
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal influence on the iso- and hetero-selectivity of complexes of bipyrrolidine derived salan ligands for the polymerisation of rac-lactide†
Matthew D.
Jones
*a,
Lauren
Brady
a,
Paul
McKeown
ab,
Antoine
Buchard
 a,
Pascal M.
Schäfer
a,
Lynne H.
Thomas
a,
Mary F.
Mahon
c,
Timothy J.
Woodman
a and
John P.
Lowe
a
a,
Pascal M.
Schäfer
a,
Lynne H.
Thomas
a,
Mary F.
Mahon
c,
Timothy J.
Woodman
a and
John P.
Lowe
a
aDepartment of Chemistry, University of Bath, Claverton Down, Bath BA2 7AY, UK. E-mail: mj205@bath.ac.uk; Fax: +44 (0)1225 386231; Tel: +44 (0)1225 384908
bDoctoral Training Centre in Sustainable Chemical Technologies, University of Bath, Bath BA2 7AY, UK
cBath Chemical Crystallography Unit, Department of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK
First published on 17th June 2015
Abstract
In this paper we have prepared a series of Ti(IV), Hf(IV) and Al(III) complexes based on bipyrrolidine salan pro-ligands. The Hf(IV) complexes have all been characterised in the solid-state, the chiral ligands coordinate to Hf(IV) in an α-cis manner whereas the meso ligand coordinates in a β-cis geometry. The Hf(IV) complexes are all active for the ROP of rac-lactide in the melt, with the fluxional meso complex affording a strong isotactic bias Pm = 0.84. As expected Hf(3)(OiPr)2 polymerised L-LA faster than rac-LA (kapp = 5.9 × 10−3 min−1vs. 3.8 × 10−3 min−1). For Ti(IV) complexes atactic PLA was formed. The salan pro-ligands have also been complexed to Al(III), and the novel Al–Me and Al-OiPr complexes were characterised in the solid and solution state. Al(1)(OiPr) was fluxional on the NMR timescale, whereas Al(3)(OiPr) was locked in solution with no exchange. Interestingly, the Al(III) complexes of 3H2 produce PLA with a very strong heterotactic bias Pr upto 0.87, whereas atactic PLA is produced with 1H2. For Al(3)(OiPr) a linear relationship is observed with Mn and conversion. Experiments with the addition of an equivalent of rac-LA to the selective initiators have also been performed and are discussed.
Introduction
Over recent years there has been a concerted effort to find iso-selective initiators for the ROP of rac-LA.1 Isotactic PLA from rac-LA has an enhanced melting point over PLA produced from the ROP of enantiopure monomers.2 This coupled with the desirable biodegradation and the fact that the monomer can be prepared from annually renewable resources has spurred research in this area.3 The majority of iso-selective initiators are based on aluminium – for example early work of Feijen and Spassky employing chiral salen complexes.1i,1j,4 However, in recent years yttrium and lanthanide complexes have also shown promise in this area.5 Moreover, we have recently shown that a zirconium complex of a bipyrrolidine salan ligand is able to produce PLA with a strong isotactic bias (Pm = 0.84).6 In this paper we have expanded further on this study with Ti(IV), Hf(IV) complexes and Al(III) systems.It is difficult to predict the resulting stereochemistry of the polymer that an initiator may produce. Al(III) complexes have a propensity to produce highly isotactic PLA.7 For example, in 2004 Gibson prepared aluminium salan complexes based on an N,N′-dimethylethylendiamine with H-substituents on the phenoxide and this afforded moderate isotactic PLA (Pm = 0.68).1c However, when this was changed to a Cl-substituted phenolate heterotactic PLA was produced, Pr of 0.88. However, with the N,N′-dibenzylethylendiamine version with H-substituents highly isotactic PLA (Pm = 0.79) was observed.1c We have shown that with Al(III) salalen complexes the tacticity can be changed from moderate heterotacticity to moderate isotacticity by altering the substituent on the amine nitrogen.1l In 2012 Williams prepared a series of yttrium phosphasalen initiators and it was observed that on varying the backbone either isotactic or heterotactic PLA could be produced.5a Kol and Okuda have, with a series of ONSO ligands complexed to Zr(IV), shown that the selectivity can be dependent upon the fluxionality of the complex, with rigid ligands forming isotactic PLA and fluxional catalysts heterotactic PLA.8 However, it has recently been reported that with phenylene ONSO ligands the tacticity was determined by the substituents on the phenolate and not fluxionality.9 There are limited examples