
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorescent carbon dot–molecular salt hydrogels†
Angelina
Cayuela
a,
Stuart R.
Kennedy
 b,
M. Laura
Soriano
a,
Christopher D.
Jones
b,
Miguel
Valcárcel
*a and
Jonathan W.
Steed
*b
b,
M. Laura
Soriano
a,
Christopher D.
Jones
b,
Miguel
Valcárcel
*a and
Jonathan W.
Steed
*b
aDepartment of Analytical Chemistry, Marie Curie Building, Campus de Rabanales, University of Córdoba, E-14071 Córdoba, Spain. E-mail: qa1vacam@uco.es; Tel: +34 957 218616
bDepartment of Chemistry, University of Durham, South Road, DH1 3LE, UK. E-mail: jon.steed@durham.ac.uk; Fax: +44 (0)191 384 4737; Tel: +44 (0)191 334 2085
First published on 29th July 2015
Abstract
The incorporation of functionalised carbon nanodots within a novel low molecular weight salt hydrogel derived from 5-aminosalicylic acid is reported. The carbon dots result in markedly enhanced gelation properties, while inclusion within the hydrophobic gel results in a dramatic fluorescence enhancement for the carbon nanomaterials. The resulting hybrid CD gels exhibit a useful sensor response for heavy metal ions, particularly Pb2+.
Introduction
Impressive progress has been made in photoluminescent (PL) carbon-based dots (CDs) since their discovery in 2004,1 in many applications such as bioimaging, drug delivery and analytical sensing. Particular applications include engineering bright nanoprobes towards pollutants and metal ions2–7 because of their stability, high water solubility, biocompatibility low toxicity, easy surface modification and excellent PL properties. To date CDs have been used in aqueous solution, however incorporating them into a gel medium offers particular promise in stabilising and immobilising the nanoparticles, and may result in interesting effects on the properties of the gel–nanomaterial hybrid itself. Moreover, shielding the CDs within a more hydrophobic environment may result in considerable enhancement of the CD fluorescence. Recently, a few researchers have incorporated carbon nanomaterials in ionic liquids or into agarose hydrogels to modify their electrochemical properties,8 as part of a sensing system for heavy metal ions9 or as a vehicle for drug delivery.10Gels are flexible, soft materials that behave as solids on the analytical timescale, but are composed predominantly of a fluid phase.11,12 There have been a number of interesting recent reports concerning the incorporation of nanomaterials into gel phase media.13–19 In particular, introduction of carbon nanomaterials such as carbon nanotubes and graphene has been shown to result in gel stabilization and modification of the carbon nanomaterial properties.13 There has been considerable recent interest in small-molecule supramolecular gels composed of self-assembled fibrillar networks (SAFINs) derived from relatively well-defined interactions between low molecular weight gelators (LMWG).11,20–29 These small-molecule SAFINs offer the opportunity to design desirable gel properties. Gelator preparation is generally straightforward allowing considerable synthetic versatility and variability, and the gels often exhibit readily reversible gelation behaviour.12 The range of structure types of LMWG is very broad, comprising amides, nucleobases, fatty acids and dendrimers, for example.12 Within this context, bis(urea) derivatives30–34 are particularly versatile because the gel-forming tendency of the urea groups is tolerant of variations in the spacer between them and the peripheral substituents, allowing the design of tailored gelators for targeted applications such as separations,35 drug delivery,36–38 polymer templating39,40 and as media to control crystal growth.41–44
In this work we report the preparation, properties and ion sensing applications of novel carbon dot–hydrogel hybrid nanomaterials using LMWGs designed to provide a hydrophobic environment suitable for fluorescent ion sensing within an aqueous medium. While polymer gels have been used in conjunction with CDs,9,18,19 to the best of our knowledge this work represents the first incorporation of CDs within an LMWG-based gel medium.
Results and discussion
Synthesis and characterization of bis(urea) gels
The bis(urea) gelators of type 1 are based on a bolaamphiphile design45 that incorporates a hydrophobic diphenylmethane-derived central spacer (which we have found commonly results in robust gelation characteristics in bis(urea) systems46) in conjunction with ionisable salicylic acid based peripheral substituents capable of imparting water solubility. Related urea and amide carboxylic acid systems have been studied previously.47–54 The bis(2-ureido-2-propyl)-m-phenylene analogues of type 2 represent comparator examples that are expected to be poor gelators as a result of the steric hindrance around the urea carbonyl group. As a result compounds with this spacer group are frequently more crystalline than most bis(ureas). In previous work we have found they act as useful model compounds allowing structural insight into the system because they are easier to isolate in a form suitable for single crystal X-ray diffraction studies.The synthesis of the salt gelators 1a and 2a was carried out in a one-step reaction between the desired isocyanate and 5-aminosalicylic acid in the presence of triethylamine base. The presence of the triethylammonium cations and the loss of the acid proton in salts 1a and 2a was clearly evident in the 1H NMR spectra and analytical data of the materials, and was confirmed by X-ray crystallography in the case of 2a (vide infra), Fig. 1. The analogous free acid 1b was prepared by sonicating 1a in 1 M hydrochloric acid for 30 minutes. Compound 2b was prepared by dissolving 2a in water followed by the addition of 1 M hydrochloric acid upon which the compound precipitated.
