Py position and 33% deuterium incorporation in the benzylic moiety. This result suggests that, as previously observed in the RhIII-promoted outcome, the regioselectivity of the reaction is controlled not by the site of the C–H cyclometalation but by the rate at which the two potential isomeric Rh-complexes undergo subsequent alkyne insertion, which turns out to be opposite in the RhI or RhIII pathways. The reasons behind the lower reactivity of complex B in the mixture DCE/D2O are not fully understood at the present time.
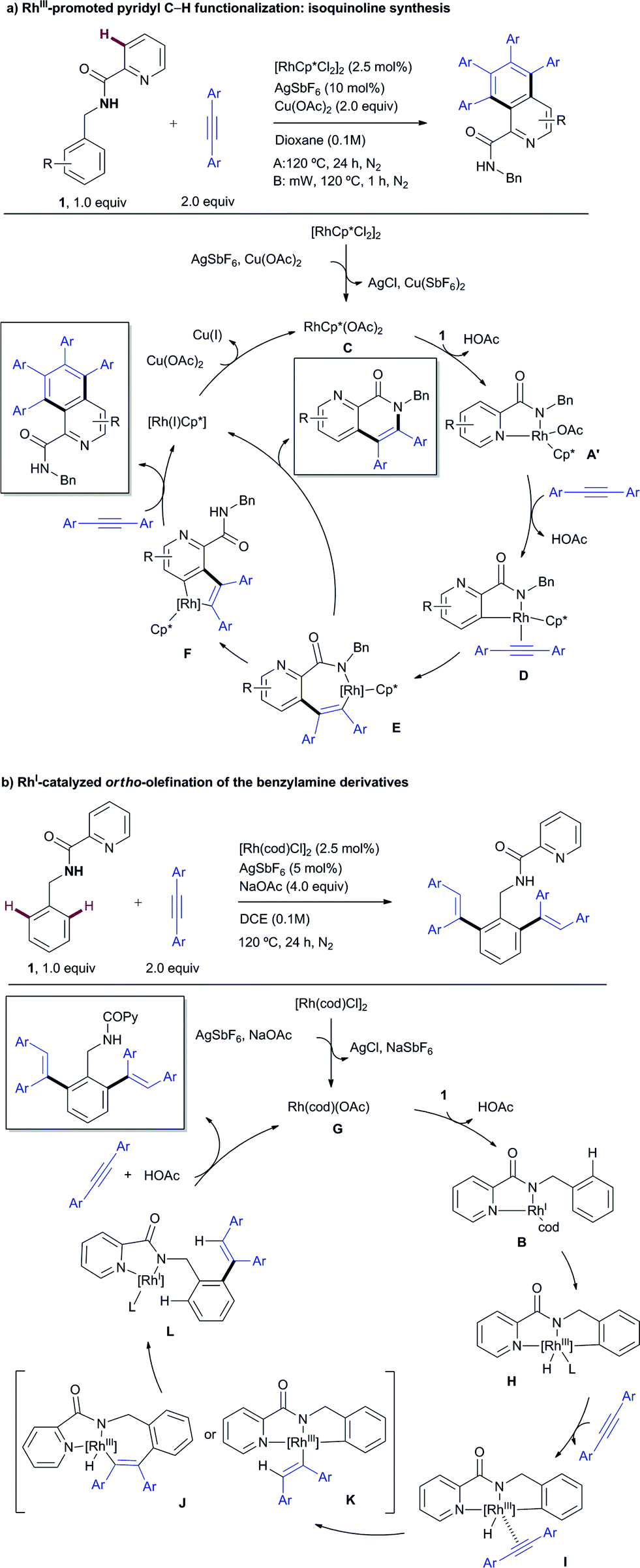 | ||
| Scheme 12 Simplified plausible mechanistic pathways. | ||
The RhIII catalytic pathway depicted in Scheme 12a is proposed to start by forming the highly soluble presumed active catalyst RhCp*(OAc)2 (C) via ligand exchange from [RhCp*Cl2]2 in the presence of an excess of acetate ions. Then, displacement of an acetate from C by the substrate (1) would lead to intermediate A′, analogous to the X-ray characterized complex A. A subsequent “rollover” cyclometalation23via pyridine decomplexation and rotation around the carbonyl–Py bond and then C–H bond activation, presumably by an acetate-assisted concerted metalation-deprotonation (CMD) pathway with concomitant loss of a second molecule of acetic acid, followed by an alkyne coordination affords D. 1,2-Migration of the rhodium–carbon bond across the alkyne results in the formation of the seven-membered rhodacycle E, which presumably triggers a second intramolecular C–H activation leading to a more stable five-membered Rh complex F. After the coordination and migratory insertion of a second alkyne molecule, a reductive elimination step releases the isoquinoline product while the concomitantly formed RhI species is oxidized by CuII acetate to regenerate the RhIIICp*catalyst. Alternatively, if formation of complex F from E is hampered (for instance by steric crowding next to the reactive C–H site), the direct formation of the carbon–nitrogen bond from Evia reductive elimination becomes more favorable to afford the mono-insertion product (previously referred to as the “interrupted” pathway), at which time the metal catalyst is reduced to RhI and further oxidized to RhIII by CuII acetate. In the case of using β-alkyl acetylenic esters (such as ethyl 2-pentynoate) as the coupling partner, the formation of the 6,7-dihydro-5H-pyrrolo[3,4-b]pyridine skeleton (product 53) may arise from a fast proto-demetalation of the complex type E followed by either an intramolecular hydroamination and subsequent oxidation or an oxidative cyclization through electrophilic activation of the olefin, C–N bond formation and subsequent β-hydride elimination.
The first step in the catalytic cycle proposed for the RhI-catalyzed ortho-olefination (Scheme 12b) would likely involve the formation of the catalytically active Rh–acetate complex Gvia chloride displacement of an acetate ion from the RhI-chloride precatalyst [Rh(cod)Cl]2. Coordination of substrate 1 in a bidentate fashion would lead to complex B,13 which has been isolated and structurally characterized by X-ray diffraction analysis. Complex B might undergo a reversible oxidative addition of an ortho aromatic C–H bond to the RhI to form hydrometallacycle H. Upon metal-coordination of the alkyne to afford complex I, a further syn-insertion to the rhodium–carbon or rhodium–hydride bond would afford J or K, respectively. Subsequent reductive elimination from J or K delivers the mono-alkenylation RhI complex L, primed for subsequent oxidative insertion at the other ortho C–H bond followed by alkyne insertion and reductive elimination to afford the di-alkenylated benzylamine product while regenerating the RhI catalyst.