in the literature of ligands which when complexed to different metals afford different tacticities. Ma has shown a switch in stereoselectivity with a series of ONN derived salan ligands complexed to either zinc or magnesium.10 With magnesium a hexa-coordinated active site is postulated and heterotactic PLA is formed, however, for zinc complexes a penta-coordinate active site is observed and isotactic PLA is isolated. Recently, Williams has prepared a series of phosphasalen complexes based on La(III) (covalent radius 2.07 Å) and Lu(III) (covalent radius 1.87 Å), with the larger La(III) affording heterotactic PLA and the smaller Lu(III) isotactic PLA.5b
Results and discussion
Ligands and complex preparation
The ligands were prepared by a modified Mannich reaction as detailed elsewhere.6,11 The complexes in this study were prepared as shown in Scheme 1.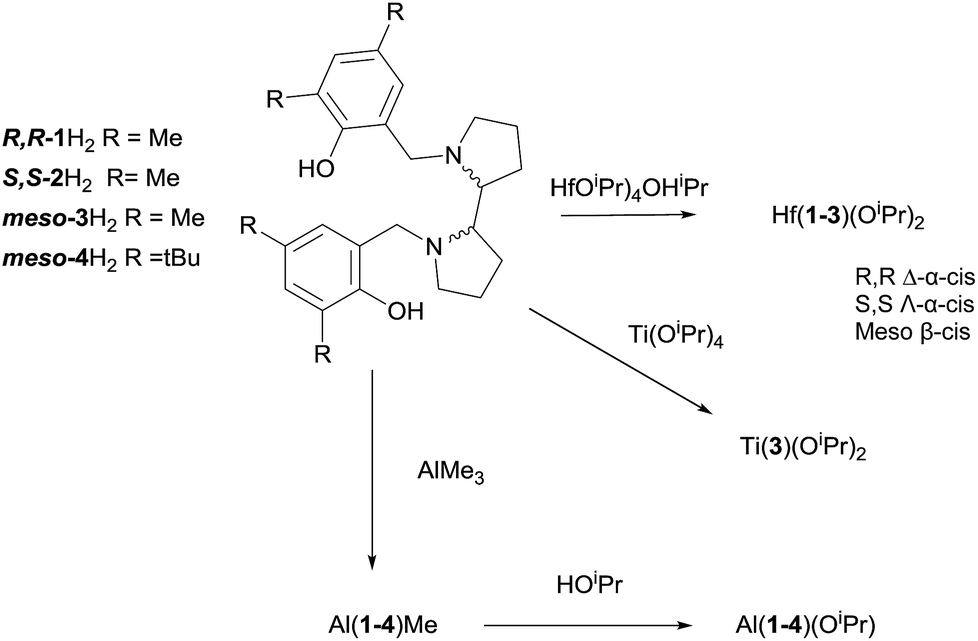 | ||
| Scheme 1 Ligands and complexes used in this study. | ||
The Hf(IV) complexes, Fig. 1, are analogues to the previously reported zirconium complexes.6 As before the meso ligand is coordinated in a β-cis geometry and the chiral species as the α-cis isomers.6,12 Solution state NMR spectroscopic measurements are in agreement with the solid state structures being maintained in solution. For the enantiopure complexes the ligands are “locked” in position as indicated by discrete doublets for the methylene CH2 moieties and there are no exchange peaks observed in the NOESY/EXSY spectrum at 298 K (CDCl3). However, from NOESY/EXSY experiments on Hf(3)(OiPr)2, there is evidence of ligand exchange on the NMR timescale. There are exchanges processes not only of the aliphatic back bone but also in the aromatic region of the spectrum, for instance the resonance at 6.51 ppm interchanges with that at 6.67 ppm. Furthermore, DOSY NMR (CDCl3) was performed on the complex further suggesting one species in solution with a diffusion coefficient of 7.8 × 10−10 m2 s−1 being observed, this equates to a hydrodynamic radii of ca. 5.1 Å. The meso-3H2 ligand was also reacted with Ti(OiPr)4 to form Ti(3)(OiPr)2 which was recrystallised in hexane. As expected the β-cis geometry was observed in the solid-state. Solution state NMR spectroscopy confirmed this was the solution structure with two distinct isopropoxide methine resonances.
 | ||
| Fig. 1 Solid state structure of Hf(3)(OiPr)2. Ellipsoids are shown at the 30% probability level, hydrogen and disordered moieties have been removed for clarity. | ||
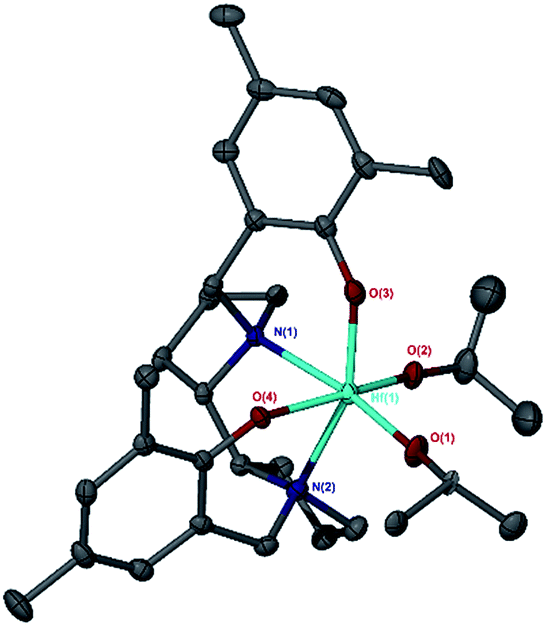
Initially the proligands 1H2 and 3H2 were reacted with AlMe3 to generate Al(1 or 3)Me.7c The solid-state structures have been determined for Al(1 or 3)Me and are shown in Fig. 2.7c The tau value for Al(1)Me is 0.97 indicating a strong preference for the R,R-ligand to form a trigonal bipyramidal structure, this is exemplified by the O(1)–Al(1)–N(2) and O(2)–Al(1)–C(1) angles of 168.66(6)° and 122.09(8)° respectively.13 There is a clear difference between Al(1)Me and A(3)Me with the bipyrrolidine rings of the meso ligand forming a “bowl” shape around the metal centre, this is observed in the space filling model (see ESI†). For Al(3)Me there are three crystallographically independent aluminium centres, with tau values of 0.37, 0.28 and 0.76 indicating there is no clear preference for either square pyramidal or trigonal bipyramidal in this case. This is exemplified by the O(1)–Al(1)–N(2) and O(2)–Al(1)–C(1) angles of 154.81(6)° and 113.08(6)° for one of the Al(3)Me moieties cf. 136.55(6)/105.95(8)° and 162.79(6)/119.09(7)° for the analogous angles in the other crystallographic unique Al(III) centres. The solution state 1H NMR spectrum (298 K, C6D5CD3) for Al(3)Me showed two aromatic resonances and two sharp doublets for the methylene Ar–CH2–N moieties. Furthermore, six aromatic, four CH2 and one CH resonance were observed in the 13C{1H} NMR, indicating that both aryl rings and pyrrolidine rings are chemically equivalent. This is in contrast to the solution state NMR spectrum of Al(1)(Me) which was very broad and fluxional at 298 K. However, at 233 K the spectrum sharpened and four aromatic resonances where observed in the 1H NMR spectrum, in the 13C{1H} spectrum eight CH2, two CH and four Ar-CH3 resonances where observed indicating that, once coordinated, each half of the ligand are no longer equivalent.