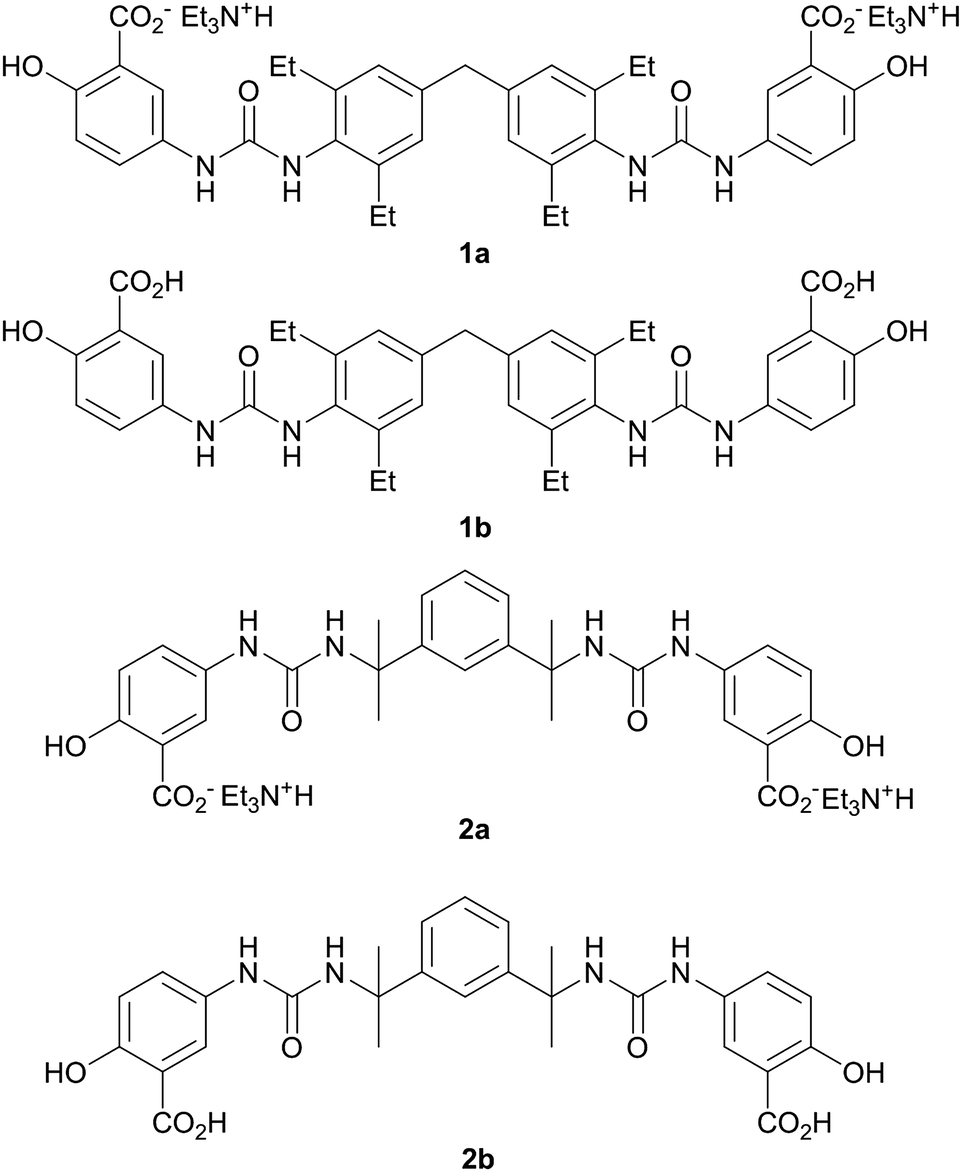 | ||
| Fig. 1 The triethylammonium salt gelators 1a and 2a, and the analogous free acids 1b and 2b. | ||
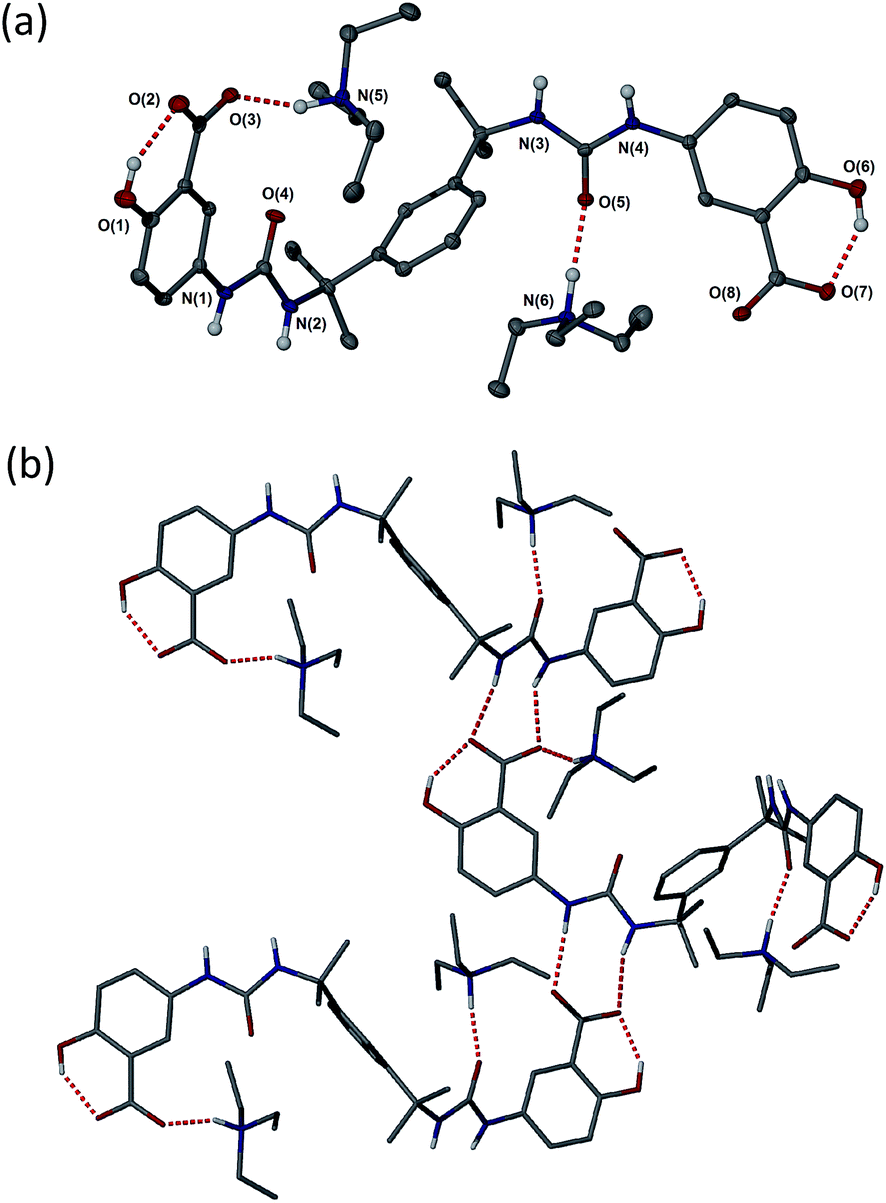 | ||
| Fig. 2 (a) Molecular structure of 2a·propan-1-ol (disordered propan-1-ol and CH hydrogen atoms omitted). (b) Hydrogen bonding in the structure of 2a·propan-1-ol. Selected hydrogen bonding distances (Å): O1⋯O2 2.577(5), N1⋯O8 2.811(6), N4⋯O3 2.832(5), N5⋯O3 2.700(6), O6⋯O7 2.505(5) and N6⋯O5 2.775(5). | ||
Compounds of type 1 and 2 as both free acids and triethylammonium salts were screened for gelation and crystallisation in a wide range of solvents. All gelation experiments were carried out at 1 wt% using 5 mg of 1a in 0.5 mL of solvent. Samples were sonicated for 30 seconds followed by careful heating in a sealed vial until the solid dissolved. Gelation was observed on cooling to ambient temperature.
Compound 1 proved to be a versatile gelator in both salt (1a) and neutral (1b) forms. Salt 1a forms gels in a variety of solvents outlined in Table S1 (ESI†). Gels formed upon heating and subsequent cooling in a total of 26 out of 50 solvents studied and partial gels were also observed in 4 solvents. The compound gels a range of polar solvents with both hydrogen bond donor (e.g. alcohols) and acceptor character (e.g. pyridine derivatives). The neutral acid form 1b gels in 12 of the 50 solvents studied and partial gels were also observed in 10 solvents outlined in Table S2 (ESI†). Interestingly, room temperature gelation of 1a on sonication was also observed in a number of instances (Table S3†).55,56 Comparison between room temperature sonogels and gels produced by a combination of ultrasound and thermal methods revealed that the thermally treated materials are more robust and longer lived, although some minor degradation was evident in the colouration of some gels (Tables S1 and S3,†Fig. 3). Some of the gels formed by the sonication-only process appeared to collapse more readily and in the case of ethanol, only persist for around one hour.
 | ||
| Fig. 3 Comparison between gels of 1a formed by a combination of ultrasound and heat treatment (labelled 1) and room temperature sonogels (2) at 1 wt%. | ||
While compound 1b gels only organic solvents, the behaviour of salt 1a in water is particularly intriguing with no gelation observed for several days after dissolution at 1 wt%. However from 72 hours onwards partial gels began to form and after 10 days robust, transparent self-supporting gels form implying very slow fibre assembly within the competitive aqueous medium (ESI, Fig. S5†). Increasing the concentration to 2 wt% results in gelation in less than one day. Gelation is also markedly accelerated by lowering the pH (gelation at 1 wt% is rapid in the presence of 1 μL of concentrated sulphuric acid). Subsequent addition of base in the form of NEt3 results in collapse of the gel.
As anticipated, compound 2 with its bulky spacer did not form gels in any solvents (Tables S4 and S5†), an outcome attributed to steric hindrance of the urea carbonyl group preventing urea tape formation (in comparison the aromatic groups of 1 may rotate to be orthogonal to the urea plane). Crystals of 2a suitable for single crystal X-ray structure determination were obtained by slow evaporation of a solution in propan-1-ol. While not from a gel, the crystal structure serves to provide insight into the kinds of interactions possible between gelators bearing these functional groups, the triethyl ammonium counter cation and the solvent. The crystals proved to be a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 solvate with two crystallographically unique HNEt3+ cations hydrogen bonded to one carboxylate group and one urea carbonyl group of a single unique bis(urea) molecule in the presence of a highly disordered molecule of propan-1-ol. The solvent molecule hydrogen bonds to a phenolic hydroxyl group. Typically gels of bis-ureas arise from the formation of α-tape hydrogen bonding interactions between the urea groups in neighbouring molecules based on a six-membered hydrogen bonded ring.34,57,58 In the present structure however, the Et3NH+ cations effectively inhibit urea tape formation by hydrogen bonding to one of the urea groups and sterically blocking the other. Instead, the strong hydrogen bond acceptor nature of the carboxylate groups results in the formation of an alternative, twisted non-planar 8-membered hydrogen-bonded ring between the urea functionalities and the carboxylate groups (Fig. 2b). The phenolic OH groups are both involved in intramolecular hydrogen to the carboxylate acceptors.