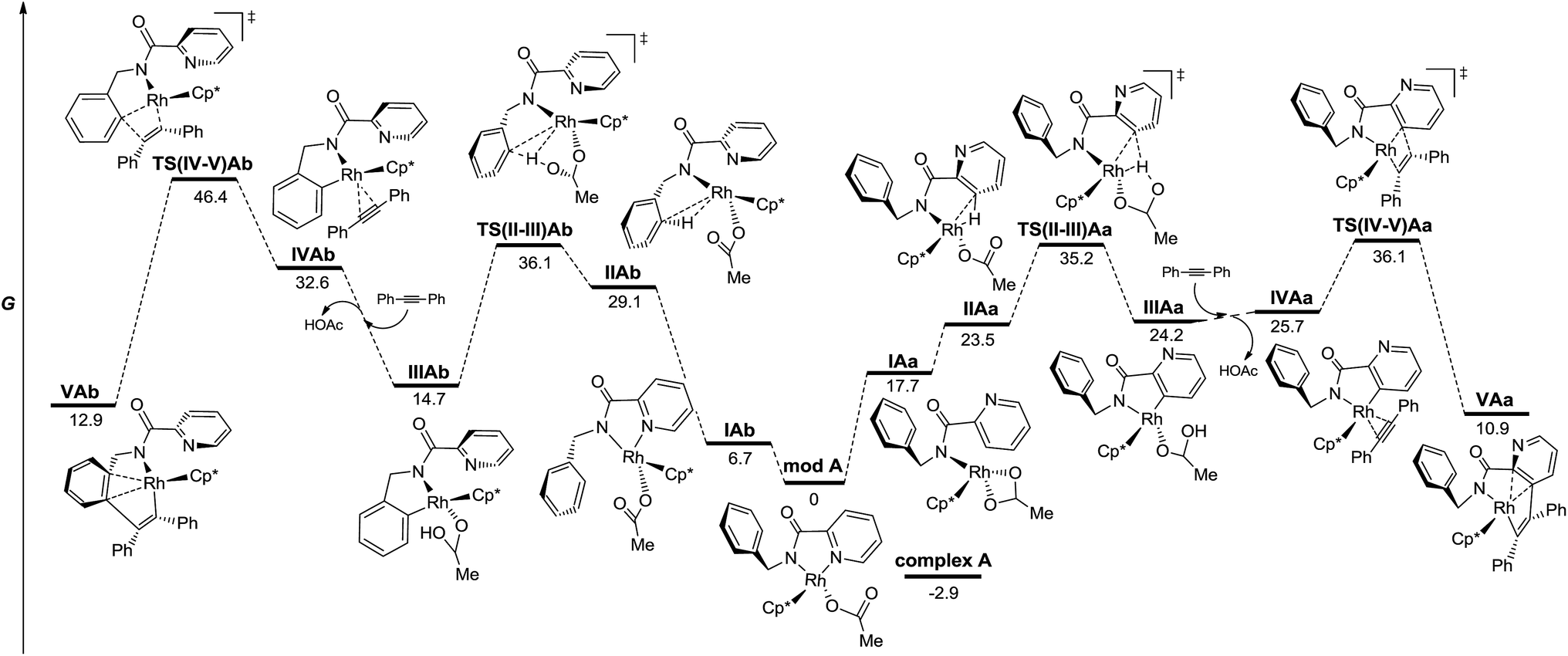 | ||
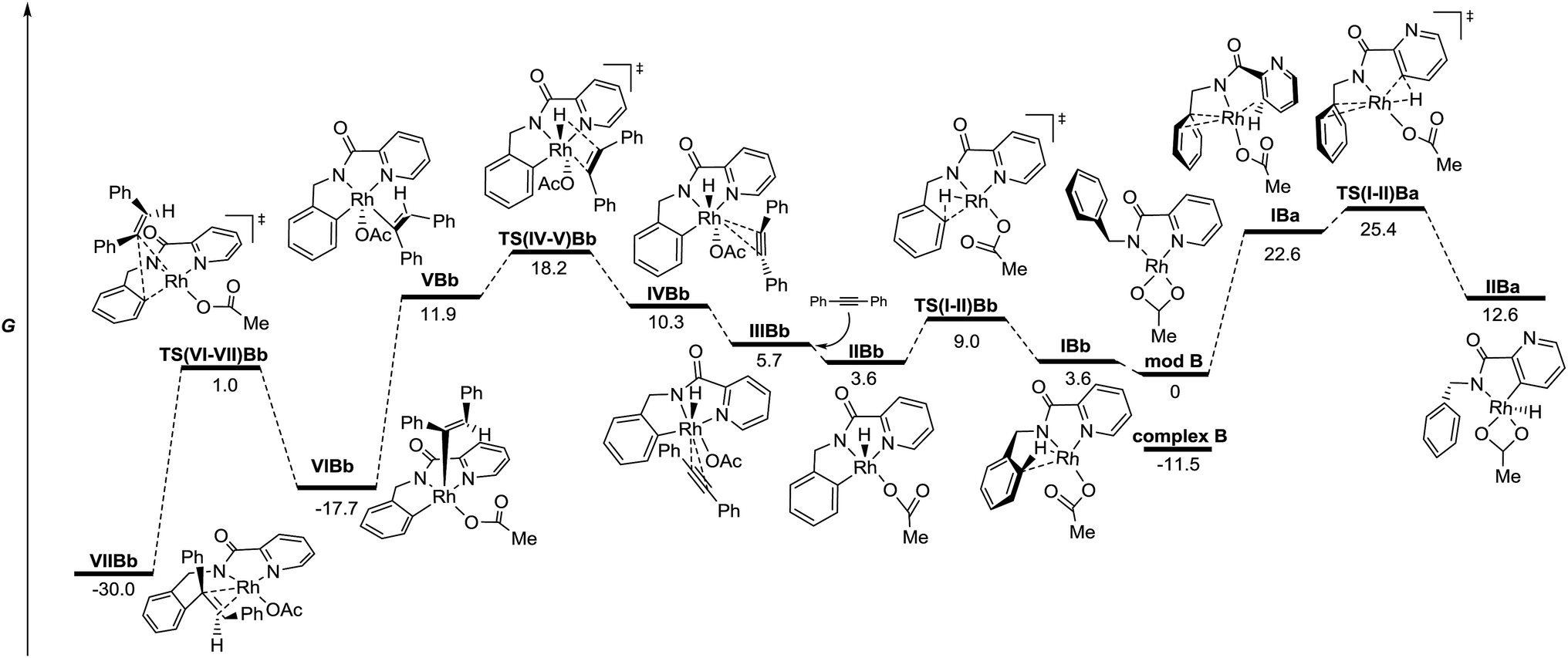
| Fig. 3 Energy profile for the C–H functionalization pathways of 1 with diphenylacetylene from neutral model RhIII complexes in the gas phase (M06/6-311+G(d,p)(C,H,N,O),SDD (Rh)//6-31G(d)(C,H,N,O), LANL2DZ(Rh). The relative G values are in kcal mol−1 at 298 K). | ||
 | ||
| Fig. 4 Energy profile for the C–H functionalization pathways of 1 with diphenylacetylene from anionic model RhI complexes in the gas phase (M06/6-311+G(d,p)(C,H,N,O),SDD (Rh)//6-31G(d)(C,H,N,O), LANL2DZ(Rh). The relative G values are in kcal mol−1 at 298 K). For simplicity, the negative charge has been omitted. | ||
Fig. 3 depicts the calculated lowest energy profile for the postulated CMD mechanism assisted by the acetate ion when using the RhIII catalyst. All species show an almost tetrahedral coordination around the Rh atom similar to that found in the solid state for complex A. The C–H activation of either the pyridine ring or the arene unit (intermediates IIA and TS(II-III)A) requires a lack of the stabilizing interaction between the Rh atom and the pyridinic nitrogen and implies an important and quite similar activation barrier (36.1 and 35.2 kcal mol−1 for benzyl and pyridyl rings respectively). However, the key step that really determines the selective functionalization of the pyridine ring with the RhIII catalyst is the insertion step for the diphenylacetylene unit. The activation barrier to reach that point after the benzyl C–H activation (TS(IV-V)Ab) is almost 10 kcal mol−1 higher than that after the pyridyl C–H activation (TS(IV-V)Aa). Once species VAa is formed, it will be involved in a second C–H activation–insertion sequence to afford the final product.