 | ||
| Fig. 2 Solid state structures for the aluminium complexes, ellipsoids are shown at the 30% probability level, all hydrogen atoms and disorder have been removed for clarity. Top left: Al(1)Me, Top right: Al(3)Me, bottom left Al(1)(OiPr) bottom right Al(3)(OiPr). | ||
To generate species that are capable of acting as ROP initiators in the melt the Al–Me complexes were reacted with HOiPr to afford discrete alkoxide species.14 Al(1)(OiPr) crystallises in the orthorhombic space group P212121. The aluminium centre is in a pseudo trigonal bipyramidal motif with a tau value of 0.70. Al(3)(OiPr) crystallises in the cubic space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) and the aluminium centre is in a pseudo trigonal bipyramidal motif with a tau value of 0.64. The solution state behaviour mirrors that of the alkyl complexes. At 298 K (CDCl3) Al(3)(OiPr) the solution state 1H NMR spectrum consisted of two aryl resonances and discrete resonances for the Ar-CH2–N moieties. Cooling to 233 K (CDCl3) or heating to 353 K (C6D5CD3) had no effect on the spectrum. Furthermore, there were no exchange peaks observed in the NOESY/EXSY spectrum. However, resonances for Al(1)(OiPr) at 298 K (CDCl3) were broad, upon cooling to 253 K these sharpened significantly, as with Al(1)Me, both halves of the ligand are not equivalent once coordinated. NOESY/EXSY at 253 K and 298 K show the presence of exchange peaks. Furthermore, DOSY NMR (CDCl3) was performed on Al(3)(OiPr) suggesting one major species in solution with a diffusion coefficient of 7.6 × 10−10 m2 s−1 being observed. DFT studies were also in agreement with the isolated structure being the most stable form, see ESI.† It should also be noted that there is the possibility of one phenoxide arm orienting itself towards or away from isopropoxide, see ESI.† The difference between the two is small (ΔG = 2.4 kcal mol−1) but adds further to the complexity of the system. The complex Al(4)(OiPr) was also prepared, its solid state structure is similar to Al(3)(OiPr) and its NMR is consistent with the desired structure being dominant in solution. However, it must be noted that it was difficult to fully convert Al(4)Me into the isopropoxide analogue; even with heating and five equivalents of isopropanol, ca. 5% of the Al(4)Me complex was co-isolated. This was not the case with the Me-substituted ligand and presumably reflects the steric difference between the two systems.
and the aluminium centre is in a pseudo trigonal bipyramidal motif with a tau value of 0.64. The solution state behaviour mirrors that of the alkyl complexes. At 298 K (CDCl3) Al(3)(OiPr) the solution state 1H NMR spectrum consisted of two aryl resonances and discrete resonances for the Ar-CH2–N moieties. Cooling to 233 K (CDCl3) or heating to 353 K (C6D5CD3) had no effect on the spectrum. Furthermore, there were no exchange peaks observed in the NOESY/EXSY spectrum. However, resonances for Al(1)(OiPr) at 298 K (CDCl3) were broad, upon cooling to 253 K these sharpened significantly, as with Al(1)Me, both halves of the ligand are not equivalent once coordinated. NOESY/EXSY at 253 K and 298 K show the presence of exchange peaks. Furthermore, DOSY NMR (CDCl3) was performed on Al(3)(OiPr) suggesting one major species in solution with a diffusion coefficient of 7.6 × 10−10 m2 s−1 being observed. DFT studies were also in agreement with the isolated structure being the most stable form, see ESI.† It should also be noted that there is the possibility of one phenoxide arm orienting itself towards or away from isopropoxide, see ESI.† The difference between the two is small (ΔG = 2.4 kcal mol−1) but adds further to the complexity of the system. The complex Al(4)(OiPr) was also prepared, its solid state structure is similar to Al(3)(OiPr) and its NMR is consistent with the desired structure being dominant in solution. However, it must be noted that it was difficult to fully convert Al(4)Me into the isopropoxide analogue; even with heating and five equivalents of isopropanol, ca. 5% of the Al(4)Me complex was co-isolated. This was not the case with the Me-substituted ligand and presumably reflects the steric difference between the two systems.
