
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhancing electron affinity and tuning band gap in donor–acceptor organic semiconductors by benzothiadiazole directed C–H borylation†
D. L.
Crossley
a,
I. A.
Cade
a,
E. R.
Clark
a,
A.
Escande
a,
M. J.
Humphries
b,
S. M.
King
b,
I.
Vitorica-Yrezabal
a,
M. J.
Ingleson
*a and
M. L.
Turner
*a
aSchool of Chemistry, University of Manchester, Manchester, M13 9PL, UK. E-mail: Michael.ingleson@manchester.ac.uk; Michael.turner@manchester.ac.uk
bCambridge Display Technology Limited, Unit 3, Cardinal Park, Cardinal Way, Godmanchester, PE29 2XG, UK
First published on 12th June 2015
Abstract
Electrophilic borylation using BCl3 and benzothiadiazole to direct the C–H functionalisation of an adjacent aromatic unit produces fused boracyclic materials with minimally changed HOMO energy levels but significantly reduced LUMO energy levels. In situ alkylation and arylation at boron using Al(alkyl)3 or Zn(aryl)2 is facile and affords boracycles that possess excellent stability towards protic solvents, including water, and display large bathochromic shifts leading to far red/NIR emission in the solid state with quantum yields of up to 34%. Solution fabricated OLEDs with far red/NIR electroluminescence are reported with EQEs > 0.4%.
Introduction
Control of the frontier orbital energy levels in organic semi-conductors is crucial to improve the performance of devices such as organic light emitting diodes (OLEDs), organic photovoltaics (OPVs) and organic field effect transistors (OFETs).1 Materials with low lying LUMO energy levels are particularly desirable for use as n-type or ambipolar semi-conductors and for generating low band gap materials.2 An important class of low band gap materials are donor–acceptor (D–A) π-conjugated systems.3 The ability to tune the frontier orbital energy levels using simple synthetic approaches is highly attractive for optimising the properties of D–A systems, particularly if it enables access to materials with low lying LUMOs. One recent development in this area involved binding Lewis acids to a nucleophilic site in the acceptor moiety to increase its electron deficiency. For example, the coordination of Lewis acids to nitrogen in pyridyl and benzothiadiazole (BT, Fig. 1, left) containing D–A oligomers and polymers produced a considerable decrease in the energy of the LUMO and a modest decrease in that of the HOMO.4–7 This methodology was used to modulate the absorption, luminescence and charge mobility properties of D–A materials.4–9 Whilst the Lewis acid binding strategy represents a significant innovation the resultant materials are sensitive to the cleavage of the N → Lewis acid dative bond by moisture and Lewis bases. Coupling of dative bond formation with C–B bond formation, herein termed borylative fusion, will give chelated Lewis acid adducts with enhanced stability and extended π-conjugation provided by locking neighbouring aromatic units co-planar (Fig. 1, right). Extended π-conjugation will further lower the LUMO energy, whilst raising the HOMO energy level, counteracting the HOMO energy level reduction normally observed on Lewis acid binding to N.4–9 Thus borylative fusion represents a simple methodology for selectively modulating the LUMO energy and reducing the band gap of a material.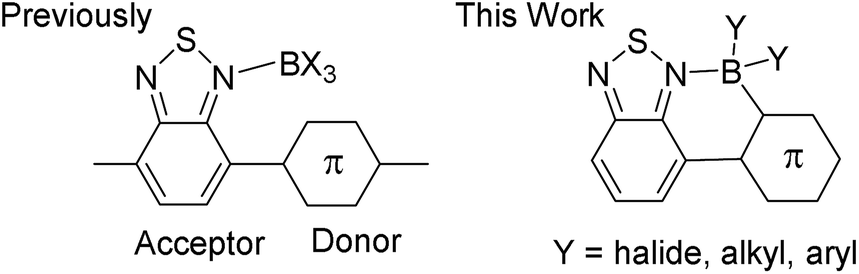 | ||
| Fig. 1 Design strategy for moisture stable, low LUMO energy D–A materials. | ||
The modulation of frontier orbital energies using borylative fusion to form four coordinate at boron C3BN units was pioneered by Yamaguchi and co-workers. They generated fused boracycles containing 1,3-thiazole acceptor moieties that displayed red-shifted absorption/fluorescence and a significantly lower energy LUMO relative to the non-borylated precursor.10 Recently, Liu and co-workers extended this approach to prepare borylatively fused thienyl–thiazole conjugated polymers with high electron affinities.11 However, the synthetic route to these materials proceeds by the cross-coupling of polyhalogenated thiophenes (e.g., 2-bromo-3-iodo-thiophene whose regiopure synthesis requires multistep procedures) and subsequent lithiation/quenching with a boron electrophile. Intramolecular electrophilic borylation is an attractive method for forming C–B bonds directly from C–H moieties and provides access to borylatively fused materials more efficiently. However, electrophilic borylation has been underutilised for the synthesis of fused boracycles and is limited to date to the borylative fusion of aryl substituted pyridyls.12 Furthermore, systematic studies into the effect of varying exocyclic boron substituents on key properties (e.g., frontier orbital energies) in these materials are also extremely limited.13 To the best of our knowledge borylative fusion has not been applied to D–A materials containing the significantly less nucleophilic (relative to pyridyls) benzothiadiazole (BT), which is a particularly strong acceptor moiety ubiquitous in organic electronics. Intramolecular electrophilic borylation of BT containing oligomers will result in lower LUMO energy levels, significantly red shifted absorption/emission and improved ambient stability (relative to non-fused Lewis adducts). Related tetra-coordinate organoboranes are widely used as highly emissive materials,14 with boron dipyrromethenes (BODIPYs) being the exemplar.15 In these materials boron acts as a light atom rigidifying unit that decreases non-radiative relaxation processes and results in larger quantum yields. Whilst BODIPYs and derivatives have found widespread applications their fluorescence is severely quenched in the solid state, unless substituted with extremely bulky groups.16 Herein we report the synthesis of fused borylated BT containing D–A materials that have significantly reduced LUMO energies relative to unborylated precursors. A number of these materials show excellent solid state fluorescence quantum yields (>30%) in the red/near infrared region. Un-optimised solution processed OLED devices show good external quantum efficiencies (EQE > 0.4%) for emission in this spectral region.