:1 solvate with two crystallographically unique HNEt3+ cations hydrogen bonded to one carboxylate group and one urea carbonyl group of a single unique bis(urea) molecule in the presence of a highly disordered molecule of propan-1-ol. The solvent molecule hydrogen bonds to a phenolic hydroxyl group. Typically gels of bis-ureas arise from the formation of α-tape hydrogen bonding interactions between the urea groups in neighbouring molecules based on a six-membered hydrogen bonded ring.34,57,58 In the present structure however, the Et3NH+ cations effectively inhibit urea tape formation by hydrogen bonding to one of the urea groups and sterically blocking the other. Instead, the strong hydrogen bond acceptor nature of the carboxylate groups results in the formation of an alternative, twisted non-planar 8-membered hydrogen-bonded ring between the urea functionalities and the carboxylate groups (Fig. 2b). The phenolic OH groups are both involved in intramolecular hydrogen to the carboxylate acceptors.
Carbon dot hybrid gels
Despite the slow assembly of hydrogels of 1a the final gels proved to be robust and transparent and hence were investigated as host media for fluorescent carbon dots. Four different types of carbon dot were investigated. High luminescence CDs (c-CDs) were prepared from microcrystalline cellulose (MCC) whereas low luminescence CDs were obtained from multi-walled carbon nanotubes (MWCNTs). The nanotube-derived CDs were passivated with acetone (p-CDs) and in some cases subsequently functionalised to give thiol rich (t-CDs) or amine rich surfaces (a-CDs) to improve their PL properties and ability to interact with the gel and with potential analytes. CD-gel hybrids were prepared by sonicating 0.5 to 4 wt% of 1a in aqueous solutions of the carbon dots ranging from 1–60 mg mL−1. The critical gelation concentration proved to be 1 wt% with solutions of 0.5 wt% not forming self-supporting gels.Remarkably, unlike the hydrogels which form over a period of 10 days at 1 wt%, gelation of all of the CD–gel hybrids occurred in a matter of minutes. This fortuitous result renders the hybrid gels much more convenient to prepare and study than the pure hydrogels and hence is of marked benefit in a practical sensing context. While the mechanism for gelation is not known the CDs may well act as a hydrophobic nucleation and cross-linking node for SAFIN assembly interacting with the fibres by hydrophobic and π–π stacking interactions. The effect is not related to the increased gelation propensity on acidification since the CD solutions are slightly basic, pH 8–9. Gels containing high concentrations of CDs proved to be opaque because of the very intense absorption of the CDs, however dilution to 1 wt% solutions containing 1 mg mL−1 CDs gave transparent gels suitable for fluorescence study, although at this low CD concentration gelation was less readily reproducible than at higher concentrations. We postulate that hydrogen bonding to the solubilising Et3NH+ cations may interfere with the gel assembly process and accordingly we investigated counter cation metathesis by adding a water soluble ionic liquid 1-butyl-3-methylimidazolium tetrafluoroborate (BMIM-BF4). The BMIM cation does not possess hydrogen bond donor functionality and hence should not interfere with gelation. Pure ionic liquids such as BMIM-BF4 are of interest as ionogels for hosting magnetic nanoparticles, for example, and silicon-functionalised carbon dots have previously been incorporated into ionic liquid based ionogels.59–61 Compound 1a does not gel pure BMIM-BF4; however, addition of 1–40 wt% of the ionic liquid to the gel–CD mixture gave robust, reproducible gels for all types of CDs. The metathesis with BMIM cations proved highly effective with the resulting hybrid 1a–CD–BMIM hydrogels forming rapidly at an optimum concentration of 2 wt% BMIM-BF4 to give robust, transparent hybrid nanomaterials materials suitable for fluorescence study.
The optimised nanomaterials of composition 1 wt% 1a in 500 μL of CD solution at 1 mg mL−1 containing 2% BMIM-BF4 are shown in Fig. 4. The hybrid gels were characterised by frequency and stress sweep rheometry and by SEM. The rheological properties of the four different CD hybrid materials were similar to one another with all exhibiting an elastic modulus G′ greater than the viscous modulus G′′, with G′ invariant with frequency, Fig. 5. At the concentration used the gels are relatively weak with G′ values of 100–300 Pa and yield stress around 1–10 Pa. The gels containing the amine-functionalised CDs proved generally to be considerably more robust that the unfunctionalised c-CDs which may indicate a specific hydrogen bonding interaction between the amine groups and the carboxylate functionalities of the gelators. The SEM images of the dried gels showed a fibrous structure consistent with similar SAFIN-based hydrogels62 with small fibres of average diameter of less than 30 nm consistent with the transparent appearance of the gels. In comparison the hydrogels before introduction of CDs give slightly larger, more tape-like fibres (ESI Fig. S4†).