Polymerisation
In this study we have simply used recrystallised monomer to mimic more industrially relevant conditions. The results for the solution and melt ROP of rac-LA, with the Hf(IV) and Ti(IV) complexes are shown in Table 1. As observed for the Zr(IV) complexes isotactic PLA was produced, with Hf(IV). Analysis of the microstructure showed a small contribution from the sis tetrad and the sii, iis and isi are approximately 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1 indicating that a chain end control mechanism is operative which would lead to a stereoblock structure of the PLA.1f,5a DSC analysis of the PLA produced from Hf(3)(OiPr)2 in solution showed a melting point of 181 °C (Table 1 entry 8) and 173 °C (Table 1 entry 7) indicating some crystallinity. Whereas, the PLA produced with Hf(1/2)(OiPr)2 (Pm = 0.71) from bulk polymerisation had no defined melting point. The Ti(3)(OiPr)2 complex produced atactic PLA and was significantly slower.
:1:1 indicating that a chain end control mechanism is operative which would lead to a stereoblock structure of the PLA.1f,5a DSC analysis of the PLA produced from Hf(3)(OiPr)2 in solution showed a melting point of 181 °C (Table 1 entry 8) and 173 °C (Table 1 entry 7) indicating some crystallinity. Whereas, the PLA produced with Hf(1/2)(OiPr)2 (Pm = 0.71) from bulk polymerisation had no defined melting point. The Ti(3)(OiPr)2 complex produced atactic PLA and was significantly slower.
| Initiator | Time (h) | Conv. (%)d | M n | PDIe | P m |
|---|---|---|---|---|---|
| a Conditions: [M]/[I] = 300, 130 °C, solvent free. b Conditions: [M]/[I] = 100, toluene, T = 70 °C. c T = 50 °C. d As determined via1H NMR spectroscopy. e Determined from GPC (in THF) referenced to polystyrene. f P m is the probability of isotactic enchainment, calculated from the 1H homonuclear decoupled NMR spectra. | |||||
| Hf(1)(OiPr)2a | 1 | 57 | 33700 |
1.09 | 0.71 |
| Hf(2)(OiPr)2a | 1 | 55 | 33250 |
1.07 | 0.71 |
| Hf(3)(OiPr)2a | 0.17 | 92 | 33000 |
1.27 | 0.68 |
| Ti(3)(OiPr)2a | 24 | 48 | 15700 |
1.06 | 0.50 |
| Hf(1)(OiPr)2b | 4 | 10 | — | — | — |
| Hf(2)(OiPr)2b | 4 | 10 | — | — | — |
| Hf(3)(OiPr)2b | 4 | 91 | 16200 |
1.12 | 0.80 |
| Hf(3)(OiPr)2c | 4 | 87 | 14100 |
1.06 | 0.84 |
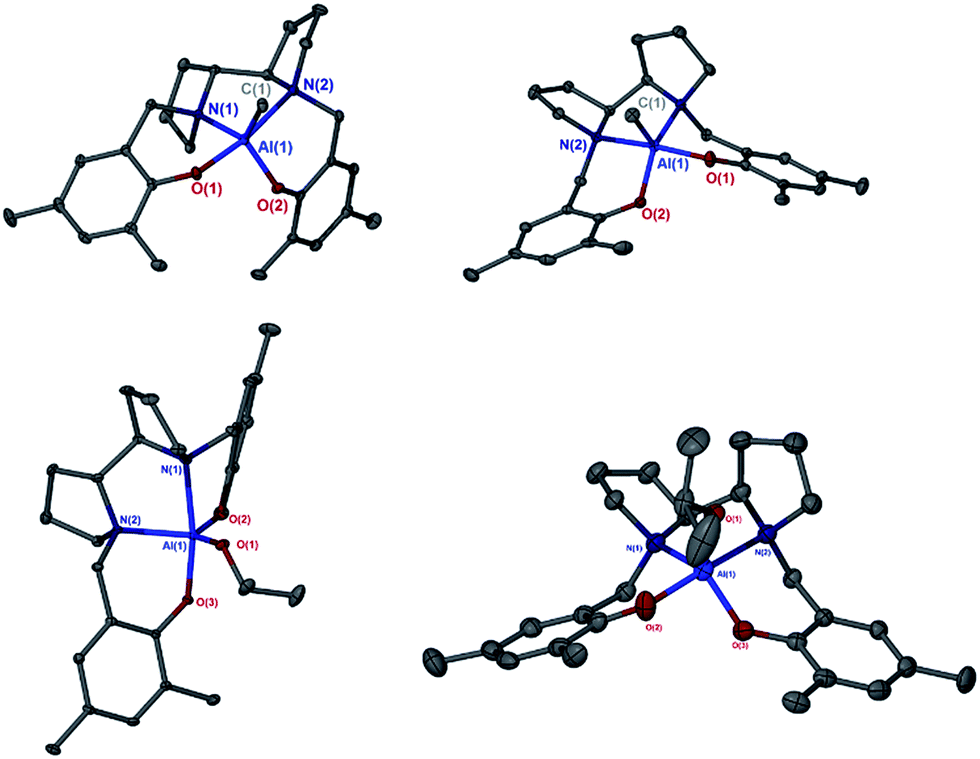
Hf(3)(OiPr)2 was significantly faster than the chiral initiators, which is exemplified by the polymerisation tests at 70 °C. The kinetics for L-LA and rac-LA polymerisation were investigated with Hf(3)(OiPr)2, Fig. 3. There is a first order dependency on the monomer, upto a conversion of 90%, and L-LA is faster than rac-LA. MALDI-ToF analysis on the PLA formed from solution (50 °C) shows a major polymer series with a repeat unit of 144 g mol−1 and a minor series with a 72 g mol−1 difference, indicating a small degree of transesterification. This also demonstrated the presence of an OiPr end group as expected from the classical coordination insertion mechanism. The mechanism for the polymerisation is presumably similar to that of the zirconium complex, which we have previously postulated is a ligand mediated chain end control mechanism with complementary chirality being maintained by virtue of the complex fluxionality. This is a “self-correcting” method to produce isotactic PLA. If the wrong isomer inserts then the initiator “switches” from Δ ⇔ Λ, Fig. 4.
