
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceC–H bond activation induced by thorium metallacyclopropene complexes: a combined experimental and computational study†
Bo
Fang‡
a,
Lei
Zhang‡
a,
Guohua
Hou
a,
Guofu
Zi
*a,
De-Cai
Fang
*a and
Marc D.
Walter
*b
aDepartment of Chemistry, Beijing Normal University, Beijing 100875, China. E-mail: gzi@bnu.edu.cn; dcfang@bnu.edu.cn; Fax: +86-10-58802075; Tel: +86-10-58806051
bInstitut für Anorganische und Analytische Chemie, Technische Universität Braunschweig, Hagenring 30, 38106 Braunschweig, Germany. E-mail: mwalter@tu-bs.de; Fax: +49-531-3915387; Tel: +49-531-3915312
First published on 17th June 2015
Abstract
Inter- and intramolecular C–H bond activations by thorium metallacyclopropene complexes were comprehensively studied. The reduction of [η5-1,2,4-(Me3C)3C5H2]2ThCl2 (1) with potassium graphite (KC8) in the presence of internal alkynes (PhC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CR) yields the corresponding thorium metallacyclopropenes [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph(R)) (R = Ph (2), Me (3), iPr (4), C6H11 (5)). Complexes 3–5 derived from phenyl(alkyl)acetylenes are very reactive resulting in an intramolecular C–H bond activation of the 1,2,4-(Me3C)3C5H2 ligand. In contrast, no intramolecular C–H bond activation is observed for the diphenylacetylene derived complex 2, but it does activate α-C–H bonds in pyridine or carbonyl derivatives upon coordination. Density functional theory (DFT) studies complement the experimental studies and provide additional insights into the observed reactivity.
CR) yields the corresponding thorium metallacyclopropenes [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph(R)) (R = Ph (2), Me (3), iPr (4), C6H11 (5)). Complexes 3–5 derived from phenyl(alkyl)acetylenes are very reactive resulting in an intramolecular C–H bond activation of the 1,2,4-(Me3C)3C5H2 ligand. In contrast, no intramolecular C–H bond activation is observed for the diphenylacetylene derived complex 2, but it does activate α-C–H bonds in pyridine or carbonyl derivatives upon coordination. Density functional theory (DFT) studies complement the experimental studies and provide additional insights into the observed reactivity.
Introduction
Highly strained metallacyclopropenes are reactive functionalities and can serve as precursors for the synthesis of highly functionalized organic molecules and heterocyclic main group compounds.1–3 In this context metallacyclopropenes of group 4 metallocenes have been of particular interest1,2 and are readily prepared by the reaction of Cp′2M (Cp′ = (un)substituted η5-cyclopentadienyl) with alkynes or by the reduction of Cp′2MCl2 in the presence of a suitable alkyne.1b However, group 4 metallacyclopropenes derived from differently substituted alkynes are exceptionally rare,2j–m see e.g. [η5-C5H5]2Zr(η2-RC2SiMe2H) (R = tBu, Ph, SiMe3).2j One reason for this is that especially for the heavier (and therefore larger) group 4 metals the use of less sterically demanding alkynes generally produces the more stable metallacyclopentadienes, and therefore detailed investigations on metallacyclopropenes have been limited to bulky and symmetrically substituted alkynes such as PhCCPh and Me3SiCCSiMe3.1b,2n,o In addition, the metallacyclopropenes derived from Me3SiCCSiMe3 are also more susceptible to substitutions and to participate in C–H bond activation processes.1b,2n,o Nevertheless, in contrast to the rich group 4 chemistry, actinide metallacyclopropenes have remained rare,4 and only recently the first stable metallacyclopropene [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2) has been prepared.5 Several studies have now established that in actinide chemistry the 5f orbitals have significant influence on the reactivity.6 Thorium with its 7s26d2 ground state stands on the borderline between group 4 metals and the actinides and it is therefore a very attractive element for further investigations. Complex 2 reacts with a variety of hetero-unsaturated molecules such as aldehydes, ketones, CS2, carbodiimides, nitriles, isothiocyanates, organic azides, and diazoalkane derivatives.5 The Th(η2-PhCCPh) moiety in complex 2 shows no reactivity towards additional alkynes to form metallacyclopentadienes and no exchange with added alkynes. Therefore it is of interest to explore the reduction of [η5-1,2,4-(Me3C)3C5H2]2ThCl2 (1) in the presence of unsymmetrically substituted alkynes such as PhCCR to prepare novel thorium metallacyclopropenes that can be tuned in their steric and electronic properties and to investigate their ability to participate in C–H bond activation processes that are a highly topical field in organoactinide research7 and also to correlate this reactivity to group 4 metal chemistry. These studies are described in this article.
Experimental
General methods
All reactions and product manipulations were carried out under an atmosphere of dry dinitrogen with rigid exclusion of air and moisture using standard Schlenk or cannula techniques, or in a glove box. All organic solvents were freshly distilled from sodium benzophenone ketyl immediately prior to use. KC8,8 [η5-1,2,4-(Me3C)3C5H2]2ThCl2 (1)9 and [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2)5 were prepared according to literature methods. All other chemicals were purchased from Aldrich Chemical Co. and Beijing Chemical Co. and used as received unless otherwise noted. Infrared spectra were recorded in KBr pellets on an Avatar 360 Fourier transform spectrometer. 1H and 13C{1H} NMR spectra were recorded on a Bruker AV 400 spectrometer at 400 and 100 MHz, respectively. All chemical shifts are reported in δ units with reference to the residual protons of the deuterated solvents, which served as internal standards, for proton and carbon chemical shifts. Melting points were measured on an X-6 melting point apparatus and were uncorrected. Elemental analyses were performed on a Vario EL elemental analyzer.Syntheses
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e001.gif) CHiPr] (7).
KC8 (1.20 g, 8.80 mmol) was added to a toluene (20 mL) solution of [η5-1,2,4-(Me3C)3C5H2]2ThCl2 (1; 2.00 g, 2.6 mmol) and PhCCiPr (0.38 g, 2.6 mmol) with stirring at room temperature. After this solution was stirred one day at 80 °C, the solvent was removed. The residue was extracted with benzene (10 mL × 3) and filtered. The volume of the filtrate was reduced to 10 mL, colorless crystals of 7 were isolated when this solution was kept at room temperature for two days. Yield: 1.64 g (75%) (found: C, 64.08; H, 8.43. C45H70Th requires C, 64.11; H, 8.37%). M.p.: 202–204 °C. 1H NMR (C6D6): δ 7.36 (t, J = 7.7 Hz, 2H, phenyl), 7.21 (d, J = 7.2 Hz, 2H, phenyl), 6.97 (t, J = 7.4 Hz, 1H, phenyl), 6.49 (d, J = 3.4 Hz, 1H, ring CH), 6.10 (d, J = 3.5 Hz, 1H, ring CH), 5.92 (d, J = 3.4 Hz, 1H, ring CH), 5.75 (d, J = 3.5 Hz, 1H, ring CH), 5.41 (d, J = 7.2 Hz, 1H, C
CHiPr] (7).