 | ||
| Fig. 4 Optimised ionic-liquid CD–Gel hybrid materials of composition 1 wt% 1a in 500 μL of CD solution at 1 mg mL−1 containing 2% BMIM-BF4 (left-to-right c-CD, p-CD, t-CD and a-CD). | ||
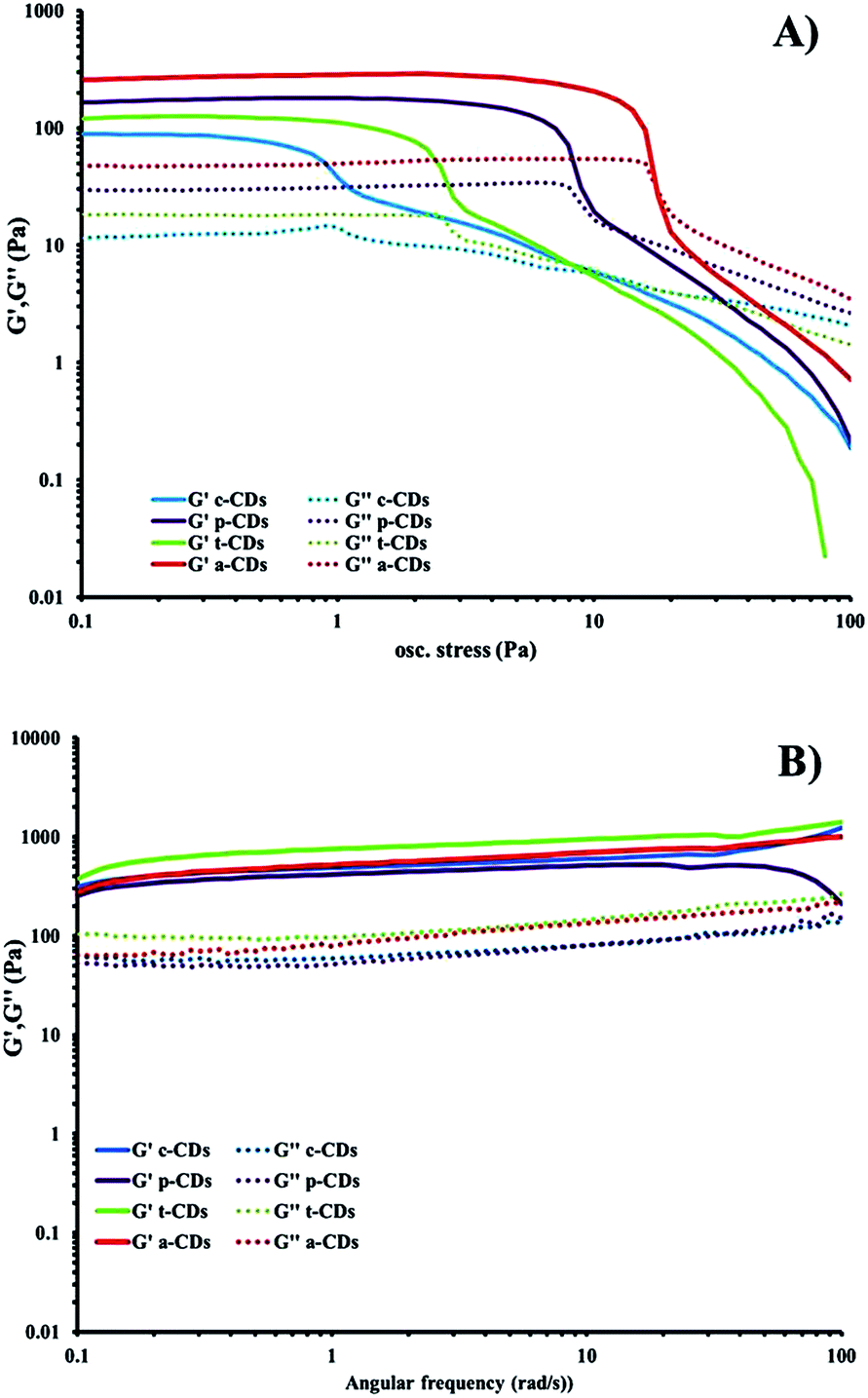 | ||
| Fig. 5 (a) Stress sweep and (b) frequency-sweep rheology of dilute ionic-liquid CD–gel hybrid materials of composition 1 wt% 1a in 500 μL of CD solution at 1 mg mL−1 containing 2% BMIM-BF4. | ||
CD-gel fluorescence
All the CDs in aqueous solution were fluorescent with a full width at half maximum (FWHM) about 100 nm which indicates their homogeneity. The bottom-up synthesised c-CDs proved to be the most fluorescent and are known to possess a higher quantum yield.63 The maximum excitation wavelength was 370 nm, shifting to 365 for the p-CDs, t-CDs and a-CDs. Fig. S1† shows the fluorescent spectra of all the CD solutions. Consistent with previous reports,64–66 the functionalised CDs were more fluorescent than the passivated p-CDs (Fig. 6). | ||
| Fig. 6 SEM micrograph of the chromium coated dried (xerogel) ionic-liquid a-CD–gel hybrid materials based on 1a. The carbon dots of average radius 3 nm are too small to be observed. | ||
Incorporation of the CDs into the gels resulted in a striking enhancement of the CD emission intensity by at least an order of magnitude for all types of CDs studied. It is well-known that the PL intensity varies significantly with the size and core structure due to the synthetic route and precursor used, the surface functionalities of CDs, as well as with the environment. As the nanodots in this case are all of similar size, the strong emission enhancement of CDs comes from the interaction of the CDs with the gelator instead of a quantum size effect. Fluorescence spectra for 1 wt% gels of 1 mg mL−1 CD solutions in the absence of the ionic liquid are shown in Fig. 7. The ionic liquid itself is also fluorescent, complicating the spectra, however the marked emission enhancement is also evident in the ionic liquid CD–gel hybrid materials, Fig. S2 (ESI†). The hydrophobic nature of the gels is likely to significantly reduce solvent quenching and the fact that the emission enhancement is so marked suggests significant interaction between the gel fibres and the CDs in a synergistic way, consistent with the marked effect the CDs have on the gelation properties.
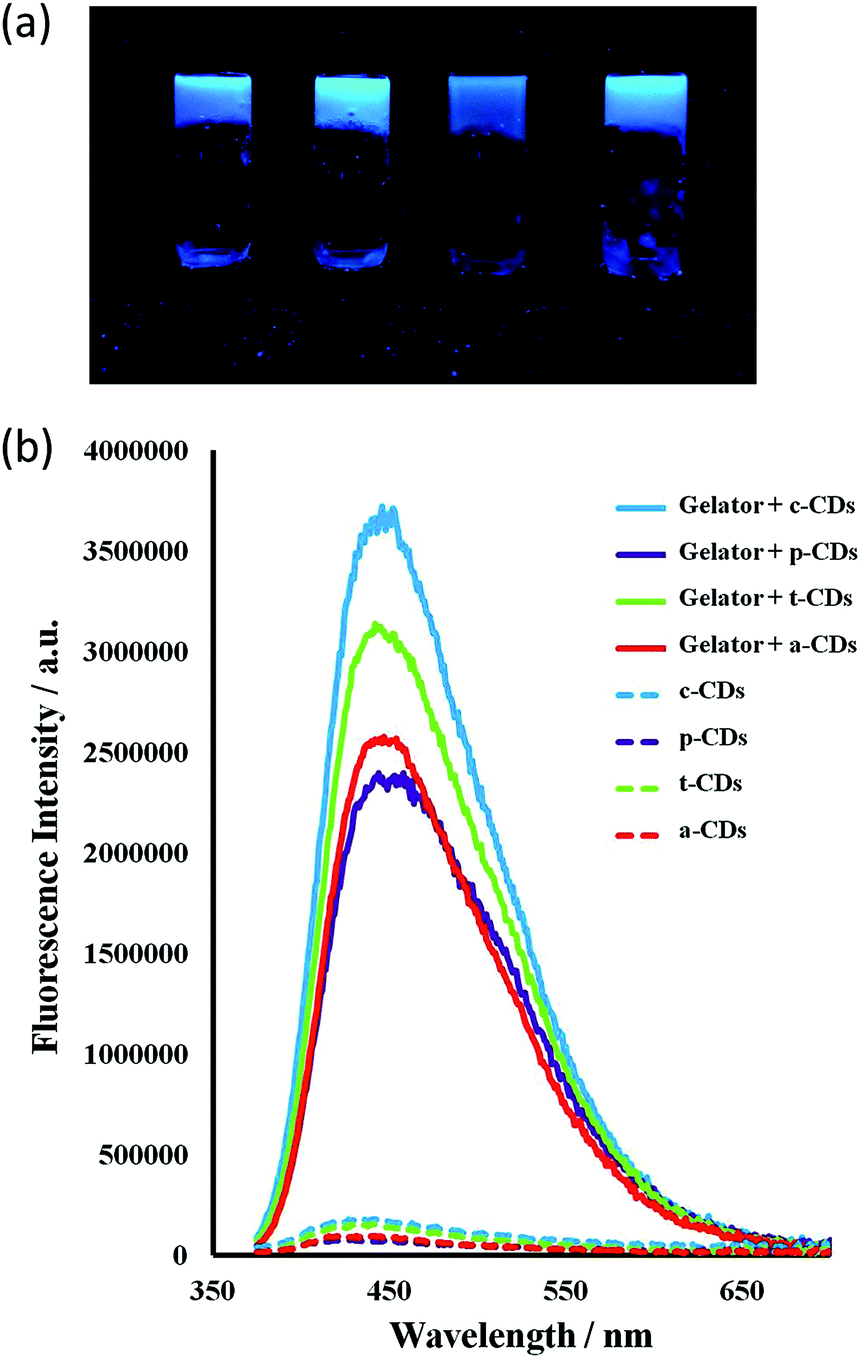 | ||
| Fig. 7 (a) a-, c-, p- and t-CD gels 1 wt% under 365 nm UV irradiation. (b) Fluorescence emission spectra of aqueous CD solutions (1 mg mL−1, λex = 365 nm for c-CDs and 370 nm for the others) and for 1 wt% CD-gels with 1a at the same CD concentration. Slits at 2 nm. | ||
The high intensity fluorescence of these novel CD gel hybrids and the possibility of tuning the properties of the materials by modification of the gelator, imidazolium cation and the size and surface functionality of the CDs offers interesting possibilities for their application as luminescent sensors, e.g. as environmental nanoprobes. CDs have been shown to be an excellent sensing platform for heavy metals,63,64 and the interaction of metal ions with the CD surface functionality may well be significantly influenced by the inclusion of the CDs within a LMWG hydrogel medium. In order to establish the feasibility of heavy metal sensing using hybrid CD gels the effect of added lead(II) and mercury(II) on the PL of the new hybrid gel system was investigated. The fluorescence of aqueous p-CD, t-CD and a-CDs solutions is moderately quenched by 10 μg mL−1 of Hg2+ and Pb2+ (I0/I = 1.1–1.25) while the luminescence of c-CDs is not affected. In the hybrid CD gel system, in addition to the marked increase in overall PL intensity, the response to Hg2+ and Pb2+ changes differentially. The response to Pb2+ of the gels containing p-CDs and t-CDs is markedly attenuated such that the luminescence is essentially unaffected, whereas the system containing the a-CDs exhibits a marked enhancement in its Pb2+ response in the gel phase (I0/I increases from 1.13 to 1.32), Fig. 8. In contrast the