Results and discussion
Synthesis of fused boracycles
Previously, the addition of BCl3 to 1 and related polymers only resulted in reversible Lewis adduct formation (Fig. 2, eqn (1)) with no C–H borylation reported.4 Based on our studies we surmised that the significant steric bulk around the borylation site in 1 was hindering borylation.17 Therefore the borylation of a related D–A–D type structure containing a less hindered thienyl borylation position, 2 (Fig. 2 eqn (2)), was investigated. 2 is readily synthesised from the Stille coupling of dibromobenzothiadiazole with 2-tributylstannyl-5-octyl-thiophene. The addition of BCl3 to a DCM solution of 2 resulted in coordination of boron to a nitrogen site on BT and electrophilic C–H borylation to form 2-BCl2. Borylation was quantitative (by 1H NMR spectroscopy) in an open system with HCl released as a gaseous by-product, whereas in a closed system borylation is reversible and requires a hindered base to sequester the protic by-product from SEAr (preventing the back reaction, protodeboronation) and drive the reaction to completion. Importantly, addition of pyridine to 2-BCl2 resulted in no cleavage of the dative bond confirming the enhanced stability of 2-BCl2 relative to 1-BCl3 provided by borylative fusion. The borylation reactivity disparity between 1 and 2 is attributed to the presence of two proximate alkyl chains in 1 providing significant steric hindrance adjacent to the borylation site.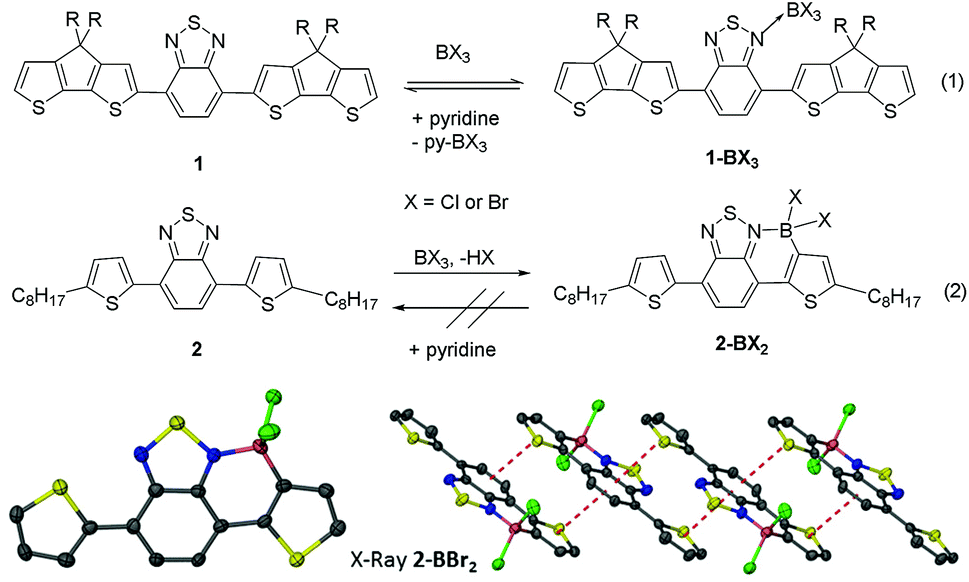 | ||
| Fig. 2 The reactivity of D–A–D structures 1 and 2 towards BX3. Bottom left the solid state structure of 2-BBr2 (ellipsoids at the 50% probability level, hydrogens and octyl chains omitted for clarity). Bottom right the extended solid state structure of 2-BBr2, dashed red lines indicate intermolecular π–π interactions. Blue = nitrogen, yellow = sulfur, green = bromine, pink = boron, grey = carbon. | ||
Borylation is not limited to the use of BCl3 as the electrophile, addition of one equivalent of PhBCl2 to 2 also resulting in borylation to yield 2-B(Ph)Cl, albeit at a slower rate than with BCl3 due to the reduced electrophilicity of PhBCl2. Compound 2 was also borylated using BBr3 to generate 2-BBr2 which was crystallographically characterised (Fig. 2, bottom). This confirmed the expected connectivity with formation of an essentially planar six membered boracycle. The short distance between the planes of the fused aromatic cores of adjacent molecules (interplane distance 3.4 Å) in the extended structure of 2-BBr2 indicates significant π–π interactions (Fig. 2, bottom right). Whilst mono-borylation was facile with a range of electrophiles the diborylation of 2 to generate the fully fused structure with both nitrogen atoms coordinated to boron proved unsuccessful under a range of conditions including using highly electrophilic borocations. Instead halide abstraction occurs preferentially, for example addition of the borylating mixture (DMT)BCl3/AlCl3 to 2-BCl2 (DMT = N,N-dimethyl-p-toluidine) resulted in the formation of (DMT)BCl3 and [2-BCl][AlCl4]. This was shown by 11B and 27Al NMR spectroscopy,17 and the significant downfield shift of the 1H resonances for the benzothiadiazole/thienyl protons consistent with the formation of the borocation. The lack of any double borylation with a range of boron electrophiles is attributed to the initial borylation reducing the nucleophilicity of the other nitrogen atom in benzothiadiazole preventing coordination of a second boron Lewis acid.18
The borylated halide species 2-BX2 and 2-B(Ph)Cl are stable to non-protic Lewis bases, however they still undergo slow hydrolysis. In situ functionalisation of boron in 2-BCl2 was facile using zinc and aluminium organometallic nucleophiles to install exocyclic aryl and alkyl groups in good to excellent yields (69–96%, Fig. 3). Furthermore, unsymmetrically substituted boron analogues, e.g., 2-B(Ph)Me, are also readily accessible simply by addition of AlMe3 to 2-B(Ph)Cl. All the materials in which boron is substituted with two exocyclic hydrocarbyl groups are stable for months in wet solvent (and for at least 12 months in the solid state under ambient atmosphere) and could be isolated by flash column chromatography on silica, further exemplifying their robust nature. Single crystals suitable for diffraction were obtained for 2-BPh2 and 2-BMe2 although the crystal quality of the former (and thus data quality) was consistently poor precluding detailed analysis. The solid state structure of 2-BPh2 does confirm connectivity (Fig. 3) and the planarity of the fused components and boracycle, whilst the extended structure contains a 1D π-stacked arrangement with interplane distances between adjacent fused π-systems of 3.5 Å. The intermolecular interactions involving the fused aromatic cores are comparable in 2-BPh2 and 2-BBr2, apart from the relative orientation of the boron moieties which are all on one face in 2-BPh2, but alternate in 2-BBr2. The structure of 2-BMe2 is more disparate containing a non-planar boracycle in which the boron atom is displaced by 0.3 Å out of the plane of the five other atoms in the ring. With similar bond distances for the boracycles in 2-BPh2 and 2-BMe2, the non-planarity in 2-BMe2 is attributed to packing forces. Specifically, a hydrogen bonding interaction is observed between a BC–H and a BT nitrogen (highlighted in green in Fig. 3). This results in adjacent molecules having fused aromatic portions that are less efficiently offset leading to a longer distance between the fused π systems (3.85 Å) in 2-BMe2. The non-planarity of the boracycle may serve to maximise both C–H⋯N and π–π attractive intermolecular interactions. It should be noted that in 2-BBr2 and 2-BPh2 other non-covalent intermolecular interactions will also impact on intermolecular distances, with short Br![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) S and S-aromatic intermolecular interactions observed.
S and S-aromatic intermolecular interactions observed.