KC8 (1.20 g, 8.80 mmol) was added to a toluene (20 mL) solution of [η5-1,2,4-(Me3C)3C5H2]2ThCl2 (1; 2.00 g, 2.6 mmol) and PhCCiPr (0.38 g, 2.6 mmol) with stirring at room temperature. After this solution was stirred one day at 80 °C, the solvent was removed. The residue was extracted with benzene (10 mL × 3) and filtered. The volume of the filtrate was reduced to 10 mL, colorless crystals of 7 were isolated when this solution was kept at room temperature for two days. Yield: 1.64 g (75%) (found: C, 64.08; H, 8.43. C45H70Th requires C, 64.11; H, 8.37%). M.p.: 202–204 °C. 1H NMR (C6D6): δ 7.36 (t, J = 7.7 Hz, 2H, phenyl), 7.21 (d, J = 7.2 Hz, 2H, phenyl), 6.97 (t, J = 7.4 Hz, 1H, phenyl), 6.49 (d, J = 3.4 Hz, 1H, ring CH), 6.10 (d, J = 3.5 Hz, 1H, ring CH), 5.92 (d, J = 3.4 Hz, 1H, ring CH), 5.75 (d, J = 3.5 Hz, 1H, ring CH), 5.41 (d, J = 7.2 Hz, 1H, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHiPr), 2.95 (m, 1H, CH(CH3)2), 1.71 (s, 3H, C(CH3)2), 1.66 (s, 3H, C(CH3)2), 1.57 (s, 9H, C(CH3)3), 1.50 (s, 9H, C(CH3)3), 1.48 (s, 9H, C(CH3)3), 1.47 (s, 9H, C(CH3)3), 1.22 (s, 9H, C(CH3)3), 1.06 (m, 7H, ThCH2 and CH(CH3)2), −0.01 (d, J = 13.0 Hz, 1H, ThCH2) ppm. 13C{1H} NMR (C6D6): δ 219.6 (ThCPh), 151.5 (phenyl C), 143.9 (phenyl C), 142.2 (phenyl C), 140.4 (phenyl C), 139.5 (ring C), 138.3 (ring C), 128.5 (ring C), 124.6 (ring C), 124.2 (ring C), 124.1 (CCHiPr), 123.7 (ring C), 116.9 (ring C), 115.5 (ring C), 114.0 (ring C), 112.0 (ring C), 49.8 (ThCH2), 35.8 (C(CH3)3), 35.5 (C(CH3)3), 35.0 (C(CH3)3), 34.9 (C(CH3)3), 34.7 (C(CH3)3), 34.4 (C(CH3)3), 34.3 (C(CH3)3), 34.2 (CH2C(CH3)2), 34.0 (C(CH3)3), 33.9 (C(CH3)3), 32.6 (C(CH3)3), 30.4 (CH2C(CH3)2), 28.5 (CH2C(CH3)2), 23.5 (CH(CH3)2), 23.4 (CH(CH3)2) ppm. IR (KBr, cm−1): ν 2954 (s), 1589 (m), 1485 (s), 1456 (s), 1384 (s), 1357 (s), 1238 (s), 1165 (s), 1070 (s), 1028 (s), 904 (m), 813 (s).
CH(C6H11)] (8).
This compound was prepared as colorless crystals from the reaction of [η5-1,2,4-(Me3C)3C5H2]2ThCl2 (1; 2.00 g, 2.6 mmol) and PhCC(C6H11) (0.48 g, 2.6 mmol) in the presence of KC8 (1.20 g, 8.80 mmol) in toluene (20 mL) at 100 °C and recrystallization from a benzene solution by a similar procedure as in the synthesis of 7. Yield: 1.84 g (80%) (found: C, 65.30; H, 8.43. C48H74Th requires C, 65.28; H, 8.45%). M.p.: 180–182 °C. 1H NMR (C6D6): δ 7.36 (t, J = 7.6 Hz, 2H, phenyl), 7.24 (d, J = 7.4 Hz, 2H, phenyl), 6.96 (t, J = 7.2 Hz, 1H, phenyl), 6.49 (d, J = 3.4 Hz, 1H, ring CH), 6.10 (d, J = 3.5 Hz, 1H, ring CH), 5.93 (d, J = 3.4 Hz, 1H, ring CH), 5.78 (d, J = 3.5 Hz, 1H, ring CH), 5.48 (d, J = 7.2 Hz, 1H, CCHCy), 2.74 (m, 1H, cyclohexyl-CH), 1.83 (br s, 2H, cyclohexyl-CH2), 1.72 (s, 3H, C(CH3)2), 1.71 (s, 3H, C(CH3)2), 1.58 (s, 9H, C(CH3)3), 1.51 (s, 9H, C(CH3)3), 1.50 (s, 18H, C(CH3)3), 1.44 (m, 4H, cyclohexyl-CH2), 1.22 (s, 9H, C(CH3)3), 1.07 (m, 5H, ThCH2 and cyclohexyl-CH2), 0.01 (d, J = 13.0 Hz, 1H, ThCH2) ppm. 13C{1H} NMR (C6D6): δ 220.3 (ThCPh), 151.6 (phenyl C), 144.0 (phenyl C), 142.2 (phenyl C), 140.4 (phenyl C), 139.6 (ring C), 138.4 (ring C), 128.5 (ring C), 124.2 (CCHCy), 124.1 (ring C), 123.7 (ring C), 123.6 (ring C), 117.0 (ring C), 115.6 (ring C), 114.0 (ring C), 112.1 (ring C), 49.7 (ThCH2), 38.2 (CH), 35.8 (C(CH3)3), 35.5 (C(CH3)3), 35.4 (C(CH3)3), 35.1 (C(CH3)3), 34.9 (C(CH3)3), 34.7 (C(CH3)3), 34.5 (C(CH3)3), 34.4 (CH2), 34.1 (C(CH3)3), 34.0 (CH2C(CH3)2), 33.9 (C(CH3)3), 33.4 (CH2C(CH3)2), 32.6 (C(CH3)3), 32.5 (CH2), 30.6 (CH2C(CH3)2), 26.2 (CH2), 26.1 (CH2), 26.0 (CH2) ppm. IR (KBr, cm−1): ν 2955 (s), 2925 (s), 1599 (m), 1448 (s), 1360 (s), 1260 (s), 1096 (s), 1028 (s), 808 (s).