 | ||
| Fig. 3 Semi-logarithmic plots of the polymerisation of L-LA and rac-LA ([LA]0 = 0.56 mol dm−3) [LA]:[Init] 100:1 init = Hf (3)(OiPr)2. CDCl3 298 K. | ||
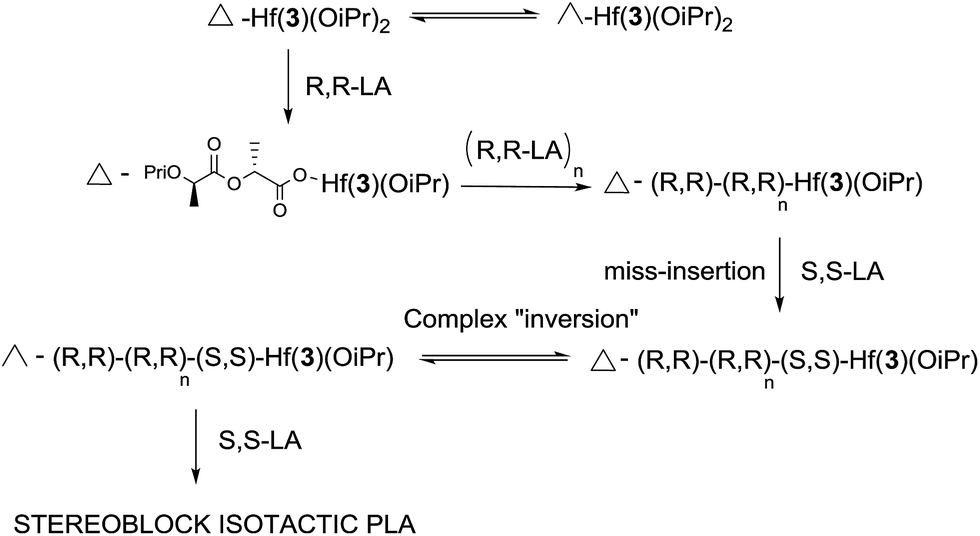 | ||
| Fig. 4 Proposed mechanism to illustrate the stereocontrolled nature of the polymerisation using the Hf(3)(OiPr)2 initiator.6 | ||
When Hf(3)(OiPr)2 is reacted with 1 equivalent of rac-LA a complex mixture is observed (see 1H NMR ESI†). However, the aromatic resonances are in a roughly 2:1:1 relationship which is indicative of the Hf(IV) centre remaining in a six coordinate octahedral β-cis geometry (as seen in the parent initiator). DOSY NMR spectroscopic analysis gave a diffusion coefficient of 6.4 × 10−10 m2 s−1. As expected this is reduced from the parent initiator due to the increased resistance caused by the ring opened lactide moiety. There appeared to be only one species in solution, which was presumably Hf(3)(OiPr)(OC(HMe)C(O)OC(HMe)C(O)OiPr). NOESY/EXSY NMR shows a similar exchange pattern as for the parent initiator.
It is well known in the literature that aluminium complexes have a strong tendency to produce isotactic PLA.1c,4a However, heterotactic PLA was produced with Al(3)(OiPr) and atactic with Al(1)(OiPr), both in solution and in the melt, Table 2. The aluminium alkyl complexes required the addition of benzyl alcohol as co-initiator to form active species. PLA exhibiting a narrow dispersity was formed, with the meso complex affording a strong heterotactic bias, Pr = 0.87. Analysis of the PLA prepared with Al(3)Me with the addition of HOCH2Ph via MALDI-ToF MS indicated the presence of the H– and –OCH2Ph end groups as expected. Furthermore, the major series has a repeat unit of 144 g mol−1 (a minor series with 72 g mol−1 is also detected) indicating little inter-molecular transesterification is occurring. However, at low mass (ca. 2000 Da) a minor series of cyclic PLA is observed (up to 16 LA units) with a repeat unit of 72 g mol−1 implying a degree of intra-molecular transesterification was occurring.