a-CD gel exhibits essentially no response to Hg2+. This differential behaviour suggests that the CD gels may provide a basis for discriminating closely related heavy metal ions in the environment. As a control the metal ion response was also examined in CD solution in the presence of the gelator without gel formation. The presence of the gelator in solution had no effect on the t-CD response, attenuated the response of the a-CD system and enhanced the response of the p-CDs. The significant and differential difference in sensor behaviour in the gel state is an interesting phenomenon. To further probe selectivity the response of a-CD-containing hydrogels (a-CDGs) as sensing probe towards a range of metal ions, namely Pb2+, Hg2+, Co2+, Fe3+ and Mg2+ was examined (ESI, Fig. S3†). The system proved to be clearly Pb2+ selective in the hydrogel state. The a-CDGs thus represent a new format of selective sensor for the detection of lead(II). In future work we will systematically explore the fluorescent response of these novel hybrid nanomaterials to a broader range of analytes. A further interesting application platform could also involve the use of freeze dried hybrid CD hydrogels coated with TiO2 allowing them to float on water as reporter medium.67
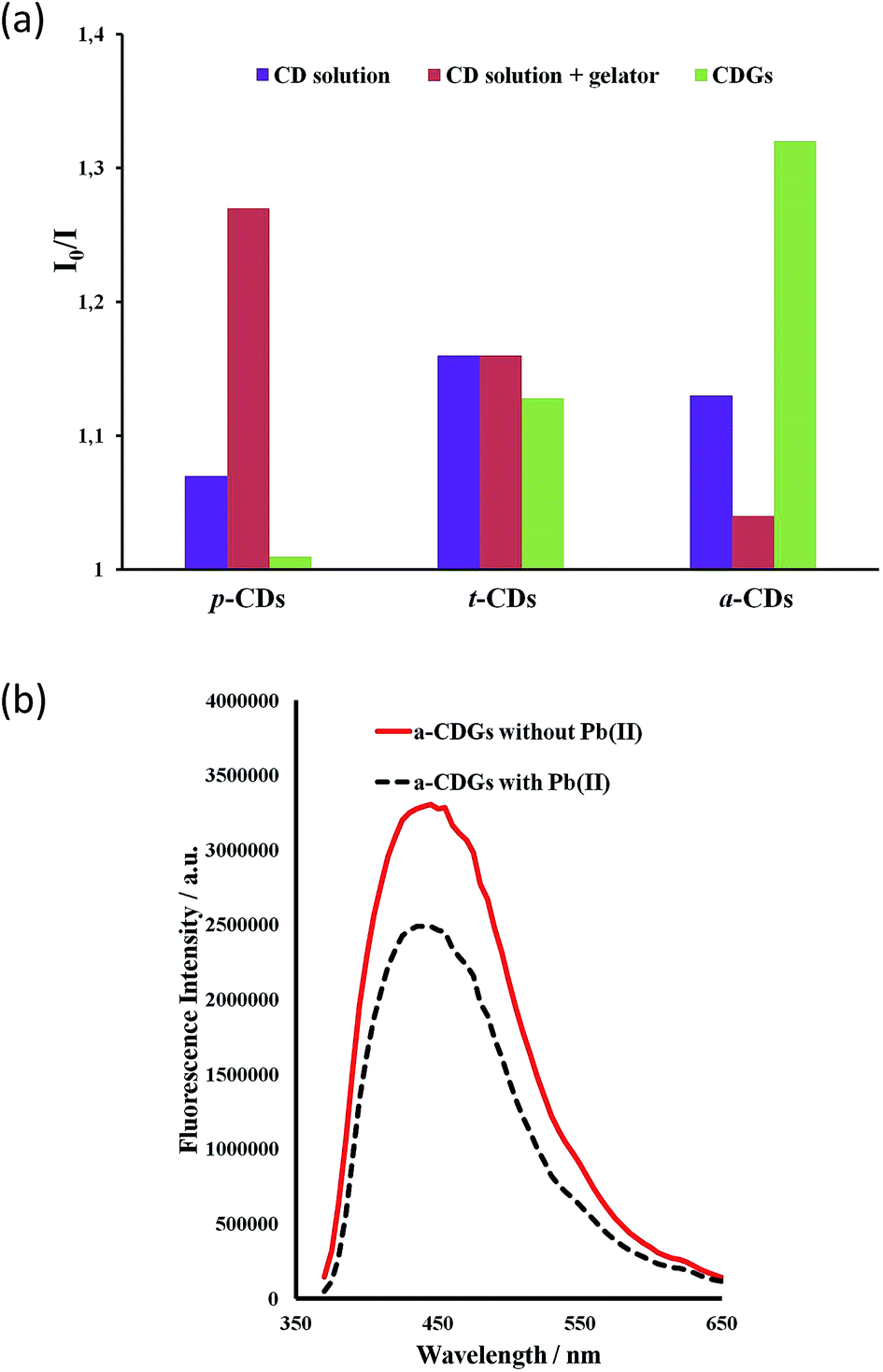 | ||
| Fig. 8 (a) Photoluminescent response to 10 μg mL−1 of Pb2+ to CD solutions, CD-containing hydrogels (CDGs) and CD solutions in the presence of dissolved gelator (1 mg mL−1, λex = 370 nm) and for 1 wt% CD–gels with 1a at the same CD concentration. (b) Fluorescence emission spectra for a-CD gel samples with and without Pb2+ (10 μg mL−1). | ||
Conclusions
Inclusion of luminescent carbon dots within a hydrophobic low molecular weight hydrogel results in a dramatic enhancement of their luminescence intensity as well as an unexpected strengthening of the gels and marked increase in their rate of self-assembly. In the gel phase the CDs exhibit a differential photoluminescent response to the presence of heavy metal ions. The novel hybrid CD–gel nanostructures reported herein offer interesting potential as highly tunable luminescent media with possible applications in environmental and biomedical heavy metal ion sensing. The potential biocompatibility of these nanostructures which are derived from non-toxic precursors may open a window for their use in medical applications such as diagnosis, drug activation or slow release.Experimental
Reagents and instrumental
Sulphuric acid (95–98%), nitric acid (69%), ethanol (99%) and chloroform were purchased from PANREAC, S.A.U. (Barcelona, Spain). N,N′-Diisopropylcarbodiimide (DIC, 98%), N-hydroxysuccinimide (NHS, 97%), triethylamine (TEA, 99%), cysteamine hydrochloride (98%), N-Boc-ethylenediamine (Boc-EDA, 98%), cellulose microcrystalline (50 μm particle size), acetone (99.5%), 1-butyl-3-methylimidazolium tetrafluoroborate (BMIM-BF4, 97%), dimethyl sulfoxide (DMSO, 99.9%), sodium carbonate (99.95%). 5-Aminosalicylic acid and the diisocyanates purchased from Sigma–Aldrich, while the multi walled carbon nanotubes were purchased from Bayer (Germany). NMR spectra were measured on a Bruker Avance-400.Synthesis
:ethanol (15 mL:1.5 mL). Triethylamine (0.78 mL, 5.59 mmol) to give a solution that was heated under reflux for 3 h. The resulting suspension was then filtered and washed with chloroform (100 mL) to give 1a as an off-white solid (0.95 g, 1.09 mmol, 67%). 1H NMR (400 MHz, DMSO-d6) δ 8.57 (s, 2H, O![[H with combining low line]](https://www.rsc.org/images/entities/char_0048_0332.gif) ), 7.68 (s, 2H, N), 7.62 (s, 2H, N) 7.31 (d, 3J = 8.7, Hz, 2H, Ar), 6.98 (2s, 6H, Ar), 6.58 (d, 3J = 8.6 Hz, 2H, Ar), 3.85 (s, 2H, C2), 3.07 (q, J = 7.3 Hz, 12H, NC2CH3), 2.55 (q, 3J = 7.7, Hz, 8H, ArC2CH3), 1.17 (t, 3J = 7.1 Hz, 18H, NCH2C3), 1.11 (t, 3J = 7.5 Hz, 12H, ArCH2C3); 13C{1H} NMR (400 MHz, DMSO-d6) δ 172.09 (
), 7.68 (s, 2H, N), 7.62 (s, 2H, N) 7.31 (d, 3J = 8.7, Hz, 2H, Ar), 6.98 (2s, 6H, Ar), 6.58 (d, 3J = 8.6 Hz, 2H, Ar), 3.85 (s, 2H, C2), 3.07 (q, J = 7.3 Hz, 12H, NC2CH3), 2.55 (q, 3J = 7.7, Hz, 8H, ArC2CH3), 1.17 (t, 3J = 7.1 Hz, 18H, NCH2C3), 1.11 (t, 3J = 7.5 Hz, 12H, ArCH2C3); 13C{1H} NMR (400 MHz, DMSO-d6) δ 172.09 (![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) O2H), 156.28 (
O2H), 156.28 (![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 142.20 (Ar), 140.02 (Ar), 132.64 (Ar), 132.19 (Ar), 126.62 (Ar), 117.43 (Ar), 112.54 (Ar), 41.20 (H2), 24.67 (H2), 15.11 (H3); ESI (m/z): 770 [M − NEt3 + H]+; analysis calc. for C49H70O8N6: C 67.56, H 8.1, N 9.65%; found: C 66.73, H 7.88, N 9.18%.