CCH3 (0.30 g, 2.6 mmol) in the presence of KC8 (1.20 g, 8.80 mmol) in toluene (20 mL) at 70 °C and recrystallization from a benzene solution by a similar procedure as in the synthesis of 7. Yield: 1.97 g (85%) (found: C, 65.88; H, 8.16. C49H72Th requires C, 65.89; H, 8.13%). M.p.: 216–218 °C. 1H NMR (C6D6): δ 7.37 (d, J = 7.8 Hz, 2H, phenyl), 7.25 (t, J = 7.7 Hz, 2H, phenyl), 7.15 (s, 6H, C6H6), 7.00 (t, J = 7.3 Hz, 1H, phenyl), 6.67 (m, 1H, PhCHCH), 6.10 (d, J = 3.4 Hz, 1H, ring CH), 6.01 (d, J = 3.5 Hz, 1H, ring CH), 5.94 (d, J = 3.4 Hz, 1H, ring CH), 5.30 (d, J = 3.5 Hz, 1H, ring CH), 4.64 (d, J = 15.6 Hz, 1H, PhCHCH), 2.59 (m, 1H, ThCH2CHCHPh), 2.47 (m, 1H, ThCH2CHCH), 1.53 (br s, 12H, C(CH3)3 and C(CH3)2), 1.52 (s, 9H, C(CH3)3), 1.40 (s, 9H, C(CH3)3), 1.33 (s, 9H, C(CH3)3), 1.29 (s, 9H, C(CH3)3), 1.03 (s, 3H, C(CH3)2), 0.27 (d, J = 12.7 Hz, 1H, ThCH2), −0.09 (d, J = 12.7 Hz, 1H, ThCH2) ppm. 13C{1H} NMR (C6D6): δ 142.9 (phenyl C), 142.0 (phenyl C), 140.2 (phenyl C), 139.8 (phenyl C), 139.6 (ring C), 139.3 (ring C), 129.3 (ring C), 128.7 (ring C), 128.5 (ring C), 128.0 (C6H6), 124.9 (ring C), 124.7 (ring C), 123.4 (ring C), 114.3 (ring C), 112.2 (ring C), 111.8 (PhCHCH), 100.8 (PhCHCH), 66.1 (ThCH2CHCHPh), 45.6 (ThCH2), 35.4 (C(CH3)3), 35.2 (C(CH3)3), 35.0 (C(CH3)3), 34.9 (C(CH3)3), 34.8 (C(CH3)3), 34.3 (C(CH3)3), 34.0 (C(CH3)3), 33.6 (C(CH3)3), 33.5 (C(CH3)3), 33.4 (CH2C(CH3)2), 32.9 (C(CH3)3), 32.8 (CH2C(CH3)2), 30.2 (C(CH3)2) ppm. IR (KBr, cm−1): ν 2956 (s), 2904 (s), 1473 (s), 1460 (s), 1386 (s), 1361 (s), 1238 (s), 1070 (s), 1022 (s), 812 (s).
CHiPr), 2.95 (m, 1H, CH(CH3)2), 1.71 (s, 3H, C(CH3)2), 1.66 (s, 3H, C(CH3)2), 1.57 (s, 9H, C(CH3)3), 1.50 (s, 9H, C(CH3)3), 1.48 (s, 9H, C(CH3)3), 1.47 (s, 9H, C(CH3)3), 1.22 (s, 9H, C(CH3)3), 1.06 (m, 7H, ThCH2 and CH(CH3)2), −0.01 (d, J = 13.0 Hz, 1H, ThCH2) ppm. 13C{1H} NMR (C6D6): δ 219.6 (ThCPh), 151.5 (phenyl C), 143.9 (phenyl C), 142.2 (phenyl C), 140.4 (phenyl C), 139.5 (ring C), 138.3 (ring C), 128.5 (ring C), 124.6 (ring C), 124.2 (ring C), 124.1 (CCHiPr), 123.7 (ring C), 116.9 (ring C), 115.5 (ring C), 114.0 (ring C), 112.0 (ring C), 49.8 (ThCH2), 35.8 (C(CH3)3), 35.5 (C(CH3)3), 35.0 (C(CH3)3), 34.9 (C(CH3)3), 34.7 (C(CH3)3), 34.4 (C(CH3)3), 34.3 (C(CH3)3), 34.2 (CH2C(CH3)2), 34.0 (C(CH3)3), 33.9 (C(CH3)3), 32.6 (C(CH3)3), 30.4 (CH2C(CH3)2), 28.5 (CH2C(CH3)2), 23.5 (CH(CH3)2), 23.4 (CH(CH3)2) ppm. IR (KBr, cm−1): ν 2954 (s), 1589 (m), 1485 (s), 1456 (s), 1384 (s), 1357 (s), 1238 (s), 1165 (s), 1070 (s), 1028 (s), 904 (m), 813 (s).
CH(C6H11)] (8).
This compound was prepared as colorless crystals from the reaction of [η5-1,2,4-(Me3C)3C5H2]2ThCl2 (1; 2.00 g, 2.6 mmol) and PhCC(C6H11) (0.48 g, 2.6 mmol) in the presence of KC8 (1.20 g, 8.80 mmol) in toluene (20 mL) at 100 °C and recrystallization from a benzene solution by a similar procedure as in the synthesis of 7. Yield: 1.84 g (80%) (found: C, 65.30; H, 8.43. C48H74Th requires C, 65.28; H, 8.45%). M.p.: 180–182 °C. 1H NMR (C6D6): δ 7.36 (t, J = 7.6 Hz, 2H, phenyl), 7.24 (d, J = 7.4 Hz, 2H, phenyl), 6.96 (t, J = 7.2 Hz, 1H, phenyl), 6.49 (d, J = 3.4 Hz, 1H, ring CH), 6.10 (d, J = 3.5 Hz, 1H, ring CH), 5.93 (d, J = 3.4 Hz, 1H, ring CH), 5.78 (d, J = 3.5 Hz, 1H, ring CH), 5.48 (d, J = 7.2 Hz, 1H, CCHCy), 2.74 (m, 1H, cyclohexyl-CH), 1.83 (br s, 2H, cyclohexyl-CH2), 1.72 (s, 3H, C(CH3)2), 1.71 (s, 3H, C(CH3)2), 1.58 (s, 9H, C(CH3)3), 1.51 (s, 9H, C(CH3)3), 1.50 (s, 18H, C(CH3)3), 1.44 (m, 4H, cyclohexyl-CH2), 1.22 (s, 9H, C(CH3)3), 1.07 (m, 5H, ThCH2 and cyclohexyl-CH2), 0.01 (d, J = 13.0 Hz, 1H, ThCH2) ppm. 13C{1H} NMR (C6D6): δ 220.3 (ThCPh), 151.6 (phenyl C), 144.0 (phenyl C), 142.2 (phenyl C), 140.4 (phenyl C), 139.6 (ring C), 138.4 (ring C), 128.5 (ring C), 124.2 (CCHCy), 124.1 (ring C), 123.7 (ring C), 123.6 (ring C), 117.0 (ring C), 115.6 (ring C), 114.0 (ring C), 112.1 (ring C), 49.7 (ThCH2), 38.2 (CH), 35.8 (C(CH3)3), 35.5 (C(CH3)3), 35.4 (C(CH3)3), 35.1 (C(CH3)3), 34.9 (C(CH3)3), 34.7 (C(CH3)3), 34.5 (C(CH3)3), 34.4 (CH2), 34.1 (C(CH3)3), 34.0 (CH2C(CH3)2), 33.9 (C(CH3)3), 33.4 (CH2C(CH3)2), 32.6 (C(CH3)3), 32.5 (CH2), 30.6 (CH2C(CH3)2), 26.2 (CH2), 26.1 (CH2), 26.0 (CH2) ppm. IR (KBr, cm−1): ν 2955 (s), 2925 (s), 1599 (m), 1448 (s), 1360 (s), 1260 (s), 1096 (s), 1028 (s), 808 (s).