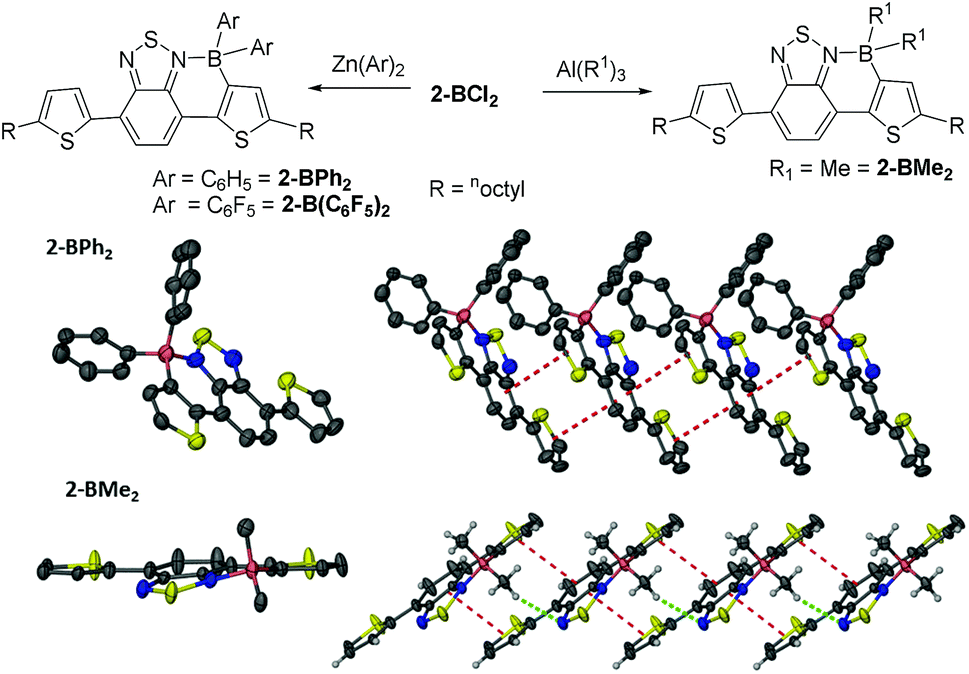 | ||
| Fig. 3 Top in situ functionalisation of 2-BCl2 with organometallic nucleophiles. Middle left the solid state structure of 2-BPh2. Middle right the extended solid state structure of 2-BPh2. Bottom left the solid state structure of 2-BMe2, bottom right the extended structure of 2-BMe2 (ellipsoids at the 50% probability level, hydrogens and octyl chains omitted for clarity). Dashed red lines indicate intermolecular π–π interactions and dashed green lines indicate C–HN hydrogen bonding interactions. | ||
To determine the degree of electronic control in benzothiadiazole directed C–H borylation the unsymmetric D–A–D compound 3 was synthesized (Fig. 4). Compound 3 contains two distinct aromatic moieties both amenable to electrophilic borylation, with the more electron rich aromatic thiophene expected to react more rapidly in electrophilic borylation.17 The addition of BCl3 to 3 at 20 °C resulted in borylative fusion, and after arylation at boron using Zn(C6F5)2, 3-B(C6F5)2 was isolated as the major product. Approximately 10% of a minor product was also observed (by 1H NMR spectroscopy) which was assigned as the product derived from competitive borylation of fluorene. Confirmation that borylative fusion onto a fluorene moiety was possible was forthcoming from borylation of 4 (Fig. 4, bottom). Compound 4 was borylated with BCl3 and BBr3 with borylation proceeding exclusively at the less sterically hindered C3 position. No borylation was observed at the more hindered C1 position further indicating the sensitivity of directed electrophilic borylation to the steric environment. Again whilst addition of pyridine resulted in no cleavage of the dative bond the halide complexes 4-BX2 (X = Cl or Br) were still sensitive to hydrolysis.
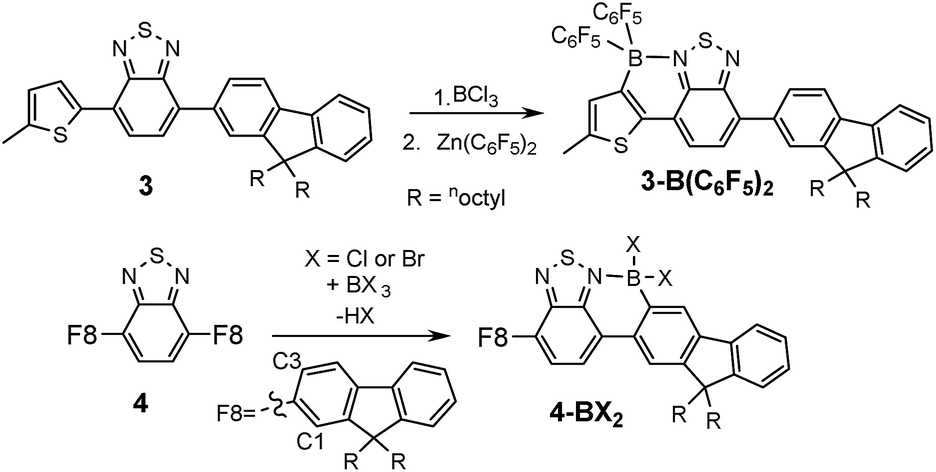 | ||
| Fig. 4 Borylation of fluorene containing D–A–D structures. | ||
The sensitivity of 4-BX2 (and 2-BX2) to protic oxo-species allows for the facile formation of the respective pinacol boronate esters. This is achieved by addition of non-purified THF (“wet THF”) to 2/4-BCl2 followed by addition of pinacol, to form 2-BPin and 4-BPin, respectively. In both pinacol boronate esters the boron centre is three coordinate with no N → B interaction as indicated by a signal in the 11B NMR spectrum at 29 ppm. Benzothiadiazole directed borylation therefore opens up access to substitution patterns that are hard to access by conventional/non-directed methods e.g., 2,3-disubstituted fluorene structures (Fig. 5).
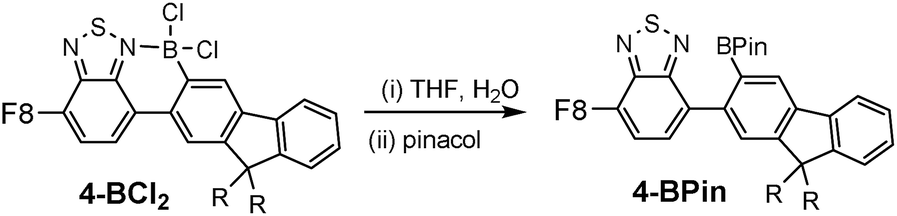 | ||
| Fig. 5 Formation of pinacol boronate esters from –BCl2 precursors. | ||
Hydrocarbyl functionalisation of 4-BCl2 also was readily achieved in situ, analogously to 2-BCl2, to yield 4-BMe2, 4-BPh2 and 4-B(C6F5)2. Both aryl substituted congeners of 4-BR2 were stable to moisture (for at least 9 months in wet solvents) and could be isolated in excellent yield (>90%) however, 4-BMe2 underwent slow hydrolysis in wet solvent. This sensitivity is in contrast to 2-BMe2 which shows excellent stability to protic media (e.g., stable in refluxing EtOH for at least 30 min). This disparity is also contrary to the relative stability of thienyl and fluorenyl boronic acids and boronate esters, where the more electron rich thienyl congeners are generally more prone to protodeboronation.17,19 In our hands crystalline material suitable for analysis by single crystal X-ray diffraction was not obtained for any of the 4-BR2 series therefore calculations were performed at the M06-2X/6-311G (d,p) level with PCM solvation (DCM) on a simplified model of 4-BMe2, where octyls have been replaced for methyl groups and the non-borylated F8 is omitted. This revealed that borylative fusion planarises the aromatic core and leads to a short HH distance (Fig. 6, H1–H2 = 2.03 Å) between a benzothiadiazole and a fluorene C–H. This presumably provides a degree of strain which may weaken the B–N bond and result in more facile B–N cleavage. B–N cleavage has previously been shown to be the initial step in the hydrolysis of related boracycles.20 No close H–H contact is observed in the structure of 2-BMe2 due to a sulfur atom being located in the position adjacent to the equivalent benzothiadiazole C–H. Notably, the calculated structure of a methyl substituted borylatively fused thienyl–BT (Fig. 6, left) system has a planar boracycle. This is consistent with the observed non-planarity in 2-BMe2 being due to packing forces (other metrics are closely comparable between the calculated structure and solid state structure of 2-BMe2). The enhanced stability of the aryl substituted analogues, e.g., 4-BPh2, is attributed to the greater steric bulk affording sufficient kinetic stabilisation to prevent protodeboronation. Due to the H2O sensitivity further studies on borylated F8 systems utilised only exocyclic aryl substituents.