), 8.87 (s, 2H, O), 7.95 (s, 2H, N), 7.64 (s, 2H, N), 7.49 (d,3J = 8.9, Hz, 2H, Ar), 6.98 (s, 6H, Ar), 6.87 (d, 3J = 8.9 Hz, 2H, ArH), 3.85 (s, 2H, C2), 2.53 (q, 3J = 7.5 Hz, 8H, ArC2CH3), 1.10 (t, 3J = 7.5 Hz, 12H, ArCH2C3); 13C{1H} NMR (400 MHz, DMSO-d6) δ 172.30 (O2H), 157.40 (O), 154.91 (Ar), 142.42 (Ar), 139.78 (Ar), 132.63 (Ar), 130.15 (Ar), 126.66 (Ar), 119.91 (Ar), 115.69 (Ar), 45.93 (H2), 24.99 (H2), 15.17 (H3), 9.10 (H3); ESI (m/z): 667 [M − H]−; analysis calc. for C37H40O8N4: C 66.45, H 6.03, N 8.38%; found: C 63.54, H 5.67, N 7.82%. The difference can be accounted for by calculating the formula with a 0.3 molar ratio of CHCl3.
:ethanol (15 mL:1.5 mL). Triethylamine (0.78 mL, 5.59 mmol) to give a solution that was heated under reflux for 4 h. The resulting suspension was filtered and washed with chloroform (100 mL) to give 2a as an off-white solid (1.19 g, 1.59 mmol, 97%). 1H NMR (400 MHz, DMSO-d6) δ 8.14 (s, 2H, N), 7.61 (s, 2H, N), 7.46 (s, 1H, Ar) 7.26–7.13 (m, 5H, Ar), 6.53 (d, 3J = 8.6 Hz, 2H, Ar), 6.45 (s, 2H, Ar), 3.07 (q, 3J = 7.2 Hz, 12H, NC2CH3), 1.59 (s, 12H, CC3), 1.17 (t, 3J = 7.0 Hz, 18H, NCH2C3); 13C{1H} NMR (400 MHz, DMSO-d6) δ 172.64 (O2H), 157.36 (O), 157.36 (Ar), 148.70 (Ar), 130.51 (Ar), 123.75 (Ar), 122.93 (Ar), 120.91 (Ar), 119.69 (Ar), 115.79 (Ar), 54.88 (CH3), 45.81 (H2) 30.41 (H3) 9.04 (H3). ESI (m/z): 652 [M − NEt3 + H]+; analysis calc. for C40H60O8N6: C 63.81, H 8.03, N 11.16%; found: C 54.61, H 6.76, N 9.32%. The difference can be accounted for by calculating the formula with a 1.3 molar ratio of CHCl3.
), 8.36 (s, 2H, N), 7.87 (s, 2H, N), 7.44 (s, 1H, Ar), 7.33 (dd, 3J = 8.9, 2.8 Hz, 2H, ArH), 7.27–7.20 (m, 3H, ArH), 6.81 (d, 3J = 8.9 Hz, 2H, Ar), 6.46 (s, 2H, Ar), 1.59 (s, 12H, CC3). 13C{1H} NMR (400 MHz, DMSO-d6) δ 172.30 (O2H) 156.13 (O) 154.78 (Ar) 148.45 (Ar) 132.69 (Ar) 127.96 (Ar) 126.77 (Ar) 122.98 (ArC) 121.49 (ArC) 119.09 (Ar) 117.47 (Ar) 112.73 (Ar) 55.01 () 30.28 (H3); ESI (m/z): 549 [M − H]−; analysis calc. for C28H30O8N4: C 61.08, H 5.49, N 10.18%; found: C 58.42, H 5.4, N 9.78%. The difference can be accounted for by calculating the formula with a 0.25 molar ratio of CHCl3.
O), 142.20 (Ar), 140.02 (Ar), 132.64 (Ar), 132.19 (Ar), 126.62 (Ar), 117.43 (Ar), 112.54 (Ar), 41.20 (H2), 24.67 (H2), 15.11 (H3); ESI (m/z): 770 [M − NEt3 + H]+; analysis calc. for C49H70O8N6: C 67.56, H 8.1, N 9.65%; found: C 66.73, H 7.88, N 9.18%.
), 8.87 (s, 2H, O), 7.95 (s, 2H, N), 7.64 (s, 2H, N), 7.49 (d,3J = 8.9, Hz, 2H, Ar), 6.98 (s, 6H, Ar), 6.87 (d, 3J = 8.9 Hz, 2H, ArH), 3.85 (s, 2H, C2), 2.53 (q, 3J = 7.5 Hz, 8H, ArC2CH3), 1.10 (t, 3J = 7.5 Hz, 12H, ArCH2C3); 13C{1H} NMR (400 MHz, DMSO-d6) δ 172.30 (O2H), 157.40 (O), 154.91 (Ar), 142.42 (Ar), 139.78 (Ar), 132.63 (Ar), 130.15 (Ar), 126.66 (Ar), 119.91 (Ar), 115.69 (Ar), 45.93 (H2), 24.99 (H2), 15.17 (H3), 9.10 (H3); ESI (m/z): 667 [M − H]−; analysis calc. for C37H40O8N4: C 66.45, H 6.03, N 8.38%; found: C 63.54, H 5.67, N 7.82%. The difference can be accounted for by calculating the formula with a 0.3 molar ratio of CHCl3.
:ethanol (15 mL:1.5 mL). Triethylamine (0.78 mL, 5.59 mmol) to give a solution that was heated under reflux for 4 h. The resulting suspension was filtered and washed with chloroform (100 mL) to give 2a as an off-white solid (1.19 g, 1.59 mmol, 97%). 1H NMR (400 MHz, DMSO-d6) δ 8.14 (s, 2H, N), 7.61 (s, 2H, N), 7.46 (s, 1H, Ar) 7.26–7.13 (m, 5H, Ar), 6.53 (d, 3J = 8.6 Hz, 2H, Ar), 6.45 (s, 2H, Ar), 3.07 (q, 3J = 7.2 Hz, 12H, NC2CH3), 1.59 (s, 12H, CC3), 1.17 (t, 3J = 7.0 Hz, 18H, NCH2C3); 13C{1H} NMR (400 MHz, DMSO-d6) δ 172.64 (O2H), 157.36 (O), 157.36 (Ar), 148.70 (Ar), 130.51 (Ar), 123.75 (Ar), 122.93 (Ar), 120.91 (Ar), 119.69 (Ar), 115.79 (Ar), 54.88 (CH3), 45.81 (H2) 30.41 (H3) 9.04 (H3). ESI (m/z): 652 [M − NEt3 + H]+; analysis calc. for C40H60O8N6: C 63.81, H 8.03, N 11.16%; found: C 54.61, H 6.76, N 9.32%. The difference can be accounted for by calculating the formula with a 1.3 molar ratio of CHCl3.
), 8.36 (s, 2H, N), 7.87 (s, 2H, N), 7.44 (s, 1H, Ar), 7.33 (dd, 3J = 8.9, 2.8 Hz, 2H, ArH), 7.27–7.20 (m, 3H, ArH), 6.81 (d, 3J = 8.9 Hz, 2H, Ar), 6.46 (s, 2H, Ar), 1.59 (s, 12H, CC3). 13C{1H} NMR (400 MHz, DMSO-d6) δ 172.30 (O2H) 156.13 (O) 154.78 (Ar) 148.45 (Ar) 132.69 (Ar) 127.96 (Ar) 126.77 (Ar) 122.98 (ArC) 121.49 (ArC) 119.09 (Ar) 117.47 (Ar) 112.73 (Ar) 55.01 () 30.28 (H3); ESI (m/z): 549 [M − H]−; analysis calc. for C28H30O8N4: C 61.08, H 5.49, N 10.18%; found: C 58.42, H 5.4, N 9.78%. The difference can be accounted for by calculating the formula with a 0.25 molar ratio of CHCl3.