CCH3 (0.30 g, 2.6 mmol) in the presence of KC8 (1.20 g, 8.80 mmol) in toluene (20 mL) at 70 °C and recrystallization from a benzene solution by a similar procedure as in the synthesis of 7. Yield: 1.97 g (85%) (found: C, 65.88; H, 8.16. C49H72Th requires C, 65.89; H, 8.13%). M.p.: 216–218 °C. 1H NMR (C6D6): δ 7.37 (d, J = 7.8 Hz, 2H, phenyl), 7.25 (t, J = 7.7 Hz, 2H, phenyl), 7.15 (s, 6H, C6H6), 7.00 (t, J = 7.3 Hz, 1H, phenyl), 6.67 (m, 1H, PhCHCH), 6.10 (d, J = 3.4 Hz, 1H, ring CH), 6.01 (d, J = 3.5 Hz, 1H, ring CH), 5.94 (d, J = 3.4 Hz, 1H, ring CH), 5.30 (d, J = 3.5 Hz, 1H, ring CH), 4.64 (d, J = 15.6 Hz, 1H, PhCHCH), 2.59 (m, 1H, ThCH2CHCHPh), 2.47 (m, 1H, ThCH2CHCH), 1.53 (br s, 12H, C(CH3)3 and C(CH3)2), 1.52 (s, 9H, C(CH3)3), 1.40 (s, 9H, C(CH3)3), 1.33 (s, 9H, C(CH3)3), 1.29 (s, 9H, C(CH3)3), 1.03 (s, 3H, C(CH3)2), 0.27 (d, J = 12.7 Hz, 1H, ThCH2), −0.09 (d, J = 12.7 Hz, 1H, ThCH2) ppm. 13C{1H} NMR (C6D6): δ 142.9 (phenyl C), 142.0 (phenyl C), 140.2 (phenyl C), 139.8 (phenyl C), 139.6 (ring C), 139.3 (ring C), 129.3 (ring C), 128.7 (ring C), 128.5 (ring C), 128.0 (C6H6), 124.9 (ring C), 124.7 (ring C), 123.4 (ring C), 114.3 (ring C), 112.2 (ring C), 111.8 (PhCHCH), 100.8 (PhCHCH), 66.1 (ThCH2CHCHPh), 45.6 (ThCH2), 35.4 (C(CH3)3), 35.2 (C(CH3)3), 35.0 (C(CH3)3), 34.9 (C(CH3)3), 34.8 (C(CH3)3), 34.3 (C(CH3)3), 34.0 (C(CH3)3), 33.6 (C(CH3)3), 33.5 (C(CH3)3), 33.4 (CH2C(CH3)2), 32.9 (C(CH3)3), 32.8 (CH2C(CH3)2), 30.2 (C(CH3)2) ppm. IR (KBr, cm−1): ν 2956 (s), 2904 (s), 1473 (s), 1460 (s), 1386 (s), 1361 (s), 1238 (s), 1070 (s), 1022 (s), 812 (s).
When the isotopically labeled alkyne PhCCCD3 was used, the resonance at δ = 4.64 ppm corresponding to PhCHCH in complex 9 disappeared, indicating that indeed a [1,3]-hydrogen migration had occurred in the PhCCHCH3 fragment resulting in the formation of 9.
CHPh](η2-C,N–C5H4N) (10).
Method A. A toluene solution (5 mL) of pyridine (20 mg, 0.25 mmol) was added to a toluene (10 mL) solution of [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2; 220 mg, 0.25 mmol) with stirring at room temperature. After the solution was stirred at room temperature four days, the solvent was removed. The residue was extracted with benzene (10 mL × 3) and filtered. The volume of the filtrate was reduced to 2 mL, colorless crystals of 10 were isolated when this solution was kept at room temperature for one week. Yield: 206 mg (86%) (found: C, 66.63; H, 7.62; N, 1.43. C53H73NTh requires C, 66.57; H, 7.70; N, 1.46%). M.p.: 192–194 °C. 1H NMR (C6D6): δ 7.90 (d, J = 7.5 Hz, 1H, pyridyl), 7.45 (m, 5H, phenyl), 7.36 (t, J = 7.6 Hz, 2H, phenyl), 7.12 (m, 2H, phenyl), 7.06 (s, 1H, C
CH), 6.98 (m, 1H, pyridyl), 6.93 (t, J = 7.4 Hz, 1H, pyridyl), 6.87 (m, 1H, phenyl), 6.58 (d, J = 3.2 Hz, 2H, ring CH), 6.38 (t, J = 6.0 Hz, 1H, pyridyl), 6.33 (d, J = 3.2 Hz, 2H, ring CH), 1.51 (s, 18H, C(CH3)3), 1.45 (s, 18H, C(CH3)3), 1.04 (s, 18H, C(CH3)3) ppm. 13C{1H} NMR (C6D6): δ 229.8 (ThCPh), 210.1 (ThCN), 154.0 (aryl C), 145.5 (aryl C), 142.7 (aryl C), 142.5 (aryl C), 141.2 (aryl C), 137.7 (aryl C), 136.3 (aryl C), 134.9 (aryl C), 133.4 (aryl C), 129.7 (aryl C), 128.5 (aryl C), 128.4 (aryl C), 126.4 (ring C), 126.3 (ring C), 124.1 (ring C), 122.8 (ring C), 118.1 (ring C), 112.6 (CCHPh), 34.9 (C(CH3)3), 34.8 (C(CH3)3), 34.5 (C(CH3)3), 34.0 (C(CH3)3), 33.0 (C(CH3)3) ppm; one C resonance of Me3C-groups overlapped. IR (KBr, cm−1): ν 2958 (s), 1590 (s), 1480 (s), 1458 (s), 1357 (s), 1237 (s), 1001 (s), 825 (s).