| Initiator | Time (h) | Temp/°C | Conv.d (%) | M n | PDI e | P r |
|---|---|---|---|---|---|---|
|
a Conditions: [M]:[I]:[BnOH] = 100:1:1, toluene.
b Conditions: [M]/[I] = 300, solvent free.
c [M]:[I] 100:1, toluene.
d As determined via1H NMR spectroscopy.
e Determined from GPC (in THF) referenced to polystyrene.
f
P
r is the probability of heterotactic enchainment, calculated from the 1H homonuclear decoupled NMR spectra.
|
||||||
| Al(1)Mea | 120 | 80 | 86 | 29150 |
1.08 | 0.49 |
| Al(3)Mea | 120 | 80 | 87 | 21550 |
1.05 | 0.87 |
| Al(1)(OiPr)b | 24 | 130 | 37 | 14700 |
1.07 | 0.50 |
| Al(3)(OiPr)b | 24 | 130 | 82 | 59650 |
1.25 | 0.69 |
| Al(4)(OiPr)b | 48 | 130 | 75 | 45500 |
1.09 | 0.50 |
| Al(3)(OiPr)c | 168 | 70 | 72 | 18850 |
1.05 | 0.87 |
| Al(3)(OiPr)c | 120 | 80 | 72 | 16279 |
1.07 | 0.82 |
| Al(3)(OiPr)c | 90 | 90 | 71 | 17900 |
1.10 | 0.79 |
| Al(3)(OiPr)c | 90 | 100 | 69 | 17150 |
1.11 | 0.76 |
As in solution the Al(3)(OiPr) initiator produced heterotactic PLA, whereas the chiral complex afforded atactic polymer under melt conditions (Table 2 entries 3 and 4). Furthermore, Al(3)(OiPr) was far more active in the melt than Al(1)(OiPr) and displayed a linear relationship between conversion and Mn, indicative of a well-controlled polymerisation, Fig. 5. When L-LA was polymerised with Al(3)(OiPr) under melt conditions a conversion of 47% (Mn = 27450 g mol−1Mw/Mn = 1.19, 24 h) was achieved which is significantly lower than that obtained with rac-LA, as expected from a heteroselective initiator. A melt test with Al(4)(OiPr) afforded atactic PLA with a high conversion only achievable after 48 h. This indicated that a combination of steric bulk and the meso chirality are key in the production of heterotactic PLA in this case.
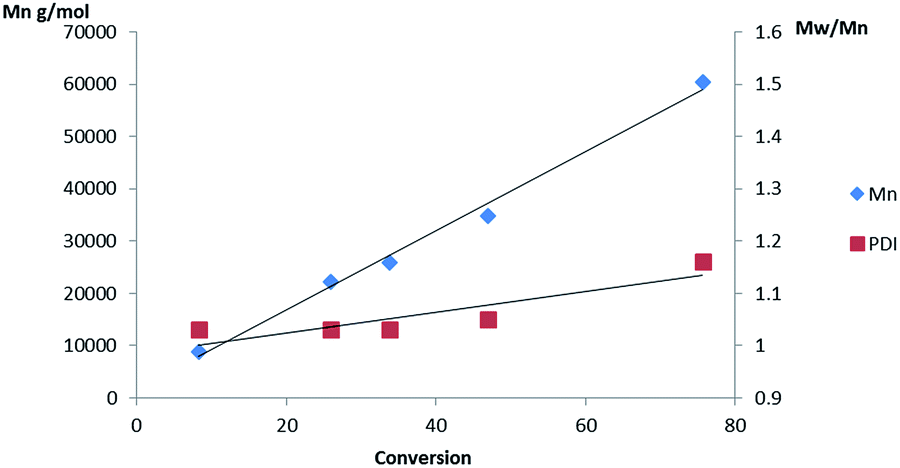 | ||
| Fig. 5 Plot of Mn and PDI vs. conversion for the melt polymerisation of rac-LA initiated with Al(3)(OiPr). | ||
In solution there is a strong preference for heterotactic PLA with Pr upto 0.87. MALDI-ToF analysis of the PLA prepared at 70 °C (Table 2 entry 6) showed a major series with the H– and –OiPr end groups and a repeat unit of 144 g mol−1 (with a minor series of 72 g mol−1), again a small amount of cyclic PLA was observed. Analysis of the PLA produced with Al(3)(OiPr) at 80 °C after 48 h afforded a Pr value of 0.85 and conversion of 25%, indicating a strong heterotacticity from initiation. Al(3)(OiPr) was also trialled for the ROP of rac-β-butyrolactone, after 8 h at 80 °C at 300:1 in the absence of solvent, a conversion of 78% was achieved (Mn = 28400 g mol−1, PDI = 1.03) unfortunately the polymer produced was atactic.