The energy profile for the reaction catalyzed by RhIvia oxidative addition across the C–H bond is depicted in Fig. 4. The different species show square planar coordination, similar to that observed in the solid state for complex B (modB and VIIBb), square pyramidal (IIBb, VBb and VIBb) or octahedral coordination (IIIBb and IVBb), depending on the number of ligands around the Rh atom in each case. The C–H activation step for the benzyl ring viaTS(I-II)Bb, which keeps the strong stabilizing interaction between the Rh atom and the pyridine nitrogen, is clearly favored over that of the pyridine ring (TS(I-II)Ba) with a lower activation barrier (9.0 compared to 25.4 kcal mol−1).25 However, analyzing the energy profile, the alkyne insertion step is again the determining step through TS(IV-V)Bb in which the new C–H bond is being formed.26 Structural reorganization gives species VIBb with a geometry suitable for the reductive elimination process (TS(VI-VII)Bb). Species VIIBb would continue the same reaction sequence: decomplexation and conformational changes to achieve cyclometalation, alkyne insertion and reductive elimination to afford the final product.
According to the energy profiles depicted in Fig. 3 and 4, the reaction catalyzed by RhIII should follow selectively route “a” to afford products coming from pyridyl C–H activation, whereas the reaction catalyzed by RhI should follow route “b” to afford the ortho-olefination of the benzyl ring. Thus, these models would explain the experimental results found in both catalytic processes: the RhIII catalyst affords products of type 2 whereas the RhI catalyst leads to products of type 3. The decrease in selectivity found in the stoichiometric reaction of complex A (Scheme 9) may be a consequence of the easy reduction of RhIII by the base27 in the absence of the usual Cu oxidant.
The results found in the H/D exchange experiments can also be rationalized on the basis of the species depicted in Fig. 3 for the reaction catalyzed by RhIII. The C–H functionalization is favored at the C3–Py position and is a reversible process. However, when RhI is used as catalyst (Scheme 11), the results found in the stoichiometric reaction pointed out the possible role of other ligands such as “cod” and the alkyne partner to reach the catalytically active species or to affect the C–H activation process. To shed some light on this point, complexes including these ligands and the corresponding C–H activation processes were studied taking complex B as the starting model (Fig. 5).
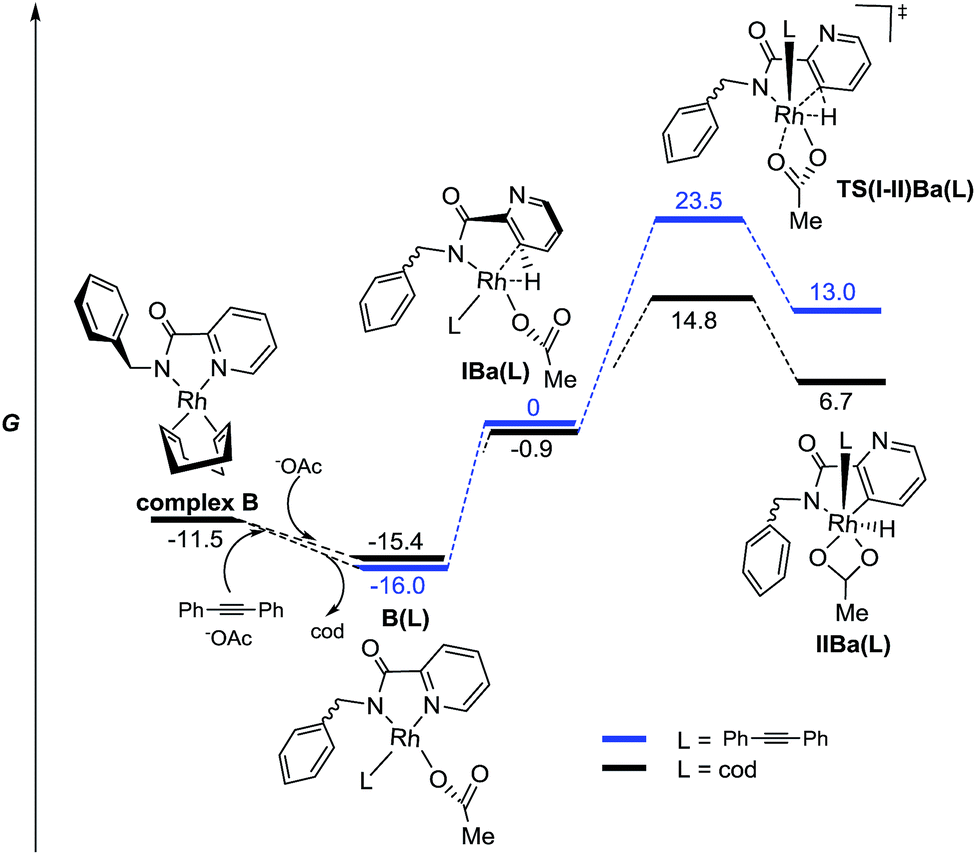 | ||
| Fig. 5 Energy profile for the C–H activation pathways of picolinamides from anionic model RhI complexes with monocoordinated “cod” or alkyne ligands in the gas phase (M06/6-311+G(d,p)(C,H,N,O),SDD (Rh)//6-31G(d)(C,H,N,O), LANL2DZ(Rh). The relative G values are in kcal mol−1 at 298 K). | ||
From this species, the coordination of an acetate ligand could shift one of the olefin units of “cod” to afford a more stable complex B(cod). Additionally, the resulting monocoordinated cod ligand could be effectively shifted by the alkyne partner to afford complex B(diphenylacetylene), which is even more stable.28 This fact could explain the crucial role of the alkyne for the ortho C–H metalation reaction to take place because otherwise the “cod” ligand would stay bonded to the Rh atom.
The complexes IBa(L), prior to the C–H activation step, resulted in quite similar energy barriers for both ligands. However, the C–H activation of the pyridyl ring through TS(I-II)Ba(L) resulted in being much more favored in the case of the “cod” ligand than in the case of the alkyne one (ΔΔG‡ = 7.8 kcal mol−1). Thus, a reversible C–H activation of the pyridine ring could be expected in the absence of the alkyne, in agreement with experimental results (Scheme 11b), whereas if the alkyne is present the evolution through modB should be favored instead of the pyridyl C–H activation. All attempts to find any intermediate keeping either “cod” or the alkyne ligand bonded to the Rh atom that would be involved in the ortho C–H activation of the benzyl ring were unsuccessful, thus reinforcing the hypothesis of modB as the catalytically active species for the benzyl ring functionalization.
The energy differences between each of the coupled key transition states, TS(IV-V)Ab/TS(IV-V)Aa and TS(IV-V)Bb/TS(IV-V)Ba, can be attributed to different steric and/or electronic interactions (Fig. 6). In the case of the reaction catalyzed by RhIII, transition states TS(IV-V)Ab and TS(IV-V)Aa show important steric differences. Whereas TS(IV-V)Aa is a late transition state that shows a shorter C1–C2 distance and longer C2–C3 with the phenyl groups spun around to avoid steric hindrance in the Z-alkene that is being formed, in TS(IV-V)Ab the pyridine ring does not allow the Ph group to reach an equivalent conformation, giving rise to an early transition state with a very distorted alkyne partner. In the case of the reaction catalyzed by RhI, there are no relevant steric interactions. However, the ligands around the Rh atoms are quite different. Whereas TS(IV-V)Bb shows an octahedral coordination with one of the ligands being the pyridine nitrogen, TS(IV-V)Ba lacks this stabilizing interaction and only five ligands (instead of six) coordinate to the Rh atom.