Method B. NMR scale. A C6D6 (0.3 mL) solution of pyridine (1.6 mg; 0.02 mmol) was slowly added to a J. Young NMR tube charged with [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2; 18 mg, 0.02 mmol) and C6D6 (0.2 mL). The NMR sample was maintained at room temperature and monitored periodically by 1H NMR spectroscopy. After one day, conversion to 10 was 40% complete, and after four days, conversion to 10 was complete.
When perdeuterated pyridine C5D5N was used, the resonance at δ = 7.06 ppm corresponding to the PhCHC fragment in 10 disappeared completely, confirming that a deuterium atom was transferred to the alkenyl group.
CHPh](η2-C,N-4-Me2N-C5H3N) (11).
Method A. This compound was prepared as colorless microcrystals from the reaction of [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2; 220 mg, 0.25 mmol) and DMAP (31 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from an n-hexane solution by a similar procedure as in the synthesis of 10. Yield: 217 mg (87%) (found: C, 66.13; H, 7.79; N, 2.83. C55H78N2Th requires C, 66.11; H, 7.87; N, 2.80%). M.p.: 176–178 °C. 1H NMR (C6D6): δ 7.55 (d, J = 7.3 Hz, 2H, phenyl), 7.48 (m, 3H, phenyl), 7.41 (t, J = 7.6 Hz, 2H, phenyl), 7.27 (s, 1H, pyridyl), 7.12 (m, 3H, phenyl and C
CH), 6.94 (t, J = 7.3 Hz, 1H, phenyl), 6.72 (d, J = 5.8 Hz, 1H, pyridyl), 6.62 (d, J = 3.2 Hz, 2H, ring CH), 6.34 (d, J = 3.2 Hz, 2H, ring CH), 5.83 (dd, J = 6.4, 2.4 Hz, 1H, pyridyl), 2.27 (s, 6H, (CH3)2N), 1.55 (s, 36H, C(CH3)3), 1.19 (s, 18H, C(CH3)3) ppm. 13C{1H} NMR (C6D6): δ 225.0 (ThCPh), 210.3 (ThCN), 154.6 (aryl C), 153.6 (aryl C), 144.6 (aryl C), 142.1 (aryl C), 142.0 (aryl C), 140.6 (aryl C), 138.0 (aryl C), 133.3 (aryl C), 129.7 (aryl C), 128.4 (aryl C), 128.3 (aryl C), 126.5 (aryl C), 126.1 (ring C), 122.5 (ring C), 117.8 (ring C), 116.9 (ring C), 112.3 (ring C), 109.2 (CCHPh), 38.6 ((CH3)2N), 35.0 (C(CH3)3), 34.8 (C(CH3)3), 34.7 (C(CH3)3), 34.6 (C(CH3)3), 34.1 (C(CH3)3), 33.1 (C(CH3)3) ppm. IR (KBr, cm−1): ν 2956 (s), 1582 (s), 1490 (s), 1434 (s), 1363 (s), 1257 (s), 1238 (s), 1165 (s), 996 (s), 825 (s).
Method B. NMR scale. A C6D6 (0.3 mL) solution of DMAP (2.5 mg; 0.02 mmol) was slowly added to a J. Young NMR tube charged with [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2; 18 mg, 0.02 mmol) and C6D6 (0.2 mL). The NMR sample was maintained at room temperature and monitored periodically by 1H NMR spectroscopy. After one day, conversion to 11 was 70% complete, and after two days, conversion to 11 was complete.
CHPh](κ2-C,O–C5H4NO) (12).
Method A. This compound was prepared as colorless crystals from the reaction of [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2; 220 mg, 0.25 mmol) and pyridine N-oxide (24 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from an n-hexane solution by a similar procedure as in the synthesis of 10. Yield: 219 mg (90%) (found: C, 65.43; H, 7.59; N, 1.43. C53H73NOTh requires C, 65.48; H, 7.57; N, 1.44%). M.p.: 136–138 °C. 1H NMR (C6D6): δ 7.46 (m, 4H, phenyl), 7.36 (t, J = 7.6 Hz, 2H, phenyl), 7.12 (t, J = 7.8 Hz, 2H, phenyl), 7.05 (t, J = 7.2 Hz, 1H, pyridyl), 6.92 (t, J = 6.6 Hz, 2H, phenyl), 6.82 (s, 1H, C
CH), 6.70 (d, J = 3.1 Hz, 2H, ring CH), 6.46 (d, J = 3.1 Hz, 2H, ring CH), 6.38 (t, J = 7.0 Hz, 1H, pyridyl), 6.29 (br s, 1H, pyridyl), 6.15 (m, 1H, pyridyl), 1.55 (s, 18H, C(CH3)3), 1.49 (s, 18H, C(CH3)3), 1.21 (s, 18H, C(CH3)3) ppm. 13C{1H} NMR (C6D6): δ 215.7 (ThCPh), 202.2 (ThCN), 152.6 (aryl C), 143.7 (aryl C), 141.2 (aryl C), 137.6 (aryl C), 136.9 (aryl C), 134.4 (aryl C), 129.8 (aryl C), 128.4 (aryl C), 128.3 (aryl C), 128.1 (aryl C), 127.9 (aryl C), 127.2 (aryl C), 127.1 (ring C), 126.5 (ring C), 122.9 (ring C), 122.4 (ring C), 117.8 (ring C), 112.9 (CCHPh), 35.2 (C(CH3)3), 34.9 (C(CH3)3), 34.6 (C(CH3)3), 34.5 (C(CH3)3), 34.4 (C(CH3)3), 33.1 (C(CH3)3) ppm. IR (KBr, cm−1): ν 2957 (s), 1590 (s), 1481 (s), 1450 (s), 1387 (s), 1237 (s), 1171 (s), 1026 (s), 821 (s).
Method B. NMR scale. A C6D6 (0.3 mL) solution of pyridine N-oxide (1.9 mg; 0.02 mmol) was slowly added to a J. Young NMR tube charged with [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2; 18 mg, 0.02 mmol) and C6D6 (0.2 mL). The color of the solution immediately changed from pale yellow to colorless, and the NMR resonances of 12 were observed by 1H NMR spectroscopy (100% conversion in 10 min).
CHPh][O–C(CH2)NMe2] (13).