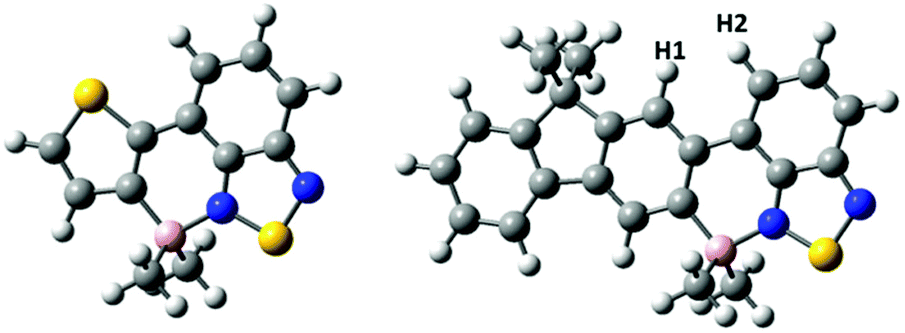 | ||
| Fig. 6 Calculated structures of methyl substituted borylatively fused thiophene–benzothiadiazole (left) and fluorene–benzothiadiazole (right). | ||
The double borylation of 4 was next attempted but was not achieved in our hands under a range of conditions with similar outcomes to that observed with attempted double borylation of 2 observed; therefore the double borylation of the A–D–A compound 5 was explored as an alternative route to a fully fused ladder structure. The incorporation of a second BT moiety will ensure a sufficiently nucleophilic donor site remains after the first borylative fusion step. In contrast to 4 compound 5 did not undergo borylation upon addition of excess BCl3 (with or without a hindered base), rather a diadduct between BCl3 and the peripheral nitrogen atoms of the benzothiadiazole units was formed (5-(BCl3)2, Fig. 7).
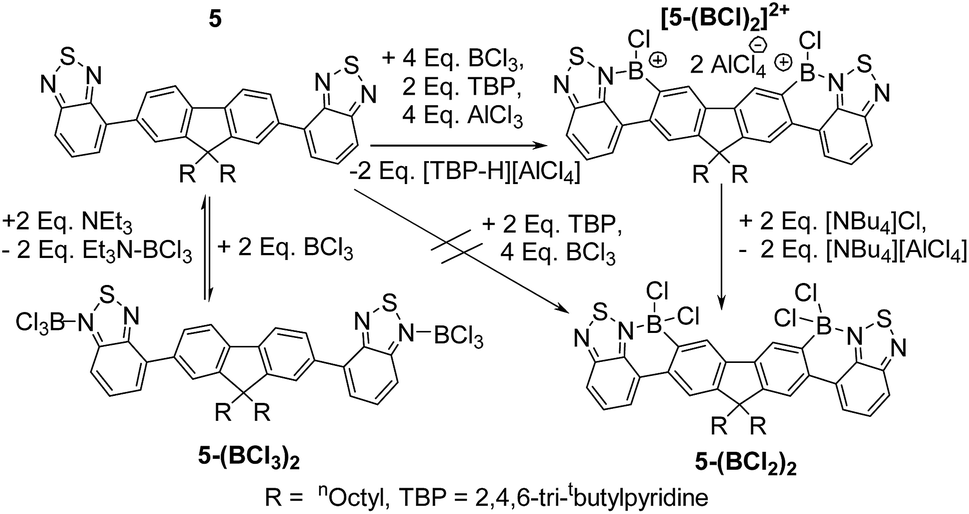 | ||
| Fig. 7 Reactivity of A–D–A compound 5 with boron electrophiles. | ||
Addition of Et3N to this compound cleaved the BT–BCl3 Lewis adduct reforming 5 as expected. Murakami's borylative fusion conditions (Et(iPr)2N/excess BBr3)12a also led to no C–H borylation. Borylative fusion of 5 was achieved by the addition of excess BCl3 (ca. 4 eq.), two equivalents of a sterically hindered base (TBP, 2,4,6-tritbutyl-pyridine) and four equivalents of AlCl3. The addition of 4 equivalents of AlCl3 is essential to ensure clean conversion to a single product, [5-(BCl)2]2+, as two equivalents of AlCl3 are consumed in competing halide abstraction reactions from the initial borylated product 5-(BCl2)2. Compound [5-(BCl)2]2+ is readily transformed to 5-(BCl2)2 by addition of [NBu4][Cl] (Fig. 7). Functionalisation of the BCl2 moieties in 5-(BCl2)2 was achieved in situ, even in the presence of the ionic by-products from C–H borylation, by addition of diaryl zinc reagents directly to the reaction mixture. Aryl substituted 5-(BPh2)2 and 5-(B(C6F5)2)2 were isolated in moderate yields (35 and 40%, respectively). These compounds show excellent moisture stability with no hydrolysis detected when exposed to moisture in solution for at least 9 months. Both 5-(B(aryl)2)2 compounds proved amenable to crystallisation and the solid state structures show the expected connectivity (Fig. 8). Both possessed a slightly curved backbone, with the angle between the benzothiadiazole planes and the plane of the central five membered ring of fluorene being 8.2 and 8.4° for 5-(BPh2)2 and 18.8° and 10.4° for 5-(B(C6F5)2)2. This slight non-planarity arises from steric hindrance in the cove region as discussed above which is supported by non-zero torsion angles, e.g., for 5-(BPh2)2 C5–C6–C7–C8 = 6.80° and C29–C30–C31–C31 = 6.15°. 5-(BPh2)2 and 5-(B(C6F5)2)2 have essentially planar six membered boracycles possessing similar structural metrics, excluding the B–N distances which are slightly shorter in 5-(B(C6F5)2)2 (B–N = 1.606(7) Å) than in 5-(BPh2)2 (1.623(5) Å). This is consistent with greater N → B σ-donation due to the enhanced Lewis acidity at boron provided by two pentafluorophenyl groups. The extended structures 5-(BPh2)2 and 5-(B(C6F5)2)2 exhibit no close π–π stacking interactions involving the fused aromatic core. This is in contrast to 2-BPh2 and is presumably due to the three quaternary-centres in 5-(B(aryl)2)2 that project considerable bulk above and below the plane precluding close approach of the delocalised ladder structure. The absence of any close π–π contacts is attractive for generating deep red/NIR solid state emissive materials, with the aryl groups in 5-(B(aryl)2)2 potentially providing sufficient bulk to disfavour aggregation induced quenching.