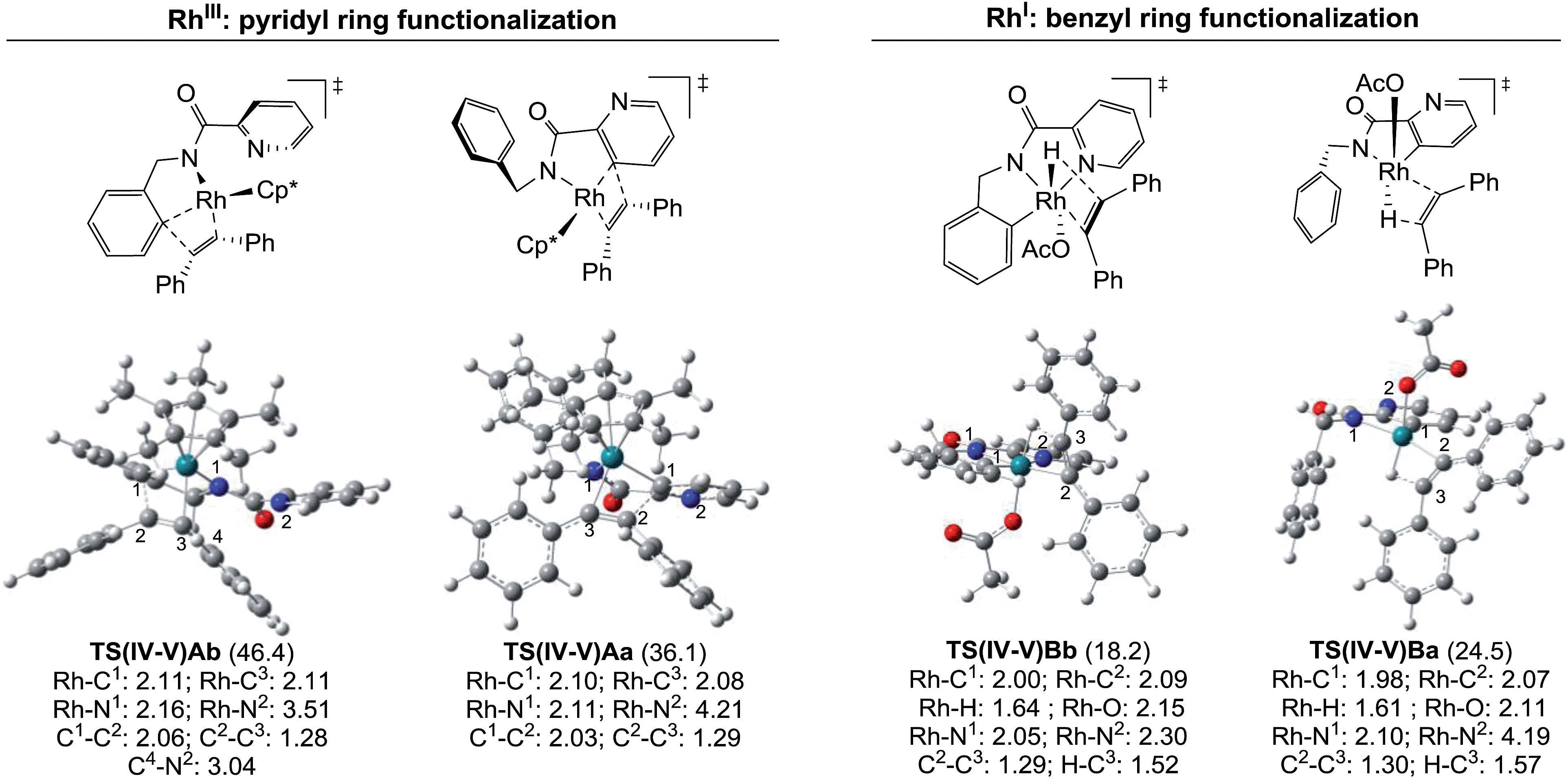 | ||
| Fig. 6 Molecular structures of the key transition states. The bond lengths are given in Å and the relative free energies with respect to modA and modB are indicated in parentheses (kcal mol−1). | ||
The stoichiometric reaction of N-benzylpicolinamide (1) with Rh(acac)(C2H4)2 in the presence of KOH in a CH2Cl2/EtOH mixture at room temperature allowed the isolation and full characterization of RhI-complex M (Scheme 13). All attempts to crystalize RhI(C2H4)2-complex M have failed so far due to its moderate stability. The activity of complex M was tested in the model reaction between N-benzylpicolinamide (1) and diphenylacetylene in otherwise standard reaction conditions. In line with our proposal, product 3 was isolated in 89% yield after only 2 h of reaction. In contrast, the reaction with catalytic amounts of RhI-complex B was notably slower, observing a similar conversion only after 12 h (see the ESI† for further details). Indeed, as evidenced in the kinetic catalytic profiles of both complexes from parallel reactions shown in Fig. 7, complex B requires an activation period (more than 1 h) prior to becoming active, whereas catalyst M promoted almost complete conversion within 1.5 h without a noticeable induction period. This stark difference between the activity of complexes B and M was ascribed to the much easier displacement of the ethylene ligands from RhI by the acetate compared to the bidentate “cod” group, thereby accelerating the catalyst turnover, along with a loss of the “cod” ligand during the course of the reaction with the generation of vacant coordination sites.