Method A. This compound was prepared as colorless crystals from the reaction of [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2; 220 mg, 0.25 mmol) and CH3CONMe2 (22 mg, 0.25 mmol) in toluene (15 mL) at room temperature and recrystallization from a benzene solution by a similar procedure as in the synthesis of 10. Yield: 202 mg (84%) (found: C, 64.75; H, 8.02; N, 1.42. C52H77NOTh requires C, 64.77; H, 8.05; N, 1.45%). M.p.: 176–178 °C. 1H NMR (C6D6): δ 7.48 (d, J = 7.1 Hz, 2H, phenyl), 7.39 (m, 3H, phenyl and C
CHPh), 7.23 (t, J = 7.7 Hz, 2H, phenyl), 7.06 (t, J = 7.7 Hz, 2H, phenyl), 6.99 (t, J = 7.3 Hz, 1H, phenyl), 6.92 (t, J = 7.4 Hz, 1H, phenyl), 6.77 (s, 4H, ring CH), 3.59 (s, 2H, OCCH2), 2.56 (s, 6H, N(CH3)2), 1.58 (s, 18H, C(CH3)3), 1.45 (s, 18H, C(CH3)3), 1.37 (s, 18H, C(CH3)3) ppm. 13C{1H} NMR (C6D6): δ 225.3 (ThCPh), 169.9 (OCC), 149.4 (phenyl C), 145.7 (phenyl C), 145.4 (phenyl C), 144.6 (phenyl C), 137.0 (phenyl C), 130.2 (phenyl C), 129.1 (phenyl C), 128.5 (phenyl C), 128.2 (ring C), 127.8 (ring C), 126.9 (ring C), 124.9 (ring C), 117.0 (ring C), 116.8 (CCHPh), 70.0 (CCH2), 40.8 (N(CH3)2), 35.3 (C(CH3)3), 35.0 (C(CH3)3), 34.8 (C(CH3)3), 34.5 (C(CH3)3), 33.2 (C(CH3)3) ppm; one C resonance of Me3C-groups overlapped. IR (KBr, cm−1): ν 2955 (s), 1610 (s), 1485 (s), 1326 (s), 1237 (s), 1194 (s), 1098 (s), 1075 (s), 1021 (s), 987 (s), 806 (s).
Method B. NMR scale. A C6D6 (0.3 mL) solution of CH3CONMe2 (1.8 mg; 0.02 mmol) was slowly added to a J. Young NMR tube charged with [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2; 18 mg, 0.02 mmol) and C6D6 (0.2 mL). The color of the solution immediately changed from pale yellow to colorless, and the NMR resonances of 13 were observed by 1H NMR spectroscopy (100% conversion in 10 min).
X-ray crystallography
Single-crystal X-ray diffraction measurements were carried out on a Bruker SMART CCD diffractometer at 100(2) K using graphite monochromated Mo Kα radiation (λ = 0.71073 Å). An empirical absorption correction was applied using the SADABS program.10 All structures were solved by direct methods and refined by full-matrix least squares on F2 using the SHELXL program package.11 All the hydrogen atoms were geometrically fixed using the riding model. Disordered solvents in the voids of 8 and 10 were modeled or removed by using the SQUEEZE program.12 The crystal data and experimental data for 7–10, 12 and 13 are summarized in Table 1. Selected bond lengths and angles are listed in Table 2.| Compound | 7 | 8 | 9·C6H6 | 10 | 12 | 13 |
|---|---|---|---|---|---|---|
| Formula | C45H70Th | C48H74Th | C49H72Th | C53H73NTh | C53H73NOTh | C52H77NOTh |
| F w | 843.05 | 883.11 | 893.10 | 956.16 | 972.16 | 964.18 |
| Crystal system | Monoclinic | Monoclinic | Triclinic | Triclinic | Monoclinic | Triclinic |
| Space group | Pc | P21/n |
P(![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) ) |
P() |
P21/c |
P() |
| a (Å) | 12.658(7) | 10.699(3) | 10.468(3) | 11.109(2) | 15.278(3) | 10.962(2) |
| b (Å) | 10.480(6) | 26.148(6) | 11.297(3) | 21.888(5) | 12.306(3) | 11.091(2) |
| c (Å) | 18.889(8) | 17.831(4) | 18.569(5) | 21.958(5) | 25.718(5) | 21.157(3) |
| α (deg) | 90 | 90 | 80.10(1) | 97.05(1) | 90 | 77.21(1) |
| β (deg) | 127.00(3) | 98.27(5) | 82.09(1) | 90.20(1) | 90.31(1) | 84.12(1) |
| γ (deg) | 90 | 90 | 78.99(1) | 90.93(1) | 90 | 75.93(1) |
| V (Å3) | 2001.2(19) | 4936(2) | 2110.9(9) | 5298.4(19) | 4835.2(17) | 2315.1(6) |
| Z | 2 | 4 | 2 | 4 | 4 | 2 |
| D calc (g cm−3) | 1.399 | 1.188 | 1.405 | 1.199 | 1.335 | 1.383 |
| μ(Mo/Kα)calc (cm−1) | 3.754 | 3.047 | 3.563 | 2.844 | 3.119 | 3.257 |
| Size (mm) | 0.10 × 0.10 × 0.10 | 0.20 × 0.10 × 0.10 | 0.20 × 0.20 × 0.15 | 0.30 × 0.20 × 0.20 | 0.40 × 0.35 × 0.30 | 0.30 × 0.25 × 0.20 |
| F(000) | 860 | 1808 | 912 | 1952 | 1984 | 988 |
| 2θ range (deg) | 3.88 to 50.50 | 3.12 to 55.22 | 3.72 to 55.28 | 3.66 to 50.50 | 3.68 to 56.57 | 3.84 to 55.40 |
| No. of reflns, collected | 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 858 858 |
33781 |
14481 |
19085 |
34068 |
16026 |
| No. of obsd reflns | 6764 | 11412 |
9716 | 19085 |
11961 |
10665 |
| No. of variables | 434 | 462 | 468 | 992 | 523 | 516 |
| Abscorr (Tmax, Tmin) | 0.75, 0.62 | 0.75, 0.62 | 0.75, 0.61 | 0.75, 0.57 | 0.75, 0.63 | 0.75, 0.64 |
| R | 0.060 | 0.056 | 0.046 | 0.082 | 0.029 | 0.054 |
| R w | 0.129 | 0.112 | 0.094 | 0.204 | 0.065 | 0.123 |
| R all | 0.078 | 0.096 | 0.058 | 0.111 | 0.038 | 0.065 |
| G of | 1.03 | 0.98 | 1.00 | 1.03 | 1.02 | 1.02 |
| CCDC | 1058993 | 1058994 | 1058995 | 1058996 | 1058997 | 1058998 |
| Compound | C(Cp)–Thb | C(Cp)–Thc | Cp(cent)–Thb | Th–X | Cp(cent)–Th–Cp(cent) | X–Th–X/Y |
|---|---|---|---|---|---|---|