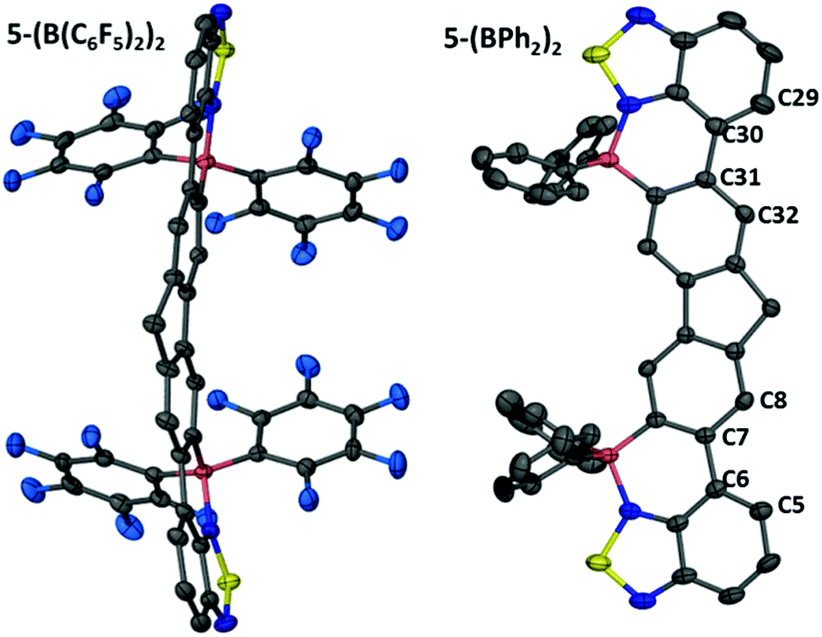 | ||
| Fig. 8 Left, the solid state structure of 5-(B(C6F5)2)2 and right, the solid state structure of 5-(BPh2)2 with ellipsoids at 50% probability (octyls, hydrogens and solvent removed for clarity in both cases). | ||
Electrochemical and photophysical properties
The borylative fusion of 2 leads to a significant red shift in the absorption spectrum of the products, with the most pronounced effect observed for C6F5 substituents on boron (Table 1). However, in each case the complexes 2-BR2 were effectively non-emissive in a range of solvents. The cyclic voltammetry (Table 1) of 2-BR2 revealed a reversible reductive process and multiple irreversible oxidation processes. Significantly, all compounds were stable to the electrolyte [nBu4N][PF6], in contrast to the B(C6F5)3 Lewis adducts of BT containing oligomers.4,5 The onset of the reduction process is significantly more positive post borylative fusion, whilst the onset of oxidation is less affected. Thus borylative fusion of 2 has minimal effect on the HOMO energy but results in a significant reduction in the LUMO energy, leading to a considerable band gap reduction and a substantial red shift in the onset of absorption. Comparison of the frontier orbital energies on varying the boron substituents in this series is notable. Alkyl substituents on boron produce the smallest reduction in the LUMO energy and the greatest (albeit still smaller than the change in the LUMO) increase in the HOMO energy. This is consistent with an enhanced electron donation from Me (relative to Ph) to boron generating a boron centre that withdraws less electron density from the benzothiadiazole moiety in 2-BMe2. The mixed phenyl/methyl derivative has frontier orbital energies between 2-BPh2 and 2-BMe2 indicating a cumulative effect of exocyclic boron substituents. Strongly electron withdrawing C6F5 groups have the most dramatic effect on the LUMO, lowering it by 0.61 eV, whilst the change in the energy of the HOMO is relatively minor (−0.07 eV). Presumably the expected minor increase in the HOMO energy derived from planarisation of the boracycle is effectively offset by the binding of the strongly Lewis acidic (thienyl)B(C6F5)2 moiety to benzothiadiazole. Whilst the low LUMO energies and reduced band gaps observed for 2-BR2 are promising for a range of applications these specific compounds are not useful for OLED devices, as they are effectively non-emissive in solution.| Compound | λ maxabs (nm) | ε (M−1 cm−1) | Optical band gapb (eV) | E onsetox (V) | E onsetred (V) | HOMOc (eV) | LUMOc (eV) | Electrochemical band gap (eV) |
|---|---|---|---|---|---|---|---|---|
| a 1 × 10−5 M solution in toluene. b Band gap estimated from onset of absorption.21 c Measured in DCM, (1 mM), with [nBu4N][PF6] (0.1 M) as the supporting electrolyte at a scan rate of 50 mV s−1, potentials are given relative to Fc/Fc+ redox couple which is taken to be 5.39 eV below vacuum.22 | ||||||||
| 2 | 471 | 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
2.29 | 0.60 | −1.66 | −6.00 | −3.73 | 2.27 |
| 2-BMe2 | 602 | 9700 | 1.73 | 0.46 | −1.33 | −5.85 | −4.06 | 1.79 |
| 2-B(Ph)Me | 611 | 9800 | 1.72 | 0.52 | −1.30 | −5.91 | −4.09 | 1.82 |
| 2-BPh2 | 617 | 12600 |
1.70 | 0.57 | −1.24 | −5.96 | −4.15 | 1.81 |
| 2-B(C6F5)2 | 641 | 7800 | 1.60 | 0.67 | −1.05 | −6.07 | −4.34 | 1.73 |
The UV-Vis absorbance spectra of the fluorene derivatives (4-BR2 and 5-BR2) were investigated next (Table 2) and these further demonstrated that borylative fusion leads to a large reduction in optical band gap (>120 nm) in all cases. As expected there is a bathochromic shift in both the λmax and the onset of the longer wavelength absorption band for C6F5 relative to C6H5 substituted analogues. The exocyclic boron substituent effect is most pronounced for 4, with only a marginal effect observed in the doubly borylated compound 5. Whilst the extinction coefficients at λmax are consistently lower post borylation much wider absorption bands are observed. Most significantly, in contrast to the thiophene derivatives borylated 4 and 5 proved to be significantly more emissive (Table 2). All four borylated fluorene compounds investigated exhibited solvatochromism of the fluorescence with the observed bathochromic shift of fluorescence with increasing solvent polarity characteristic of emission from a photoinduced-excited state with strong intramolecular charge transfer character.23 Compound 4-BPh2 showed a large Stokes shift in a range of solvents, for example in toluene the λmax emission is 702 nm, but emission continues significantly into the near-infrared spectral region. The solution quantum yield in toluene for 4-BPh2 of 10% is a respectable value for a deep red/NIR emitter due to the low band gap accelerating non-radiative processes consistent with the energy gap law.