When Al(3)(OiPr) is reacted with 1 equivalent of rac-LA at 353 K (on the NMR scale ca. 20 mg of initiator), there are significant changes in the 1H NMR spectrum (see ESI†). Four resonances (1:1:1:1) are observed for the aryl protons, a series of 1H doublets for the backbone, two clear quartets for the opened lactide and there is a slight downfield shift in the isopropoxide methine proton. There was little change in the 27Al NMR with a resonance at ca. 44 ppm being observed at 353 K. When a further 3 equivalents were added there was still no change in the 27Al NMR at 353 K implying that the aluminium centre remains 5 coordinate as the polymer chain is growing.15 Upon cooling back to 298 K the 1H NMR spectrum was sharp and showed analogous features as the 353 K spectrum, but no signal could be observed in the 27Al NMR spectrum. From analysis of the 13C{1H} NMR spectrum two carbonyl resonances were observed at 170.4 and 180.8 ppm respectively. This compares to the signal at 180.7 previously observed for a five coordinate Schiff base Al-lactate complex.15 To probe the coordination of the active species further Al(3)Me was reacted with (S)-methyl-lactate. The 27Al NMR had a broad resonance at 42.4 ppm (cf. 46.5 for Al(3)(OiPr)) which is indicative of a five coordinate Al(III) centre.15 We have also probed the complexes formed by the reaction of Al(3)Me with (S)-methyl-lactate via DFT calculations, which supports the notion of the five coordinated intermediate being favoured. However, it was clear that there were more than one species present in solution, this is exemplified by the presence of two CH3 resonances for the methoxy and methyl groups of the lactate (at room temperature). This may be due to the presence of various stereoisomers, as well as the different possible orientations of the phenoxide arms being present. DOSY NMR gave a diffusion constant of 8.3 × 10−10 m2 s−1 (cf. 7.4 × 10−10 m2 s−1 for Al(3)(OiPr)) and the NOESY NMR showed no exchange processes. These gives a hydrodynamic radii for Al(3)(OiPr) of ca. 5.3 Å (NB the solid-state diameter is ≈ 12 Å) and for the lactate complex the radii is ca. 5 Å both indicating monomeric species in solution.16 Al(1)Me was also treated with 1 equivalent of (S)-methyl-lactate at room temperature a broad spectrum was obtained, cooling to 233 K sharp signals were observed. The 27Al NMR spectrum has a resonance at 42.2 ppm, which again implies a five coordinate Al(III) centre. In an attempt to rationalise the heterotacticity the Gibbs free energies of the different possibilities for the Al(3)-OiPr complexes with growing PLA chains were calculated. Again these showed that the Al(III) centre is 5 coordinate, it is interesting to note that the isomer with the PLA chain growing alongside the phenoxide arm (see ESI†) is more stable and there is a weak van der Waal interaction between the chain and the ortho substituent. However, given the complexities of this system future work will aim to rationalise the observed stereochemisty of the PLA produced. Very recently Kol and co-workers have shown that a racemic version of the 2,2′-bipyrrolidine (Cl substituted) ligand produced heterotactic PLA, the mechanism proposed was based on polymeryl exchange between the different isomers matching the chirality of the polymer to the chirality at the metal.7c In this case there are inactive and active diastereomers, which become awoken by polymer exchanges.7c It is assumed that the same mechanism is in operation with the meso version of the ligand in this case.
Conclusions
A series of salan complexes based on a bipyrrolidine backbone have been prepared and characterised in solution and solid state. There is a switch in selectivity from highly isotactic for hafnium initiators to heterotactic for aluminium initiators. It has previously been seen with Lu(III) and La(III) that there is a switch in selectivity with the smaller atomic radii metal forming isotactic PLA.5b In this case the opposite is observed with the larger Hf(IV) producing isotactic PLA and the smaller Al(III) heterotactic PLA. Moreover, Ma has shown that the tacticity is dependent upon coordination number, which may also account for the differences observed in this study. This illustrates the rich and diverse chemistry still to be explored and understood for the ROP of rac-LA. The meso-3H2 is unique in the literature as either isotactic, heterotactic or atactic PLA can be prepared by simply changing the metal centre.Acknowledgements
We wish to thank the EPSRC (EP/G03768X/1), the EPSRC UK National Service for Computational Chemistry Software (chem752), Corbion for lactide and support of the CDT at Bath and the University of Bath for funding. We also thank the EPSRC National Mass spectrometry service in Swansea for MALDI-ToF data.Notes and references
- (a) S. Abbina and G. Du, ACS Macro Lett., 2014, 3, 689–692 CrossRef CAS PubMed; (b) A. J. Chmura, M. G. Davidson, M. D. Jones, M. D. Lunn, M. F. Mahon, A. F. Johnson, P. Khunkamchoo, S. L. Roberts and S. S. F. Wong, Macromolecules, 2006, 39, 7250–7257 CrossRef CAS; (c) P. Hormnirun, E. L. Marshall, V. C. Gibson, A. J. P. White and D. J. Williams, J. Am. Chem. Soc., 2004, 126, 2688–2689 CrossRef CAS PubMed; (d) Z. Mou, B. Liu, M. Wang, H. Xie, P. Li, L. Li, S. Li and D. Cui, Chem. Commun., 2014, 50, 11411–11414 RSC; (e) N. Nomura, R. Ishii, Y. Yamamoto and T. Kondo, Chem.–Eur. J., 2007, 13, 4433–4451 CrossRef CAS PubMed; (f) T. M. Ovitt and G. W. Coates, J. Polym. Sci., Part A-Polym. Chem., 2000, 38, 4686–4692 CrossRef CAS; (g) T. M. Ovitt and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1316–1326 CrossRef CAS PubMed; (h) A. Pilone, K. Press, I. Goldberg, M. Kol, M. Mazzeo and M. Lamberti, J. Am. Chem. Soc., 2014, 136, 2940–2943 CrossRef CAS PubMed; (i) Z. Y. Zhong, P. J. Dijkstra and J. Feijen, Angew. Chem., Int. Ed. Engl., 2002, 41, 4510–4513 CrossRef CAS; (j) Z. Y. Zhong, P. J. Dijkstra and J. Feijen, J. Am. Chem. Soc., 2003, 125, 11291–11298 CrossRef CAS PubMed; (k) E. L. Whitelaw, M. D. Jones and M. F. Mahon, Inorg. Chem., 2010, 49, 7176–7181 CrossRef CAS PubMed; (l) E. L. Whitelaw, G. Loraine, M. F. Mahon and M. D. Jones, Dalton Trans., 2011, 40, 11469–11473 RSC.