| a Cp = cyclopentadienyl ring. b Average value. c Range. d The angle of C(34)–Th(1)–C(35). e The angle of C(34)–Th(1)–C(36). f The angle of C(34)–Th(1)–C(37). g The angle of C(49)–Th(1)–N(1). h The angle of C(42)–Th(1)–N(1). i The angle of C(42)–Th(1)–C(49). j The angle of C(41)–Th(1)–C(49). k The angle of C(41)–Th(1)–O(1). l The angle of C(49)–Th(1)–O(1). | ||||||
| 7 | 2.84(3) | 2.68(1) to 3.01(3) | 2.58(3) | C(34) 2.57(2), C(41) 2.57(3) | 142.5(2) | 109.7(2) |
| 8 | 2.867(7) | 2.705(6) to 2.969(7) | 2.579(7) | C(16) 2.544(7), C(41) 2.480(6) | 142.2(2) | 112.6(2) |
| 9 | 2.852(5) | 2.693(5) to 2.987(5) | 2.583(5) | C(34) 2.545(5), C(35) 2.632(6) | 139.8(2) | 124.0(2)d, 95.9(2)e |
| C(36) 2.851(6), C(37) 2.984(6) | 73.1(2)f | |||||
| 10 | 2.904(13) | 2.832(12) to 2.964(12) | 2.640(12) | C(42) 2.555(12), C(49) 2.440(11) | 137.1(4) | 31.8(4)g, 78.0(4)h |
| N 2.422(10) | 109.6(4)i | |||||
| 12 | 2.917(3) | 2.843(3) to 3.019(3) | 2.653(3) | C(41) 2.569(3), C(49) 2.640(3) | 138.0(2) | 77.1(1)j, 130.7(1)k |
| O 2.406(2), N 2.990(2) | 53.9(1)l | |||||
| 13 | 2.870(6) | 2.834(6) to 2.917(6) | 2.603(6) | C(41) 2.537 (7), O 2.198(4) | 134.7(2) | 110.5(2) |
Computational methods
All calculations were carried out with the Gaussian 09 program (G09),13 employing the B3PW91 functional, plus a polarizable continuum model (PCM) (denoted as B3PW91-PCM), with standard 6-31G(d) basis set for C, H and N elements and Stuttgart RLC ECP from the EMSL basis set exchange (https://bse.pnl.gov/bse/portal) for Th,14 to fully optimize the structures of reactants, complexes, transition state, intermediates, and products, and also to mimic the experimental toluene-solvent conditions (dielectric constant ε = 2.379). All stationary points were subsequently characterized by vibrational analyses, from which their respective zero-point (vibrational) energy (ZPE) were extracted and used in the relative energy determinations; in addition frequency calculations were also performed to ensure that the reactant, complex, intermediate, product and transition state structures resided at minima and 1st order saddle points, respectively, on their potential energy hyper surfaces. In order to consider the dispersion effect for the reaction 2 + py, single-point B3PW91-PCM-D3 (ref. 15) calculations, based on B3PW91-PCM geometries, have been performed.Results and discussion
Reaction of 1:1 mixture of [η5-1,2,4-(Me3C)3C5H2]2ThCl2 (1) and diphenylacetylene (PhCCPh) with an excess of KC8 in toluene solution gives the metallacyclopropene, [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2) (2) (Scheme 1).5 However, under similar reaction conditions, the treatment of [η5-1,2,4-(Me3C)3C5H2]2ThCl2 (1) and phenyl(alkyl)acetylenes PhCCR (R = Me, iPr, C6H11) with KC8 does not yield the expected metallacyclopropenes [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph(R)) (R = Me (3), iPr (4), C6H11 (5)), instead, the cyclometalated alkenyl complexes [η5-1,2,4-(Me3C)3C5H2][η5,σ-1,2-(Me3C)2-4-(CH2CMe2)C5H2]Th[C(Ph)CHR] (R = iPr (7), C6H11 (8)) and [η5-1,2,4-(Me3C)3C5H2][η5,σ-1,2-(Me3C)2-4-(CH2CMe2)C5H2]Th[η3-CH(Ph)CHCH2] (9) are isolated, respectively, in good yields (Scheme 1). Moreover, in contrast to the [(η5-C5Me5)2An] (An = Th, U) fragment,16 no thorium metallacyclopentadiene complexes were isolated for the sterically more demanding 1,2,4-(Me3C)3C5H2 ligand regardless of the amount of added internal alkynes. We propose in analogy to the PhCCPh reaction that the metallacyclopropenes 3–5 are initially formed, but they are unstable and convert by an intramolecular C–H bond activation to yield [η5-1,2,4-(Me3C)3C5H2][η5,σ-1,2-(Me3C)2-4-(CH2CMe2)C5H2]Th[C(Ph)CHR] (R = Me (6), iPr (7), C6H11 (8)). However, it is noteworthy that the C–H bond activation occurs selectively at the alkyl-end of the disubstituted acetylene. Moreover, in contrast to complexes 7 and 8, the least sterically hindered complex 6 further undergoes an [1,3]-hydrogen migration to form the cyclometallated allyl complex 9 (Scheme 1).
 | ||
| Scheme 1 Synthesis of compounds 7–9. | ||
In contrast to the metallacyclopropenes 3–5, complex 2 is stable and no ligand cyclometalation was observed, even when heated at 100 °C for one week. Nevertheless, in contrast to zirconium metallacyclopropenes,1b complex 2 is capable of activating C–H bonds of different substrates, such as those of pyridine or carbonyl derivatives containing an α-H atom upon coordination. For example, treatment of complex 2 with 1 equiv of pyridine, DMAP, pyridine N-oxide or CH3CONMe2, the pyridyl alkenyl thorium complexes [η5-1,2,4-(Me3C)3C5H2]2Th[C(Ph)CHPh](η2-C,N–C5H4N) (10), [η5-1,2,4-(Me3C)3C5H2]2Th[C(Ph)CHPh](η2-C,N-4-Me2NC5H3N) (11) and [η5-1,2,4-(Me3C)3C5H2]2Th[C(Ph)CHPh](κ2-C,O–C5H4NO) (12), and enolyl alkenyl thorium complex [η5-1,2,4-(Me3C)3C5H2]2Th[C(Ph)CHPh][O–C(CH2)NMe2] (13) are formed, respectively, in quantitative conversions (Scheme 2), in which an α-H of the pyridine, DMAP, pyridine N-oxide or CH3CONMe2 is transferred to the metallacyclopropene Th(η2-C2Ph2) moiety.