| Compound | λ maxabs (nm) | ε (M−1 cm−1) | Optical band gapb (eV) | λ maxem (nm) | QYa,c (%) | E onsetox (V) | E onsetred (V) | HOMOd (eV) | LUMOd (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a 1 × 10−5 M solution in toluene. b Band gap estimated from onset of absorption. c Fluorescence quantum yield estimated by using cresyl violet as standard (QY = 54% in methanol).21 d Measured in DCM, (1 mM), with [nBu4N][PF6] (0.1 M) as the supporting electrolyte at a scan rate of 50 mV s−1, potentials are given relative to Fc/Fc+ redox couple which is taken to be 5.39 eV below vacuum.22 | |||||||||
| 4 | 419 | 21000 |
2.59 | 540 | — | 0.88 | −1.87 | −6.27 | −3.52 |
| 4-BPh2 | 559 | 12400 |
1.92 | 702 | 10 | 0.80 | −1.28 | −6.19 | −4.11 |
| 4-B(C6F5)2 | 579 | 9600 | 1.83 | 730 | 2 | 0.85 | −1.12 | −6.24 | −4.27 |
| 5 | 394 | 25200 |
2.82 | 512 | — | 0.95 | −1.87 | −6.34 | −3.52 |
| 5-(BPh2)2 | 538 | 19500 |
2.02 | 636 | 18 | 0.72 | −1.28 | −6.11 | −4.11 |
| 5-(B(C6F5)2)2 | 540 | 14700 |
2.00 | 645 | 12 | 0.94 | −1.07 | −6.33 | −4.32 |
In toluene 5-(BPh2)2 showed a λmax for emission at 636 nm with a higher intensity emission than 4-BPh2 probably due to the increased rigidity of the system. 4-B(C6F5)2 and 5-(B(C6F5)2)2 have similar emission properties to the phenyl congeners, but further red-shifted and with lower quantum yields. In all cases the excited state lifetimes are short (between 5–6 ns). The considerable reduction in band gap on borylative fusion observed by UV/Vis spectroscopy of these compounds was consistent with the computational analysis (at the M06-2x/6-311g (d,p) level with PCM solvation (DCM)) for model compounds of 5 and 5-(BPh2)2 where octyl is replaced for methyl (termed 5′ and 5′-(BPh2)2, Fig. 9). These revealed that for both 5′ and 5′-(BPh2)2 the HOMO is delocalised across the fluorene and benzothiadiazole units, whilst the LUMO is localised on the benzothiadiazole units. For 5′-(BPh2)2 the calculations indicate a significant decrease in the LUMO energy level by 0.67 eV and an increase in the HOMO energy level by 0.21 eV upon borylative fusion.
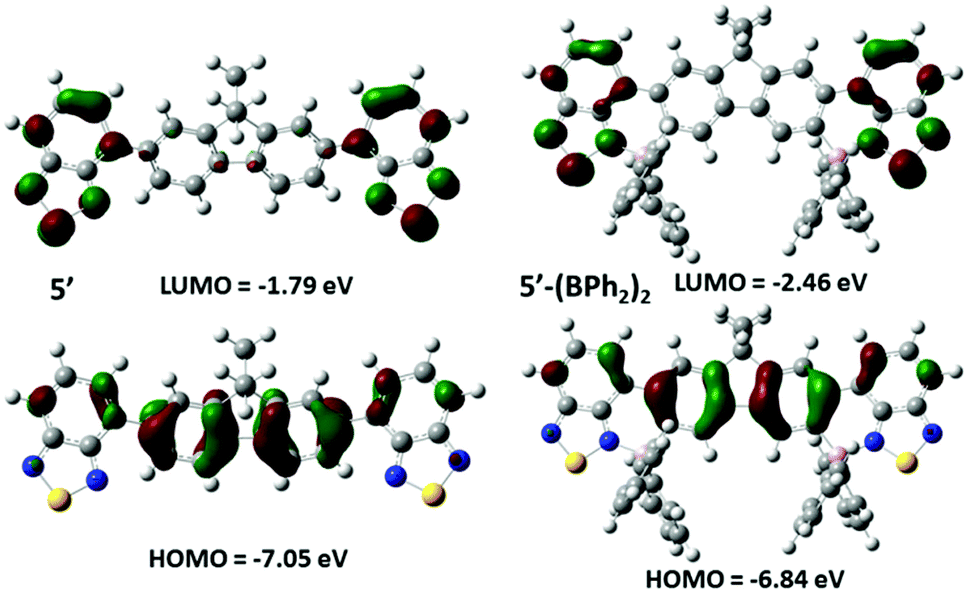 | ||
| Fig. 9 Molecular orbital diagrams (isovalue = 0.04) and calculated energy levels for the HOMO and LUMO of compounds 5′ and 5′-(BPh2)2. | ||
Cyclic voltammetry was used in order to gain further insight into the changes in frontier orbital energies on borylation. The effect of borylative fusion was similar across the 4-BAr2 and 5-(B(aryl)2)2 series. An irreversible oxidation and a single reversible reduction for all 4-BAr2 and 5-(B(aryl)2)2 compounds is observed, with the reduction shifted to a significantly less negative potential relative to the unborylated precursor in each case. The LUMO is consistently significantly lowered in energy by between 0.6 and 0.8 eV (Table 2), whilst the HOMO level is only slightly higher in energy. These energy changes are closely comparable to those observed post borylation of 2, indicating the generality of this methodology to modulate frontier orbital energies in BT containing materials. Comparing phenyl with the C6F5 substituted analogue is again noteworthy. For both borylated 4 and 5 the C6F5 congeners have lower HOMO and LUMO energies relative to the C6H5 analogues (by between 0.15–0.2 eV), again consistent with the strongly electron withdrawing nature of C6F5. The HOMO is effectively unchanged in energy for the B(C6F5)2 congeners relative to the unborylated precursors (ΔV = 0.01 and 0.03 V). Thus, borylative fusion and functionalisation with C6F5 is an effective method for drastically lowering the LUMO energy whilst the HOMO energy remains effectively unchanged. Borylative fusion provides a distinct frontier orbital energy modulation to Lewis adduct formation (which reduces both HOMO and LUMO orbital energies),4 and represents a simple method for generating modified BT containing D–A materials with increased electron affinities, lower band gaps and good stability to protic media. Significantly, both 4-B(C6F5)2 and 5-(B(C6F5)2)2 have frontier orbital energies closely comparable to PCBM (HOMO/LUMO of PCBM = −6.45 eV/−4.33 eV by cyclic voltammetry, see ESI†) indicating the potential of these compounds as low band gap electron acceptors in OPVs.