- M. J. Stanford and A. P. Dove, Chem. Soc. Rev., 2010, 39, 486–494 RSC.
- (a) R. Auras, B. Harte and S. Selke, Macromol. Biosci., 2004, 4, 835–864 CrossRef CAS PubMed; (b) O. Dechy-Cabaret, B. Martin-Vaca and D. Bourissou, Chem. Rev., 2004, 104, 6147–6176 CrossRef CAS PubMed; (c) R. E. Drumright, P. R. Gruber and D. E. Henton, Adv. Mater., 2000, 12, 1841–1846 CrossRef CAS; (d) B. J. O'Keefe, M. A. Hillmyer and W. B. Tolman, J. Chem. Soc., Dalton Trans., 2001, 2215–2224 RSC; (e) J. C. Wu, T. L. Yu, C. T. Chen and C. C. Lin, Coord. Chem. Rev., 2006, 250, 602–626 CrossRef CAS PubMed.
- (a) N. Spassky, M. Wisniewski, C. Pluta and A. LeBorgne, Macromol. Chem. Phys., 1996, 197, 2627–2637 CrossRef CAS PubMed; (b) M. Wisniewski, A. LeBorgne and N. Spassky, Macromol. Chem. Phys., 1997, 198, 1227–1238 CrossRef CAS PubMed.
- (a) C. Bakewell, T. P. A. Cao, N. Long, X. F. Le Goff, A. Auffrant and C. K. Williams, J. Am. Chem. Soc., 2012, 134, 20577–20580 CrossRef CAS PubMed; (b) C. Bakewell, A. J. P. White, N. J. Long and C. K. Williams, Angew. Chem., Int. Ed. Engl., 2014, 53, 9226–9230 CrossRef CAS PubMed; (c) P. L. Arnold, J. C. Buffet, R. P. Blaudeck, S. Sujecki, A. J. Blake and C. Wilson, Angew. Chem., Int. Ed. Engl., 2008, 47, 6033–6036 CAS.
- M. D. Jones, S. L. Hancock, P. McKeown, P. M. Schafer, A. Buchard, L. H. Thomas, M. F. Mahon and J. P. Lowe, Chem. Commun., 2014, 50, 15967–15970 RSC.
- (a) A. Alaaeddine, C. M. Thomas, T. Roisnel and J. F. Carpentier, Organometallics, 2009, 28, 1469–1475 CrossRef CAS; (b) M. Bouyahyi, T. Roisnel and J. F. Carpentier, Organometallics, 2012, 31, 1458–1466 CrossRef CAS; (c) K. Press, I. Goldberg and M. Kol, Angew. Chem., Int. Ed., 2015 DOI:10.1002/anie.201503111.
- A. Stopper, J. Okuda and M. Kol, Macromolecules, 2012, 45, 698–704 CrossRef CAS.
- A. Stopper, K. Press, J. Okuda, I. Goldberg and M. Kol, Inorg. Chem., 2014, 53, 9140–9150 CrossRef CAS PubMed.
- H. Wang, Y. Yang and H. Ma, Macromolecules, 2014, 47, 7750–7764 CrossRef CAS.
- M. Miller and E. Y. Tshuva, Eur. J. Inorg. Chem., 2014, 2014, 1485–1491 CrossRef CAS PubMed.
- E. Sergeeva, J. Kopilov, I. Goldberg and M. Kol, Chem. Commun., 2009, 3053–3055 RSC.
- D. A. Atwood, A. R. Hutchison and Y. Z. Zhang, in Group 13 Chemistry Iii: Industrial Applications, ed. H. W. Roesky and D. A. Atwood, Springer-Verlag Berlin, Berlin, 2003, vol. 105, pp. 167–201 Search PubMed.
- S. L. Hancock, M. D. Jones, C. J. Langridge and M. F. Mahon, New J. Chem., 2012, 36, 1891–1896 RSC.
- Z. Tang, X. Pang, J. Sun, H. Du and X. Chen, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 4932–4938 CrossRef CAS PubMed.
- R. Evans, Z. Deng, A. K. Rogerson, A. S. McLachlan, J. J. Richards, M. Nilsson and G. A. Morris, Angew. Chem., Int. Ed. Engl., 2013, 52, 3199–3202 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Full experimental data and crystal data in the .cif format. CCDC 1401694–1401702. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01819f |
| This journal is © The Royal Society of Chemistry 2015 |