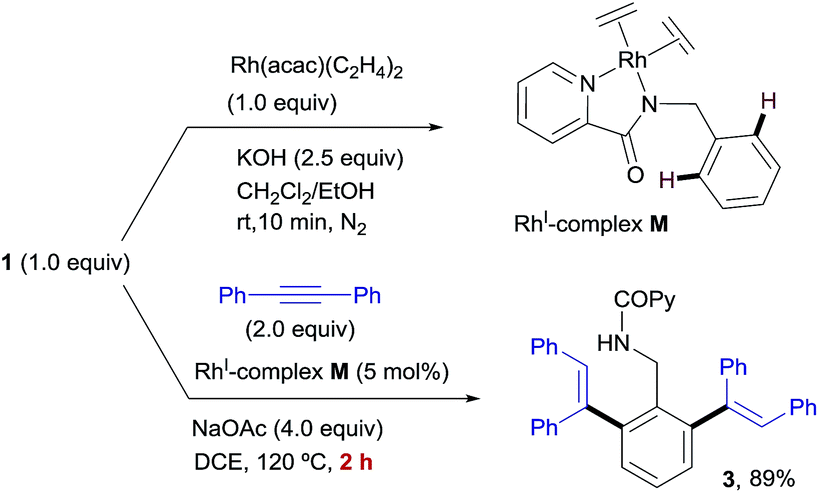 | ||
| Scheme 13 Synthesis and activity of RhI-complex M. | ||
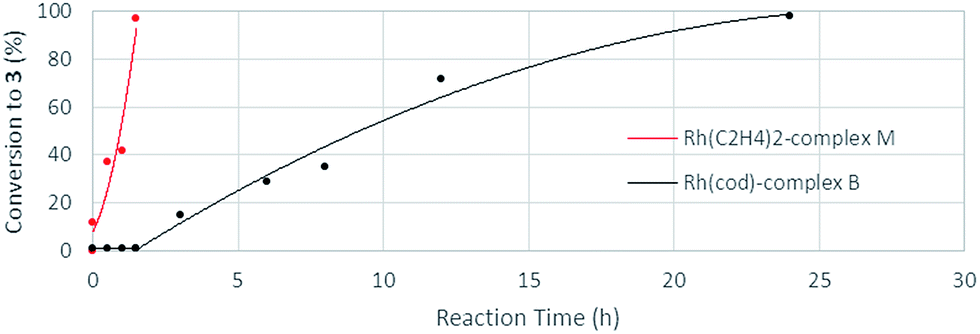 | ||
| Fig. 7 Kinetic profiles of complexes B and M as catalysts in parallel dialkenylation reactions of 1 with diphenylacetylene. | ||
Conclusions
In conclusion, divergent highly site-selective control in the direct functionalization of both aryl and heteroaryl C–H bonds of N-substituted picolinamide substrates has been cleanly achieved by simply using either a RhI or RhIII catalyst precursor, either using [RhCp*Cl2]2/AgSbF6/Cu(OAc)2 or [Rh(cod)Cl]2/AgSbF6/NaOAc. This method provides access to either isoquinoline derivatives or ortho-olefinated benzylamine and phenethylamine derivatives, respectively. Some experimental mechanistic studies based on the isolation of RhI and RhIII picolinamide complexes, stoichiometric experiments and deuterium labeling studies, as well as DFT theoretical calculations, have been performed to explain this site-selective control for both the RhI and RhIII catalytic systems and the intimate involvement of the acetate ion in the mechanism of these reactions.Acknowledgements
We thank the Spanish Government (MINECO, CTQ2012-35790) for financial support. N. R. thanks the MICINN for a Ramón y Cajal contract and the European Commission for a Marie Curie Career Integration Grant (CIG: CHAAS-304085). We also thank the Centro de Computación Científica de la UAM for their generous allocation of computer time.Notes and references
- Recent general reviews on C–H functionalization: (a) J. Wencel-Delord, T. Drçge, F. Liu and F. Glorius, Chem. Soc. Rev., 2011, 40, 4740 RSC; (b) T. Newhouse and P. S. Baran, Angew. Chem., Int. Ed., 2011, 50, 3362 CrossRef CAS PubMed; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (d) L. McMurray, F. O'Hara and M. J. Gaunt, Chem. Soc. Rev., 2011, 40, 1885 RSC; (e) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215 CrossRef CAS PubMed; (f) M. C. White, Science, 2012, 335, 807 CrossRef CAS PubMed; (g) K. M. Engle, T.-S. Mei, M. Wasa and J.-Q. Yu, Acc. Chem. Res., 2012, 45, 788 CrossRef CAS PubMed; (h) J. Yamaguchi, A. D. Yamaguchi and K. Itami, Angew. Chem., Int. Ed., 2012, 51, 8960 CrossRef CAS PubMed; (i) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed; (j) N. Kuhl, M. N. Hopkinson, J. Wencel-Delord and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 10236 CrossRef CAS PubMed; (k) D. Y.-K. Chen and S. W. Youn, Chem.–Eur. J., 2012, 18, 9452 CrossRef CAS PubMed; (l) J. Wencel-Delord and F. Glorius, Nat. Chem., 2013, 5, 369 CrossRef CAS PubMed; (m) L. Ackermann, Acc. Chem. Res., 2014, 47, 281 CrossRef CAS PubMed; (n) X.-S. Zhang, K. Chen and Z.-J. Shi, Chem. Sci., 2014, 5, 2146 RSC. See also: (o) D.-G. Yu, F. de Azambuja and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 7710 CrossRef CAS PubMed.
- For elegant examples: (a) L.-C. Campeau, D. J. Chipper and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 3266 CrossRef CAS PubMed; (b) D. J. Schipper, L.-C. Campeau and K. Fagnou, Tetrahedron, 2009, 65, 3155 CrossRef CAS PubMed; (c) D. Lapointe, T. Markiewicz, C. J. Whipp, A. Toderian and K. Fagnou, J. Org. Chem., 2011, 76, 749 CrossRef CAS PubMed; (d) J. D. Dooley, S. R. Chidipudi and H. W. Lam, J. Am. Chem. Soc., 2013, 135, 10829 CrossRef CAS PubMed; (e) X. Zhang, W. Si, M. Bao, N. Asao, Y. Yamamoto and T. Jin, Org. Lett., 2014, 16, 4830 CrossRef CAS PubMed; (f) J. D. Dooley, S. R. Chidipudi and H. W. Lam, J. Am. Chem. Soc., 2013, 135, 10829 CrossRef CAS PubMed; (g) W. B. Cross, S. Razak, K. Singh and A. J. Warner, Chem.–Eur. J., 2014, 20, 13203 CrossRef CAS PubMed; (h) W. Du, Q. Gu, Z. Li and D. Yang, J. Am. Chem. Soc., 2015, 137, 1130 CrossRef CAS PubMed; (i) R. B. Bedford, S. J. Durrant and M. Montgomery, Angew. Chem., Int. Ed., 2015, 54 DOI:10.1002/anie.201502150; (j) D. Kang and S. Hong, Org. Lett., 2015, 17, 1938 CrossRef CAS PubMed. For a review on catalytic selective synthesis: (k) J. Mahatthananchai, A. M. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954 CrossRef CAS PubMed.
- For reviews on Rh-catalyzed C–H functionalization: (a) J. C. Lewis, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2008, 41, 1013 CrossRef CAS PubMed; (b) J. Bouffard and K. Itami, Top. Curr. Chem., 2010, 292, 231 CrossRef CAS; (c) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2010, 110, 624 CrossRef CAS PubMed; (d) J. Bouffard and K. Itami, Top. Curr. Chem., 2010, 292, 231 CrossRef CAS; (e) T. Satoh and M. Miura, Chem.–Eur. J., 2010, 16, 11212 CrossRef CAS PubMed; (f) D. A. Colby, A. S. Tsai, R. G. Bergman and J. A. Ellman, Acc. Chem. Res., 2012, 45, 814 CrossRef CAS PubMed; (g) G. Song, F. Wang and X. Li, Chem. Soc. Rev., 2012, 41, 3651 RSC; (h) F. W. Patureau, J. Wencel-Delord and F. Glorius, Aldrichimica Acta, 2012, 45, 31 CAS; (i) N. Kuhl, N. Schröder and F. Glorius, Adv. Synth. Catal., 2014, 356, 1443 CrossRef CAS PubMed; (j) D. Ghorai and J. Choudhury, Acc. Chem. Res., 2015, 48, 1007 CrossRef PubMed; (k) B. Ye and N. Cramer, Acc. Chem. Res., 2015, 48, 1308 CrossRef CAS PubMed.