With the solid state structures of 5-(BPh2)2 and 5-(B(C6F5)2)2 displaying no close intermolecular π–π stacking interactions that involve the conjugated core aggregation induced fluorescence quenching should be minimised. Aggregation induced quenching is particularly prevalent in red/NIR emitters with a limited number of organic materials showing high quantum yields in the red/NIR spectral region in the solid state.24 The solid state emission properties of 4-BPh2, 5-(BPh2)2 and 5-(B(C6F5)2)2 were examined as these compounds showed the highest solution state quantum yields. These were tested by preparing thin films from a 5 wt% mixture of each compound dispersed in LUMATION™ 1300 Series green emitting polymer (L1300)25 and poly[(9,9-di-n-octylfluorenyl-2,7-diyl)-alt-(benzo[2,1,3]-thiadiazol-4,8-diyl)] (PF8-BT) polymer spin coated from toluene. On excitation at 468 nm in the PF8-BT polymer host compounds 4-BPh2, 5-(BPh2)2 and 5-(B(C6F5)2)2 emitted at a λmax of 696, 651, 673 nm with excellent (for deep red emitters) solid state quantum yields of 34, 33 and 20%, respectively (Table 3). These values suggest good exciton transfer between PF8-BT and the borylated dopants. The quantum yield values on doping into a L1300 host matrix are lower at 24, 16 and 3%, respectively, after exciting at 400 nm. This is attributed to less efficient exciton transfer between host and dopant due to a greater mismatch in frontier orbital energies of the host/dopant.26
| Compound X | PLaλmax/nm (QY/%) | Deviceb | ELcλmax/nm | V on /V | EQEe (%) |
|---|---|---|---|---|---|
|
a Photoluminescence of a film deposited from a 5/95 wt% solution of compound X/PF8-BT. Excitation at 468 nm and quantum yields determined using an integrating sphere.
b OLED device structure: ITO (45 nm)/Plexcore OC (65 nm)/PF8-TFB (22 nm)/emissive layer (100 nm 85% PF8-BT/15% PF8-TFB/5% compound X)/Ba (3.5 nm).
c Electroluminescence emission maxima.
d Turn-on voltage.
e Maximum external quantum efficiency.
f Emissive layer 95:5 wt% PF8BT/4-BPh2.
|
|||||
| 4-BPh2 | 696 (34) | 1 | 678 | 2.3 | 0.46 |
| 5-(BPh2)2 | 651 (33) | 2 | 634 | 2.1 | 0.14 |
| 5-(B(C6F5)2)2 | 673 (20) | 3 | 643 | 2.2 | 0.13 |
| 4-BPh2 | 696 (34) | 4f | 679 | 2.5 | 0.48 |
With excellent solid state fluorescence observed using PF8-BT as host the potential application of these materials in OLED devices was explored. A series of un-optimised OLED devices (devices 1–4, Table 3) were fabricated by solution processing. The emission layer (EmL) was deposited from a solution containing 5 wt% of the appropriate borylated compound, 80 wt% PF8-BT and 15 wt% PF8TFB (PF8TFB = poly[(9,9-dioctylfluorenyl-2,7-diyl)-co-(4,4′-(N-(4-sec-butylphenyl)-diphenylamine))]), with the latter used to improve hole transport. The devices were constructed as follows, ITO (45 nm)/Plexcore OC® (65 nm)/EmL (100 nm)/Ba (3.5 nm). An additional device was fabricated where the EmL was deposited from a solution containing only 5 wt% of 4-BPh2 and 95% of PF8-BT (i.e. in the absence of hole transport material PF8TFB, device 4). All devices possessed low turn-on voltages and showed electroluminescence spectra (Fig. 10) similar to their photoluminescence data in PF8-BT hosts, albeit with slightly blue shifted emission maxima (18–30 nm). Devices 1 and 4, both containing 4-BPh2 as the emitter, showed the highest maximum EQE values (0.46 and 0.48%, respectively) of the series of compounds with a λmax of 678 nm and a broad emission stretching into the NIR.
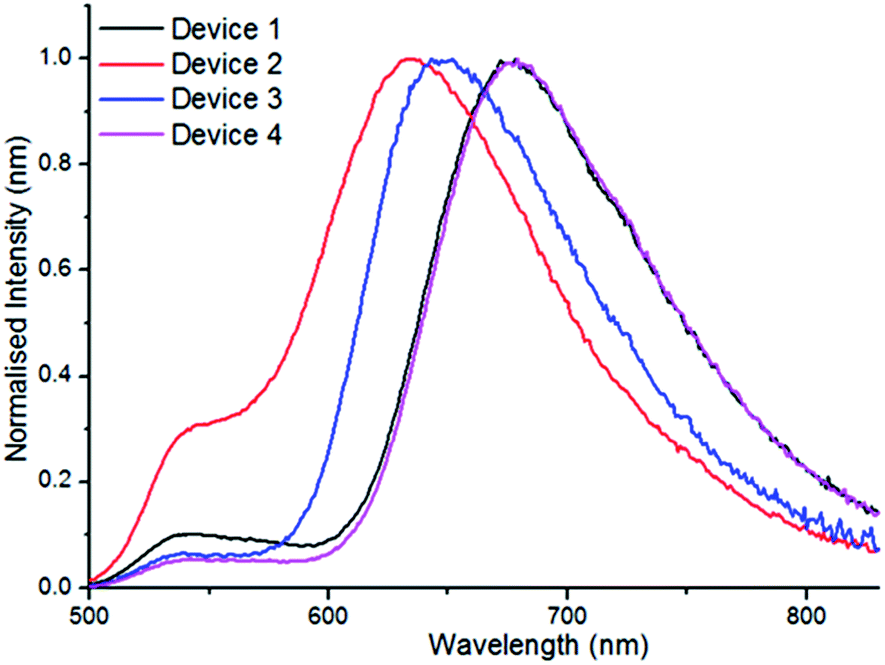 | ||
| Fig. 10 Electroluminescence from devices 1–4. | ||
Devices 1, 3 and 4 showed minimal green emission from the PF8-BT host whereas in device 2 this emission was significant. The EQE values using 4-BPh2 are among the highest reported for solution processed OLED devices emitting in the red/NIR region of the spectra.24 The simple, unoptimised device structure used to achieve these good EQE values indicates that the electrophilic borylative fusion methodology is highly attractive for constructing useful materials for generating OLED devices that emit in the deep red/NIR.
Conclusions
In summary, we report a simple strategy for modifying a range of BT containing donor–acceptor materials by directed C–H electrophilic borylation. Borylative fusion dramatically lowers the LUMO energy of these materials whilst changes to the energy of the HOMO are minimal, particularly with exocyclic C6F5 substituents on boron. The versatility of this methodology is demonstrated by: (i) functionalising both thiophene and fluorene based systems which are amongst the most common donor motifs in organic semiconductors and (ii) fine tuning frontier orbital energies and stability by variation in exocyclic boron substituents. To demonstrate further the usefulness of this methodology borylated D–A materials were synthesised that show large bathochromic shifts in absorption (relative to unborylated precursors), considerable Stokes shifts and far red/NIR fluorescence with excellent solid state quantum yields and excellent ambient/moisture stability. Un-optimised solution processed OLED devices were fabricated and showed good maximum EQE values for far-red/NIR emitters. These initial studies indicate that the borylative fusion of BT containing D–A oligomers is an attractive method for generating materials with extremely low lying LUMOs. These and other borylated D–A materials are currently under investigation in our laboratories as NIR emitters and as acceptors in OPVs.Acknowledgements
The research leading to these results has received funding from Cambridge Display Technology, the EPSRC (EP/J000973/1 and EP/K03099X/1) and the European Research Council (FP/2007-2013/ERC Grant Agreement 305868). MJI acknowledges the Royal Society (for the award of a University Research Fellowship) and MLT thanks Innovate UK for financial support of the Knowledge Centre for Material Chemistry. The authors would also like to acknowledge the use of the EPSRC UK National Service for Computational Chemistry Software (NSCCS) at Imperial College London in carrying out this work.Notes and references
- (a) A. J. Heeger, Adv. Mater., 2014, 26, 10 CrossRef CAS PubMed; (b) K. Colladet, S. Fourier, T. J. Cleij, L. Lutsen, J. Gelan, D. Vanderzande, L. H. Nguyen, H. Neugebauer, S. Sariciftci, A. Aguirre, G. Janssen and E. Goovaerts, Macromolecules, 2007, 40, 65 CrossRef CAS.