All gelation experiments were carried out at 1 wt% by dissolving 5 mg of the gelator in 0.5 mL of the desired solvent. The vial was then sealed, sonicated for 30 seconds and heated. Gels formed on standing and cooling to room temperature over a period of minutes to hours. Gel formation was determined by the inversion test.
Single crystal X-ray diffraction studies
Single crystal data was collected at 120.0(2) K on a Bruker D8Venture diffractometer (PHOTON-100 CMOS detector, IμS-microsource, focusing mirrors, λMoKα, λ = 0.71073 Å) and processed using Bruker APEX-II software. The temperature of the samples was maintained by the Cryostream (Oxford Cryosystems) open-flow nitrogen cryostat. The structure was solved by direct method and refined by full-matrix least squares on F2 for all data using Xseed, OLEX2 and SHELXTL software. All non-disordered non-hydrogen atoms were refined anisotropically, hydrogen atoms were placed in the calculated positions. Site occupation factors of the carbon atoms of disordered propan-1-ol molecule were fixed at 0.50, 0.30 and 0.20, the disordered atoms were refined isotropically.Crystals of 2a were prepared by allowing a propan-1-ol solution of the sample to slowly evaporate. Crystal data for 2a: C43H68N6O9, M = 813.03, 0.154 × 0.225 × 0.254 mm3, monoclinic, space group P21 (no. 4), a = 8.3147(6), b = 19.2037(13), c = 14.1567(10) Å, β = 102.128(2)°, V = 2210.0(3) Å3, Z = 2, Dc = 1.222 g cm−3, F000 = 880, MoKα radiation, λ = 0.71073 Å, T = 120(2) K, 2θmax = 46.0°, 18991 reflections collected, 6058 unique (Rint = 0.0783). Final GooF = 1.001, R1 = 0.0570, wR2 = 0.1421, R indices based on 5189 reflections with I > 2σ(I) (refinement on F2), 540 parameters, 7 restraints. Lp and absorption corrections applied, μ = 0.086 mm−1. Absolute structure parameter = 0.1(12).
Carbon dot solutions
Four different types of CDs were synthesised as described previously.22–25 Briefly, CDs were prepared by acid thermo-hydrolyses using microcrystalline cellulose (MCC) or multi-walled carbon nanotubes (MWCNTs) as carbon precursors. Highly luminescent dots (c-CDs) were obtained using MCC, while less fluorescent CDs were obtained from MWCNTs. Later, the last CDs were passivated with acetone (p-CDs) to boost their PL properties. The p-CDs were then surface functionalised either with cysteamine to give a thiol-rich surface (t-CDs) or N-Boc-ethylenediamine to give an amine rich surface (a-CDs). All the CDs synthesised were neutralised and purified by precipitation with ethanol at low temperatures.Preparation and optimization of the CD–salt hydrogels
Gels were prepared by dissolving a carefully weighed amount of bis(urea) 1 into an aqueous solution of the relevant CD heating until the gelator dissolved and sonicating the as-prepared mixture three times for few seconds each time. This procedure resulted in gel formation over the course of a few minutes, however at low concentrations of the gelator (≤1 wt%) in the presence of 1 mg mL−1 CD solution the gels proved relatively weak. Addition of BMIM-BF4 (2 wt%) to metathesise the hydrogen bonding triethyl ammonium cation resulted in stable, robust and transparent gels.Fluorescence studies
The differences in the fluorescence intensity and the maximum excitation wavelength between liquid and solid were studied for: c-CD, p-CD, t-CD and a-CD containing gels in comparison with aqueous solutions. Each measurement was carried out three times, λex = 365 and 370 nm, 2 nm excitation and emission slit widths. Measurements were performed with a PTI QuantaMaster™ Spectrofluorometer.Analyses of metal ions
Aliquots of standards (Pb2+, Hg2+, Co2+, Fe3+ and Mg2+ at 10 μg mL−1) were dropped onto the CD solution (before gel formation) and shaken for 1 min. In the case of CD gels as fluorescent probes, formation of the gels was performed as previously described.Rheology
Rheological measurements were carried out with an AR2000 rheometer at 10 °C. Frequency sweep experiments were performed at a constant oscillation stress of 1 for the angular frequency of 0.1–100, while the stress sweep was performed for the oscillation stress of 0.1–100.Scanning electron microscopy
SEM samples were dried in air at room temperature for 2 days, coated with 3 nm of chromium using a Cressington 328 Ultra High Resolution EM Coating System, and imaged using an FEI Helios NanoLab DualBeam microscope in immersion mode, with typical beam settings of 1.5 kV and 0.17 nA.Acknowledgements
We thank the Engineering and Physical Sciences Research Council and the Spanish Ministry of Innovation and Science for funding projects EP/J013021/1 and CTQ2011-23790, respectively. This work was also supported by the CEiA3 (Agrifood Campus of International Excellence).Notes and references
- X. Xu, R. Ray, Y. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed.
- H. M. R. Gonçalves, A. J. Duarte and J. C. G. Esteves da Silva, Biosens. Bioelectron., 2010, 26, 1302–1306 CrossRef PubMed.
- W. Lu, X. Qin, S. Liu, G. Chang, Y. Zhang, Y. Luo, A. M. Asiri, A. O. Al-Youbi and X. Sun, Anal. Chem., 2012, 84, 5351–5357 CrossRef CAS PubMed.
- F. Du, F. Zeng, Y. Ming and S. Wu, Microchim. Acta, 2013, 180, 453–460 CrossRef CAS.
- J. Wei, J. Ren, J. Liu, X. Meng, X. Ren, Z. Chen and F. Tang, Biosens. Bioelectron., 2014, 52, 304–309 CrossRef CAS PubMed.
- H. Dai, Y. Shi, Y. Wang, Y. Sun, J. Hu, P. Ni and Z. Li, Sens. Actuators, B, 2014, 202, 201–208 CrossRef CAS PubMed.
- X. Cui, L. Zhu, J. Wu, Y. Hou, P. Wang, Z. Wang and M. Yang, Biosens. Bioelectron., 2015, 63, 506–512 CrossRef CAS PubMed.
- C. T. Nguyen, Y. Zhu, X. Chen, G. A. Sotzing, S. Granados-Focil and R. M. Kasi, J. Mater. Chem. C, 2015, 3, 399–408 RSC.
- N. Gogoi, M. Barooah, G. Majumdar and D. Chowdhury, ACS Appl. Mater. Interfaces, 2015, 7, 3058–3067 CAS.
- C. T. Nguyen, T. H. Tran, X. Lu and R. M. Kasi, Polym. Chem., 2014, 5, 2774–2783 RSC.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- P. Terech and R. G. Weiss, Chem. Rev., 1997, 97, 3133–3160 CrossRef CAS PubMed.
- S. K. Samanta, K. S. Subrahmanyam, S. Bhattacharya and C. N. R. Rao, Chem.–Eur. J., 2012, 18, 2890–2901 CrossRef CAS PubMed.
- S. K. Samanta, A. Pal, S. Bhattacharya and C. N. R. Rao, J. Mater. Chem., 2010, 20, 6881–6890 RSC.
- B. Adhikari and A. Banerjee, Soft Matter, 2011, 7, 9259–9266 RSC.
- Z. Xie, F. Wang and C.-y. Liu, Adv. Mater., 2012, 24, 1716–1721 CrossRef CAS PubMed.
- E. Sotelo-Gonzalez, A. M. Coto-Garcia, M. T. Fernandez-Argüelles, J. M. Costa-Fernandez and A. Sanz-Medel, Sens. Actuators, B, 2012, 174, 102–108 CrossRef CAS PubMed.
- S. Hu, Q. Zhao, Y. Dong, J. Yang, J. Liu and Q. Chang, Langmuir, 2013, 29, 12615–12621 CrossRef CAS PubMed.