- For selected examples of RhIII-catalyzed C–H annulative coupling reactions of (hetero)arenes involving one equivalent of an alkyne: (a) K. Ueura, T. Satoh and M. Miura, Org. Lett., 2007, 9, 1407 CrossRef CAS PubMed; (b) L. Li, W. W. Brennessel and W. D. Jones, J. Am. Chem. Soc., 2008, 130, 12414 CrossRef CAS PubMed; (c) D. R. Stuart, M. Bertrand-Laperle, K. M. N. Burgess and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 16474 CrossRef CAS PubMed; (d) N. Guimond and K. Fagnou, J. Am. Chem. Soc., 2009, 131, 12050 CrossRef CAS PubMed; (e) T. Fukutani, N. Umeda, K. Hirano, T. Satoh and M. Miura, Chem. Commun., 2009, 5141 RSC; (f) D. R. Stuart, P. Alsabeh, M. Kuhn and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 18326 CrossRef CAS PubMed; (g) S. Rakshit, F. W. Patureau and F. Glorius, J. Am. Chem. Soc., 2010, 132, 9585 CrossRef CAS PubMed; (h) N. Guimond, C. Gouliaras and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6908 CrossRef CAS PubMed; (i) T. K. Hyster and T. Rovis, J. Am. Chem. Soc., 2010, 132, 10565 CrossRef CAS PubMed; (j) K. Morimoto, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2010, 12, 2068 CrossRef CAS PubMed; (k) J. Chen, G. Song, C.-L. Pan and X. Li, Org. Lett., 2010, 12, 5426 CrossRef CAS PubMed; (l) P. C. Too, Y.-F. Wang and S. Chiba, Org. Lett., 2010, 12, 5688 CrossRef CAS PubMed; (m) K. Muralirajan, K. Parthasarathy and C.-H. Cheng, Angew. Chem., Int. Ed., 2011, 50, 4169 CrossRef CAS PubMed; (n) F. W. Patureau, T. Besset, N. Kuhl and F. Glorius, J. Am. Chem. Soc., 2011, 133, 2154 CrossRef CAS PubMed; (o) N. Guimond, S. I. Gorelsky and K. Fagnou, J. Am. Chem. Soc., 2011, 133, 6449 CrossRef CAS PubMed; (p) Y.-F. Wang, K. K. Toh, J.-Y. Lee and S. Chiba, Angew. Chem., Int. Ed., 2011, 50, 5927 CrossRef CAS PubMed; (q) M. P. Huestis, L. Chan, D. R. Stuart and K. Fagnou, Angew. Chem., Int. Ed., 2011, 50, 1338 CrossRef CAS PubMed; (r) X. Zhang, D. Chen, M. Zhao, J. Zhao, A. Jia and X. Li, Adv. Synth. Catal., 2011, 353, 719 CrossRef CAS PubMed; (s) H. Wang, C. Grohmann, C. Nimphius and F. Glorius, J. Am. Chem. Soc., 2012, 134, 19592 CrossRef CAS PubMed; (t) J. Jayakumar, K. Parthasarathy and C.-H. Cheng, Angew. Chem., Int. Ed., 2012, 51, 197 CrossRef CAS PubMed; (u) M. V. Pham, B. Ye and N. Cramer, Angew. Chem., Int. Ed., 2012, 51, 10610 CrossRef CAS PubMed; (v) X. Xu, Y. Liu and C.-M. Park, Angew. Chem., Int. Ed., 2012, 51, 9372 CrossRef CAS PubMed; (w) B.-J. Li, H.-Y. Wang, Q.-L. Zhu and Z.-J. Shi, Angew. Chem., Int. Ed., 2012, 51, 3948 CrossRef CAS PubMed; (x) X. Tan, B. Liu, X. Li, B. Li, S. Xu, H. Song and B. Wang, J. Am. Chem. Soc., 2012, 134, 16163 CrossRef CAS PubMed; (y) W. Dong, L. Wang, K. Parthasarathy, F. Pan and C. Bolm, Angew. Chem., Int. Ed., 2013, 52, 11573 CrossRef CAS PubMed; (z) B. Liu, C. Song, C. Sun, S. Zhou and J. Zhu, J. Am. Chem. Soc., 2013, 135, 16625 CrossRef CAS PubMed; (a a) D. Zhao, Z. Shi and F. Glorius, Angew. Chem., Int. Ed., 2013, 52, 12426 CrossRef CAS PubMed; (a b) C. Wang, H. Sun, Y. Fang and Y. Huang, Angew. Chem., Int. Ed., 2013, 52, 5795 CrossRef CAS PubMed; (a c) C. Wang and Y. Huang, Org. Lett., 2013, 15, 5294 CrossRef CAS PubMed; (a d) D.-S. Kim, J.-W. Park and C.-H. Jun, Adv. Synth. Catal., 2013, 355, 2667 CrossRef CAS PubMed; (a e) L. Zheng and R. Hua, Chem.–Eur. J., 2014, 20, 2352 CrossRef CAS PubMed; (a f) X. G. Li, K. Liu, G. Zou and P. N. Liu, Adv. Synth. Catal., 2014, 356, 1496 CrossRef CAS PubMed; (a g) K. Muralirajana and C.-H. Cheng, Adv. Synth. Catal., 2014, 356, 1571 CrossRef PubMed; (a h) Y. Hoshino, Y. Shibata and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1577 CrossRef CAS PubMed; (a i) M. Fukui, Y. Hoshino, T. Satoh, M. Miura and K. Tanaka, Adv. Synth. Catal., 2014, 356, 1638 CrossRef CAS PubMed; (a j) L. Xing, Z. Fan, C. Hou, G. Yong and A. Zhang, Adv. Synth. Catal., 2014, 356, 972 CrossRef CAS PubMed; (a k) J. Jayakumar, K. Parthasarathy, Y.-H. Chen, T.-H. Lee, S.-C. Chuang and C.-H. Cheng, Angew. Chem., Int. Ed., 2014, 53, 9889 CrossRef CAS PubMed; (a l) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188 CrossRef CAS PubMed; (a m) T. Iitsuka, K. Hirano, T. Satoh and M. Miura, Chem.–Eur. J., 2014, 20, 385 CrossRef CAS PubMed; (a n) D.-G. Yu, F. de Azambuja, T. Gensch, C. G. Daniliuc and F. Glorius, Angew. Chem., Int. Ed., 2014, 53, 9650 CrossRef CAS PubMed; (a o) D. Y. Li, H. J. Chen and P. N. Liu, Org. Lett., 2014, 16, 6176 CrossRef CAS PubMed; (a p) A. Seoane, N. Casanova, N. Quiñones, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2014, 136, 834 CrossRef CAS PubMed; (a q) J. Jia, J. Shi, J. Zhou, X. Liu, Y. Song, H. E. Xu and W. Yi, Chem. Commun., 2015, 51, 2925 RSC; (a r) Z. Fan, S. Song, W. Li, K. Geng, Y. Xu, Z.-H. Miao and A. Zhang, Org. Lett., 2015, 17, 310 CrossRef CAS PubMed; (a s) X.-F. Yang, X.-H. Hu and T.-P. Loh, Org. Lett., 2015, 17, 1481 CrossRef CAS PubMed; (a t) D. Ghorai and J. Choudhury, ACS Catal., 2015, 5, 2692 CrossRef CAS; (a u) S. Duttwyler, C. Lu, A. L. Rheingold, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2012, 134, 4064 CrossRef CAS PubMed; (a v) R. Azpíroz, A. Di Giuseppe, R. Castarlenas, J. J. Pérez-Torrente and L. A. Oro, Chem.–Eur. J., 2013, 19, 3812 CrossRef PubMed; (a w) Q.-R. Zhang, J.-R. Huang, W. Zhang and L. Dong, Org. Lett., 2014, 16, 1684 CrossRef CAS PubMed; (a x) C.-Z. Luo, J. Jayakumar, P. Gandeepan, Y.-C. Wu and C.-H. Cheng, Org. Lett., 2015, 17, 924 CrossRef CAS PubMed; (a y) S. Zhao, R. Yu, W. Chen, M. Liu and H. Wu, Org. Lett., 2015, 17, 2828 CrossRef CAS PubMed; (b `) Y. Yang, M.-B. o. Zhou, X.-H. Ouyang, R. Pi, R.-J. Song and J.-H. Li, Angew. Chem., Int. Ed., 2015, 54, 6595 CrossRef CAS PubMed; (b a) J. Zheng, S.-B. Wang, C. Zheng and S.-L. You, J. Am. Chem. Soc., 2015, 137, 4880 CrossRef CAS PubMed; (b b) R. B. Dateer and S. Chang, J. Am. Chem. Soc., 2015, 137, 4908 CrossRef CAS PubMed; (b c) L. Qiu, D. Huang, G. Xu, Z. Dai and J. Sun, Org. Lett., 2015, 17, 1810 CrossRef CAS PubMed; (b d) Y. Zhao, F. Han, L. Yang and C. Xia, Org. Lett., 2015, 17, 1477 CrossRef CAS PubMed; (b e) Z. Qi, S. Yu and X. Li, J. Org. Chem., 2015, 80, 3471 CrossRef CAS PubMed; (b f) S. Chen, R. G. Bergman and J. A. Ellman, Org. Lett., 2015, 17, 2567 CrossRef CAS PubMed; (b g) T. Litsuka, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2015, 80, 2804 CrossRef PubMed; (b h) J. Jia, J. Shi, J. Zhou, X. Liu, Y. Song, H. E. Xu and W. Yi, Chem. Commun., 2015, 51, 2925 RSC; (b i) J.-R. Huang, L. Qin, Y.-Q. Zhu, Q. Song and L. Dong, Chem. Commun., 2015, 51, 2844 RSC.