- For a recent example see: C. Y. Chiu, H. Wang, F. G. Brunetti, F. Wudl and C. J. Hawker, Angew. Chem., Int. Ed., 2014, 53, 3996 CrossRef CAS PubMed , and references therein.
- For a recent review see: X. Guo, A. Facchetti and T. J. Marks, Chem. Rev., 2014, 114, 8943 CrossRef CAS PubMed.
- G. C. Welch and G. C. Bazan, J. Am. Chem. Soc., 2011, 133, 4632 CrossRef CAS PubMed.
- G. C. Welch, R. Coffin, J. Peet and G. C. Bazan, J. Am. Chem. Soc., 2009, 131, 10802 CrossRef CAS PubMed.
- P. Zalar, Z. B. Henson, G. C. Welch, G. C. Bazan and T. Nguyen, Angew. Chem., Int. Ed., 2012, 51, 7495 CrossRef CAS PubMed.
- P. Zalar, M. Kuik, Z. B. Henson, C. Woellner, Y. Zhang, A. Sharenko, G. C. Bazan and T. Nguyen, Adv. Mater., 2014, 26, 724 CrossRef CAS PubMed.
- E. Poverenov, N. Zamoshchik, A. Patra, Y. Ridelman and M. Bendikov, J. Am. Chem. Soc., 2014, 136, 5138 CrossRef CAS PubMed.
- (a) S. Hayashi and T. Koizumi, Chem. Lett., 2012, 41, 979 CrossRef CAS; (b) S. Hayashi, A. Asano and T. Koizumi, Polym. Chem., 2011, 2, 2764 RSC; (c) S. Hayashi, A. Asano and T. Koizumi, RSC Adv., 2013, 3, 7375 RSC.
- (a) A. Wakamiya, T. Taniguchi and S. Yamaguchi, Angew. Chem., Int. Ed., 2006, 45, 3170 CrossRef CAS PubMed; (b) A. Job, A. Wakamiya, G. Kehr, G. Erker and S. Yamaguchi, Org. Lett., 2010, 12, 5470 CrossRef CAS PubMed.
- C. Dou, Z. Ding, Z. Zhang, Z. Xie, J. Liu and L. Wang, Angew. Chem., Int. Ed., 2015, 54, 3648 CrossRef CAS PubMed.
- (a) N. Ishida, T. Moriya, T. Goya and M. Murakami, J. Org. Chem., 2010, 75, 8709 CrossRef CAS PubMed; (b) Z. Zhao, Z. Chang, B. He, B. Chen, C. Deng, P. Lu, H. Qiu and B. Z. Tang, Chem.–Eur. J., 2013, 19, 11512 CrossRef CAS PubMed; (c) J. Chen, R. A. Lalancette and F. Jäkle, Organometallics, 2013, 32, 5843 CrossRef CAS; (d) M. Enders, G. Ludwig and H. Pritzkow, Organometallics, 2002, 21, 3856 CrossRef CAS.
- One notable exception comes from Wang and co-workers who compared mesityl, C6F5 and phenyl substituents on boron, H. Amarne, C. Baik, S. K. Murphy and S. Wang, Chem.–Eur. J., 2010, 16, 4750 CrossRef CAS PubMed.
- (a) D. Li, H. Zhang and Y. Wang, Chem. Soc. Rev., 2013, 42, 8416 RSC; (b) Y.-L. Rao and S. Wang, Inorg. Chem., 2011, 50, 12263 CrossRef CAS PubMed; (c) F. Jäkle, Chem. Rev., 2010, 16, 4574 Search PubMed.
- A. Loudet and K. Burgess, Chem. Rev., 2007, 107, 4891 CrossRef CAS PubMed.
- (a) F. S. Mancilha, L. Barloy, F. S. Rodembusch, J. Dupont and M. Pfeffer, Dalton Trans., 2011, 40, 10535 RSC; (b) T. Ozdemir, S. Atilgan, I. Kutuk, L. T. Yildrim, A. Tulek, M. Baylindir and E. U. Akkaya, Org. Lett., 2009, 11, 2105 CrossRef CAS PubMed.
- V. Bagutski, A. Del Grosso, J. Ayuso Carrillo, I. A. Cade, M. D. Helm, J. R. Lawson, P. J. Singleton, S. A. Solomon, T. Marcelli and M. J. Ingleson, J. Am. Chem. Soc., 2013, 135, 474 CrossRef CAS PubMed.
- Q. Hao, S. Yu, S. Li, J. Chen, Y. Zeng, T. Yu, G. Yang and Y. Li, J. Org. Chem., 2014, 79, 459 CrossRef CAS PubMed.
- E. Tyrrell and P. Brookes, Synthesis, 2003, 35, 469 CrossRef.
- Y.-L. Rao, T. Kusamoto, R. Sakamoto, H. Nishihara and S. Wang, Organometallics, 2014, 33, 1787 CrossRef CAS.
- A. M. Brouwer, Pure Appl. Chem., 2011, 82, 2213 Search PubMed.
- C. M. Cardona, W. Li, A. E. Kaifer, D. Stockdale and G. C. Bazan, Adv. Mater., 2011, 23, 2367 CrossRef CAS PubMed.
- M. Jia, X. Ma, L. Yan, H. Wang, Q. Guo, X. Wang, Y. Wang, X. Zhan and A. Xia, J. Phys. Chem. A, 2010, 114, 7345 CrossRef CAS PubMed.
- For select recent examples of solid state fluorescent red/NIR emitters and their applications in OLEDs see: (a) D. Li, S. Huang, S. Qu, X. Liu, Q. Zhu, H. Zhang and Y. Wang, J. Mater. Chem., 2011, 21, 15298 RSC; (b) M. Shimizu, R. Kaki, Y. Takeda, T. Hiyama, N. Nagai, H. Yamagishi and H. Furutani, Angew. Chem., Int. Ed., 2012, 51, 4095 CrossRef CAS PubMed; (c) B. Stender, S. F. Voelker, C. Lambert and J. Pflaum, Adv. Mater., 2013, 25, 2943 CrossRef CAS PubMed; (d) L. Yao, S. Zhang, R. Wang, W. Li, F. Zhen, B. Yang and Y. Ma, Angew. Chem., Int. Ed., 2014, 53, 2119 CrossRef CAS PubMed; (e) G. Li, Y. Zhao, J. Li, J. Cao, J. Zhu, X. W. Sun and Q. Zhang, J. Org. Chem., 2015, 80, 196 CrossRef CAS PubMed.
- D. Poplavskyy and F. So, J. Appl. Phys., 2005, 99, 033707 CrossRef PubMed.
- D. Poplavskyy, W. Su and F. So, J. Appl. Phys., 2005, 98, 014501 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, compound characterisation data, copies of NMR spectra and crystallographic data. CCDC 1050921–1050923, 1050963 and 1062559–1062560. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01800e |
| This journal is © The Royal Society of Chemistry 2015 |