- A. Quaranta, S. Carturan, A. Campagnaro, M. Dalla Palma, M. Giarola, N. Daldosso, G. Maggioni and G. Mariotto, Thin Solid Films, 2014, 553, 188–192 CrossRef CAS PubMed.
- V. A. Mallia, P. D. Butler, B. Sarkar, K. T. Holman and R. G. Weiss, J. Am. Chem. Soc., 2011, 133, 15045–15054 CrossRef CAS PubMed.
- J. A. Foster, R. M. Edkins, G. J. Cameron, N. Colgin, K. Fucke, S. Ridgeway, A. G. Crawford, T. B. Marder, A. Beeby, S. L. Cobb and J. W. Steed, Chem.–Eur. J., 2014, 20, 279–291 CrossRef CAS PubMed.
- K. L. Morris, L. Chen, J. Raeburn, O. R. Sellick, P. Cotanda, A. Paul, P. C. Griffiths, S. M. King, R. K. O'Reilly, L. C. Serpell and D. J. Adams, Nat. Commun., 2013, 4, 1480 CrossRef PubMed.
- X. Yang, G. Zhang and D. Zhang, J. Mater. Chem., 2012, 22, 38–50 RSC.
- G. Yu, X. Yan, C. Han and F. Huang, Chem. Soc. Rev., 2013, 42, 6697–6722 RSC.
- M. George and R. G. Weiss, Acc. Chem. Res., 2006, 39, 489–497 CrossRef CAS PubMed.
- J. W. Steed, Chem. Commun., 2011, 47, 1379–1383 RSC.
- D. K. Smith, Nat. Chem., 2010, 2, 162–163 CrossRef CAS PubMed.
- A. Y.-Y. Tam and V. W.-W. Yam, Chem. Soc. Rev., 2013, 42, 1540–1567 RSC.
- P. Dastidar, Chem. Soc. Rev., 2008, 37, 2699–2715 RSC.
- M. de Loos, A. Friggeri, J. van Esch, R. M. Kellogg and B. L. Feringa, Org. Biomol. Chem., 2005, 3, 1631–1639 CAS.
- M. de Loos, B. L. Feringa and J. H. van Esch, Eur. J. Org. Chem., 2005, 3615–3631 CrossRef CAS PubMed.
- J. H. van Esch, F. Schoonbeek, M. de Loos, H. Kooijman, A. L. Spek, R. M. Kellogg and B. L. Feringa, Chem.–Eur. J., 1999, 5, 937–950 CrossRef CAS.
- J. H. van Esch, S. DeFeyter, R. M. Kellogg, F. DeSchryver and B. L. Feringa, Chem.–Eur. J., 1997, 3, 1238–1243 CrossRef CAS PubMed.
- J. W. Steed, Chem. Soc. Rev., 2010, 39, 3686–3699 RSC.
- E. Krieg, H. Weissman, E. Shirman, E. Shimoni and B. Rybtchinski, Nat. Nanotechnol., 2011, 6, 141–146 CrossRef CAS PubMed.
- S. Sutton, N. L. Campbell, A. I. Cooper, M. Kirkland, W. J. Frith and D. J. Adams, Langmuir, 2009, 25, 10285–10291 CrossRef CAS PubMed.
- P. D. Thornton, R. J. Mart, S. J. Webb and R. V. Ulijn, Soft Matter, 2008, 4, 821–827 RSC.
- K. J. C. van Bommel, M. C. A. Stuart, B. L. Feringa and J. van Esch, Org. Biomol. Chem., 2005, 3, 2917–2920 CAS.
- B. Li, L. Tang, L. Qiang and K. Chen, Soft Matter, 2011, 7, 963–969 RSC.
- Q. Wei and S. L. James, Chem. Commun., 2005, 1555–1556 RSC.
- J. A. Foster, M.-O. M. Piepenbrock, G. O. Lloyd, N. Clarke, J. A. K. Howard and J. W. Steed, Nat. Chem., 2010, 2, 1037–1043 CrossRef CAS PubMed.
- D. K. Kumar and J. W. Steed, Chem. Soc. Rev., 2014, 43, 2080–2088 RSC.
- L. A. Estroff, L. Addadi, S. Weiner and A. D. Hamilton, Org. Biomol. Chem., 2004, 2, 137–141 CAS.
- F. Aparicio, E. Matesanz and L. Sánchez, Chem. Commun., 2012, 48, 5757–5759 RSC.
- C. Dolle, P. Magrone, S. Riva, M. Ambrosi, E. Fratini, N. Peruzzi and P. Lo Nostro, J. Phys. Chem. B, 2011, 115, 11638–11649 CrossRef CAS PubMed.
- L. Meazza, J. A. Foster, K. Fucke, P. Metrangolo, G. Resnati and J. W. Steed, Nat. Chem., 2013, 5, 42–47 CrossRef CAS PubMed.
- L. A. Estroff and A. D. Hamilton, Angew. Chem., Int. Ed., 2000, 39, 3447–3450 CrossRef CAS.
- D. J. Adams and P. D. Topham, Soft Matter, 2010, 6, 3707–3721 RSC.
- D. J. Adams, M. F. Butler, W. J. Frith, M. Kirkland, L. Mullen and P. Sanderson, Soft Matter, 2009, 5, 1856–1862 RSC.
- A. Bernet, R. Q. Albuquerque, M. Behr, S. T. Hoffmann and H.-W. Schmidt, Soft Matter, 2012, 8, 66–69 RSC.
- R. C. T. Howe, A. P. Smalley, A. P. M. Guttenplan, M. W. R. Doggett, M. D. Eddleston, J. C. Tan and G. O. Lloyd, Chem. Commun., 2013, 49, 4268–4270 RSC.
- J. Li, Y. Kuang, Y. Gao, X. Du, J. Shi and B. Xu, J. Am. Chem. Soc., 2013, 135, 542–545 CrossRef CAS PubMed.
- Z. Yang, G. Liang and B. Xu, Chem. Commun., 2006, 738–740 RSC.
- S. Fleming and R. V. Ulijn, Chem. Soc. Rev., 2014, 43, 8150–8177 RSC.
- G. Cravotto and P. Cintas, Chem. Soc. Rev., 2009, 38, 2684–2697 RSC.
- C. Deng, R. Fang, Y. Guan, J. Jiang, C. Lin and L. Wang, Chem. Commun., 2012, 48, 7973–7975 RSC.
- L. S. Reddy, S. Basavoju, V. R. Vangala and A. Nangia, Cryst. Growth Des., 2006, 6, 161–173 CAS.
- M. C. Etter, Z. Urbanczyk-Lipkowska, M. Zia-Ebrahimi and T. W. Panunto, J. Am. Chem. Soc., 1990, 112, 8415–8426 CrossRef CAS.
- J. Yan, J. Liu, P. Jing, C. Xu, J. Wu, D. Gao and Y. Fang, Soft Matter, 2012, 8, 11697–11703 RSC.
- Y. Feng, H. Li, Q. Gan, Y. Wang, B. Liu and H. Zhang, J. Mater. Chem., 2010, 20, 972–975 RSC.
- Z.-L. Xie, H.-B. Xu, A Geßner, M. U. Kumke, M. Priebe, K. M. Fromm and A. Taubert, J. Mater. Chem., 2012, 22, 8110–8116 RSC.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201–1217 CrossRef CAS PubMed.
- A. Cayuela, M. L. Soriano and M. Valcárcel, Sens. Actuators, B, 2015, 207, 596–601 CrossRef CAS PubMed.
- A. Cayuela, M. Laura Soriano and M. Valcárcel, Anal. Chim. Acta, 2013, 804, 246–251 CrossRef CAS PubMed.
- A. Cayuela, M. L. Soriano, M. C. Carrión and M. Valcárcel, Anal. Chim. Acta, 2014, 820, 133–138 CrossRef CAS PubMed.
- A. Cayuela, M. L. Soriano and M. Valcárcel, Anal. Chim. Acta, 2015, 872, 70–76 CrossRef CAS PubMed.
- J. T. Korhonen, M. Kettunen, R. H. A. Ras and O. Ikkala, ACS Appl. Mater. Interfaces, 2011, 3, 1813–1816 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fluorescent emission spectra of CD systems and tables of gelation experiments and single crystal structure determination in CIF format. CCDC 1402495. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01859e |
| This journal is © The Royal Society of Chemistry 2015 |