- For RhIII-catalyzed aromatic homologation via C–H coupling with two alkyne molecules: (a) N. Umeda, H. Tsurugi, T. Satoh and M. Miura, Angew. Chem., Int. Ed., 2008, 47, 4019 CrossRef CAS PubMed; (b) G. Song, D. Chen, C.-L. Pan, R. H. Crabtree and X. Li, J. Org. Chem., 2010, 75, 7487 CrossRef CAS PubMed; (c) S. Mochida, M. Shimizu, K. Hirano, T. Satoh and M. Miura, Chem.–Asian J., 2010, 5, 847 CrossRef CAS PubMed; (d) N. Umeda, K. Hirano, T. Satoh, N. Shibata, H. Sato and M. Miura, J. Org. Chem., 2011, 76, 13 CrossRef CAS PubMed; (e) G. Song, X. Gong and X. Li, J. Org. Chem., 2011, 76, 7583 CrossRef CAS PubMed; (f) T. Fukutani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2011, 76, 2867 CrossRef CAS PubMed; (g) Z. Shi, C. Tang and N. Jiao, Adv. Synth. Catal., 2012, 354, 2695 CrossRef CAS PubMed; (h) B. Li, J. Ma, Y. Liang, N. Wang, S. Xu, H. Song and B. Wang, Eur. J. Org. Chem., 2013, 1950 CrossRef CAS PubMed; (i) D. Zhao, C. Nimphius, M. Lindale and F. Glorius, Org. Lett., 2013, 15, 4504 CrossRef CAS PubMed; (j) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2014, 16, 1188 CrossRef CAS PubMed; (k) M. V. Pham and N. Cramer, Angew. Chem., Int. Ed., 2014, 53, 3484 CrossRef CAS PubMed; (l) B. Liu, F. Hu and B.-F. Shi, Adv. Synth. Catal., 2014, 356, 2688 CrossRef CAS PubMed. See also: (m) H. Wang, Y. Wang, H. Yang, C. Tan, Y. Jiang, Y. Zhao and H. Fu, Adv. Synth. Catal., 2015, 357, 489 CrossRef CAS PubMed; (n) J. Wang, D. Qin, J. Lan, Y. Cheng, S. Zhang, Q. Guo, J. Wu, D. Wu and J. You, Chem. Commun., 2015, 51, 6337 RSC; (o) H. Wang, Y. Wang, H. Yang, C. Tan, Y. Jiang, Y. Zhao and H. Fu, Adv. Synth. Catal., 2015, 357, 489 CrossRef CAS PubMed. For an example involving an initial transmetalation from a boronic acid derivative: (p) T. Fukutani, K. Hirano, T. Satoh and M. Miura, Org. Lett., 2009, 11, 5198 CrossRef CAS PubMed.
- For C–H vinylation of (hetero)arenes with alkynes: (a) U. Dürr and H. Kisch, Synlett, 1997, 1335 CrossRef; (b) Y.-G. Lim, K.-H. Lee, B. T. Koo and J.-B. Kang, Tetrahedron Lett., 2001, 42, 7609 CrossRef CAS; (c) T. Katagiri, T. Mukai, T. Satoh, K. Hirano and M. Miura, Chem. Lett., 2009, 38, 118 CrossRef CAS; (d) D. J. Schipper, M. Hutchinson and K. Fagnou, J. Am. Chem. Soc., 2010, 132, 6910 CrossRef CAS PubMed; (e) J. Ryu, S. H. Cho and S. Chang, Angew. Chem., Int. Ed., 2012, 51, 3677 CrossRef CAS PubMed; (f) B. Liu, T. Zhou, B. Li, S. Xu, H. Song and B. Wang, Angew. Chem., Int. Ed., 2014, 53, 4191 CrossRef CAS PubMed; (g) T. Shibata and Y. Matsuo, Adv. Synth. Catal., 2014, 356, 1516 CrossRef CAS PubMed; (h) M.-B. Zhou, R. Pi, M. Hu, Y. Yang, R.-J. Song, Y. Xia and J.-H. Li, Angew. Chem., Int. Ed., 2014, 53, 11338 CrossRef CAS PubMed; (i) S. Sharma, S. Han, Y. Shin, N. K. Mishra, H. Oh, J. Park, J. H. Kwak, B. S. Shin, Y. H. Jung and I. S. Kim, Tetrahedron Lett., 2014, 55, 3104 CrossRef CAS PubMed; (j) Z.-C. Qian, J. Zhou, B. Li, F. Hu and B.-F. Shi, Org. Biomol. Chem., 2014, 12, 3594 RSC; (k) W. Zhou, Y. Yang, Z. Wang and G.-J. Deng, Org. Biomol. Chem., 2014, 12, 251 RSC; (l) B. Chen, Y. Jiang, J. Cheng and J.-T. Yu, Org. Biomol. Chem., 2015, 13, 2901 RSC; (m) K. Nobushige, K. Hirano, T. Satoh and M. Miura, Tetrahedron, 2015 DOI:10.1016/j.tet.2015.03.046.
- For RhIII-catalyzed C–H activation of heterocycles containing a basic nitrogen: (a) T. K. Hyster and T. Rovis, Chem. Commun., 2011, 47, 11846 RSC; (b) N. Schröder, F. Lied and F. Glorius, J. Am. Chem. Soc., 2015, 137, 1448 CrossRef PubMed, and ref. 5f.
- A. M. Martínez, N. Rodríguez, R. Gómez Arrayás and J. C. Carretero, Chem. Commun., 2014, 50, 6105 RSC.
- For the pioneering work of COPy-assisted C–H reactions: (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed. For selected recent examples: (b) S.-Y. Zhang, G. He, W. A. Nack, Y. Zhao, Q. Li and G. Chen, J. Am. Chem. Soc., 2013, 135, 2124 CrossRef CAS PubMed; (c) R. Odani, K. Hirano, T. Satoh and M. Miura, J. Org. Chem., 2013, 78, 11045 CrossRef CAS PubMed; (d) L.-S. Zhang, G. Chen, X. Wang, Q.-Y. Guo, X.-S. Zhang, F. Pan, K. Chen and Z.-J. Shi, Angew. Chem., Int. Ed., 2014, 53, 3899 CrossRef CAS PubMed. For reviews on directed C–H functionalizations: (e) G. Rousseau and B. Breit, Angew. Chem., Int. Ed., 2011, 50, 2450 CrossRef CAS PubMed; (f) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed.
- For rare exceptions: (a) A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797 CrossRef CAS PubMed; (b) X. Cong and X. Zeng, Org. Lett., 2014, 16, 3716 CrossRef CAS PubMed; (c) X.-F. Xia, S.-L. Zhu, Z. Gu and H. Wang, RSC Adv., 2015, 5, 28892 RSC; (d) R. Thenarukandiyil and J. Choudhury, Organometallics, 2015, 34, 1890 CrossRef CAS.
- J. Zhou, B. Li, Z.-C. Qian and B.-F. Shi, Adv. Synth. Catal., 2014, 356, 1038 CrossRef CAS PubMed.
- For Rh-catalyzed C–H functionalization of benzylamine derivatives, see: (a) K. Morimoto, K. Hirano, T. Satoh and M. Miura, Chem. Lett., 2011, 40, 600 CrossRef CAS; (b) D.-S. Kim, J.-W. Park and C.-H. Jun, Adv. Synth. Catal., 2013, 355, 2667 CrossRef CAS PubMed. For C–H functionalization of N-(COPy) phenethylamine derivatives, see: (c) E. T. Nadres and O. Daugulis, J. Am. Chem. Soc., 2012, 134, 7 CrossRef CAS PubMed. For other C–H functionalization of benzylamines and β-arylethylamines: (d) G. Cai, Y. Fu, Y. Li, X. Wan and Z. Shi, J. Am. Chem. Soc., 2007, 129, 7666 CrossRef CAS PubMed; (e) T.-S. Mei, X. Wang and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 10806 CrossRef CAS PubMed; (f) J.-J. Li, T.-S. Mei and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 6452 CrossRef CAS PubMed; (g) C. J. Vickers, T.-S. Mei and J.-Q. Yu, Org. Lett., 2010, 12, 2511 CrossRef CAS PubMed; (h) B. Haffemayer, M. Gulias and M. J. Gaunt, Chem. Sci., 2011, 2, 312 RSC.
- ESI.†.
- For a review: K. W. Bentley, Nat. Prod. Rep., 1992, 8, 365 RSC.
- Unfortunately, attempts to utilize aryl/alkyl or dialkyl internal alkynes, or terminal alkynes failed.
- RhI-catalyzed C–H coupling of arenes with alkynes has received much less attention: (a) S.-G. Lim, J. H. Lee, C. W. Moon, J.-B. Hong and C.-H. Jun, Org. Lett., 2003, 5, 2759 CrossRef CAS PubMed; (b) Y. Shibata, Y. Otake, M. Hirano and K. Tanaka, Org. Lett., 2009, 11, 689 CrossRef CAS PubMed; (c) K. Parthasarathy and C.-H. Cheng, J. Org. Chem., 2009, 74, 9359 CrossRef CAS PubMed. See also: (d) D. A. Colby, R. G. Bergman and J. A. Ellman, J. Am. Chem. Soc., 2008, 130, 3645 CrossRef CAS PubMed; (e) J. Kwak, Y. Ohk, Y. Jung and S. Chang, J. Am. Chem. Soc., 2012, 134, 17778 CrossRef CAS PubMed. For a rhodium(I)-catalyzed alkylation of aromatic amides containing an 8-aminoquinoline with α,β-unsaturated esters, see: (f) K. Shibata and N. Chatani, Org. Lett., 2014, 16, 5148 CrossRef CAS PubMed.
- Other RhI precatalysts were less effective (see the ESI† for details).
- Di-ortho-tolylacetylene proved to be completely unreactive under the optimized reaction conditions even after prolonged reaction times.
- The key directing role of the COPy unit is demonstrated by the lack of reactivity of the corresponding N-COPh protected substrates (see the ESI†).
- For examples of PdII-catalyzed olefination protocols featuring tolerance with regard to the tether length: (a) A. G. Rubia, R. Gómez Arrayás and J. C. Carretero, Angew. Chem., Int. Ed., 2011, 50, 10927 CrossRef PubMed; (b) D.-H. Wang, T.-S. Mei and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 17676 CrossRef CAS PubMed; (c) D.-H. Wang, K. M. Engle, B.-F. Shi and J.-Q. Yu, Science, 2010, 327, 315 CrossRef CAS PubMed; (d) G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391 CrossRef CAS PubMed.
- See the ESI of ref. 8 for the X-ray structure of RhIII-complex A.
- A. Di Giuseppe, R. Castarlenas, J. J. Pérez-Torrente, F. J. Lahoz, V. Polo and L. A. Oro, Angew. Chem., Int. Ed., 2011, 50, 3938 CrossRef CAS PubMed.
- For a review on “rollover” cyclometalation, see: B. Butschke and H. Schwarz, Chem. Sci., 2012, 3, 308 RSC . For a recent example of rollover cyclometalation in Rh-catalysis, see: ref. 16e.
- Complexes modA and modB resulted in being 2.9 and 11.5 kcal mol−1 less stable than the corresponding calculated complexes A and B (see the ESI†).
- With regard to the apparently very low energy barrier, it must be pointed out that the catalytically active intermediate modB has to be formed from the catalytically competent complex B (11.5 kcal mol−1 more stable) by ligand exchange between the acetate ion and “cod”. In this process, the alkyne partner may be involved, according to the results of the H/D exchange experiments (Scheme 11 and vide supra).
- The corresponding alkyne insertion transition state in the case of pyridine functionalization lies close to the previous C–H activation transition state TS(I-II)Ba (24.5 kcal mol−1, see vide supra and in the ESI†). The model transition state in which a C–C bond instead of a C–H one is formed (affording species J instead of K, Scheme 12) showed a much higher activation barrier (37.4 kcal mol−1, see the ESI†). When 2-butyne was used as a dialkylalkyne model, the activation barrier for this insertion step was also higher (24.5 kcal mol−1 instead of 18.2 kcal mol−1 for the diphenylacetylene, see the ESI†) which is in agreement with the lack of reactivity experimentally found for these substrates.
- RhI is frequently invoked as the catalyst species under basic conditions (see ref. 16e and references therein).
- The corresponding complex with 2-butyne resulted in being 4.6 kcal mol−1 less stable than complex B, indicating that in this case the “cod” ligand likely stays bonded to the Rh atom. This fact along with the higher activation barrier for the key transition state (see ref. 26) could justify the lack of reactivity observed with dialkylalkynes.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details as well as spectroscopic and analytical data for new compounds. CCDC 1027406–1027411. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01885d |
| This journal is © The Royal Society of Chemistry 2015 |