 | ||
| Scheme 2 Synthesis of complexes 10–13. | ||
Complexes 7–13 are stable in dry nitrogen atmosphere, but they are moisture sensitive. They were characterized by various spectroscopic techniques and elemental analyses. In addition, the solid-state structures of complexes 7–10, 12 and 13 were determined by single crystal X-ray diffraction analyses (Table 1). Selected bond distances and angles for these compounds are listed in Table 2. The molecular structures of 7 and 8 are shown in Fig. 1 and 2. The Th–C(CH2CMe2Cp) distance of 2.57(2) Å in 7 is comparable to that (2.544(7) Å) found in 8, but significantly longer than that in [η5-1,2,4-(Me3C)3C5H2]2ThMe2 (2.480(3) Å).9 Furthermore, the Th–C(alkenyl) distances (2.57(3) Å for 7 and 2.480(6) Å for 8) are in the range of previously reported Th–C(sp2) σ-bonds (2.420(3)–2.654(14) Å),17 but are slightly longer than that (2.395(2) Å) found in the metallacyclopropene [η5-1,2,4-(Me3C)3C5H2]2Th(η2-C2Ph2).5
 | ||
| Fig. 1 Molecular structure of 7 (thermal ellipsoids drawn at the 35% probability level). | ||
 | ||
| Fig. 2 Molecular structure of 8 (thermal ellipsoids drawn at the 35% probability level). | ||
Fig. 3 depicts the molecular structure of 9. The C–C distances of the allyl fragment are of 1.385(8) Å for C(35)–C(36) and 1.372(8) Å for C(36)–C(37). Nevertheless, the Th–C(35), Th–C(36) and Th–C(37) distances of 2.632(6) Å, 2.851(6) Å and 2.984(6) Å, respectively, become progressively longer, suggesting that the η3-coordination allyl moiety observed in the solid state is weak and that hapticity switch (η3 → η1) is likely to occur in solution. Indeed, in the 13C{1H} NMR spectrum the corresponding allyl resonances are found at δ = 66.1, 100.8 and 111.8 ppm, respectively, which would be more consistent with a η1-coordination mode in solution.18 Furthermore, while the Th–C(35) distance of 2.632(6) Å is longer than those found in [η5-1,2,4-(Me3C)3C5H2]2ThMe2 (2.480(3) Å)9 and [η5-1,2,4-(Me3C)3C5H2]2Th(CH2Ph)2 (2.521(3) and 2.527(3) Å),19 it is consistent with the value of ca. 2.73 Å found in [η3-1,3-(Me3Si)2C3H3]4Th.20 In contrast, the Th–C(34) distance of 2.545(5) Å is comparable to those found in 7 (2.57(2) Å) and 8 (2.544(7) Å).
 | ||
| Fig. 3 Molecular structure of 9 (thermal ellipsoids drawn at the 35% probability level). | ||
The solid state molecular structures of 10 and 12 are shown in Fig. 4 and 5 and for selected bond distances and angles see Table 2. The Th–C(alkenyl) distances (2.555(12) Å for 10 and 2.569(3) Å for 12) are in the same range as those found in 7 (2.57(3) Å), 8 (2.480(6) Å), and 9 (2.632(6) Å). In 10, the Th–C(pyridyl) distance is 2.440(11) Å, and the Th–N distance is 2.422(10) Å. Nevertheless, the Th–C(pyridyl) distance of 2.640(3) Å in 12 is close to that found (η5-C5Me5)2Th(CH2Ph)(κ2-C,O-ONC5H4) (2.621(3) Å).21 Furthermore, the Th–O distance (2.406(2) Å) in 12 is shorter than that expected for a dative interaction,22 but is comparable to that found in (η5-C5Me5)2Th(CH2Ph)(κ2-C,O-ONC5H4) (2.416(2) Å).21 The N–O distance (1.369(3) Å) is slightly longer than that in the free pyridine N-oxide (1.330(9) Å),23 but virtually identical to that found in (η5-C5Me5)2Th(CH2Ph)(κ2-C,O-ONC5H4) (1.360(3) Å).21
 | ||
| Fig. 4 Molecular structure of 10 (thermal ellipsoids drawn at the 35% probability level). | ||
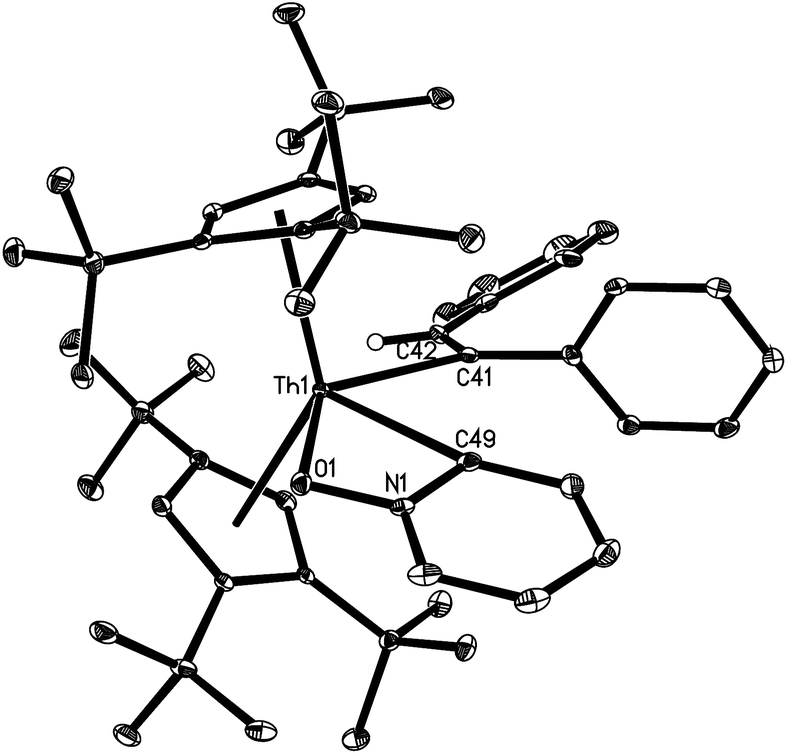 | ||
| Fig. 5 Molecular structure of 12 (thermal ellipsoids drawn at the 35% probability level). | ||
The solid state molecular structure of 13 is depicted in Fig. 6. The Th4+ ion is η5-bound to two Cp-rings and one σ-coordinate carbon atom and one oxygen atom with the average Th–C(Cp) distance of 2.870(6) Å and the angle Cp(cent)–Th–Cp(cent) of 134.7(2)°. The Th–C(41) distance (2.537(7) Å) is comparable to those found in 7 (2.57(3) Å), 8 (2.480(6) Å), 9 (2.632(6) Å), 10 (2.555(12) Å), and 12 (2.569(3) Å), and the Th–O distance (2.198(4) Å) is comparable to those found in [η5-1,2,4-(Me3C)3C5H2]2Th[O2CPh2] (2.202(3) Å),24 and [η5-1,2,4-(Me3C)3C5H2]2Th[(OCPh2)2] (2.182(2) Å).5
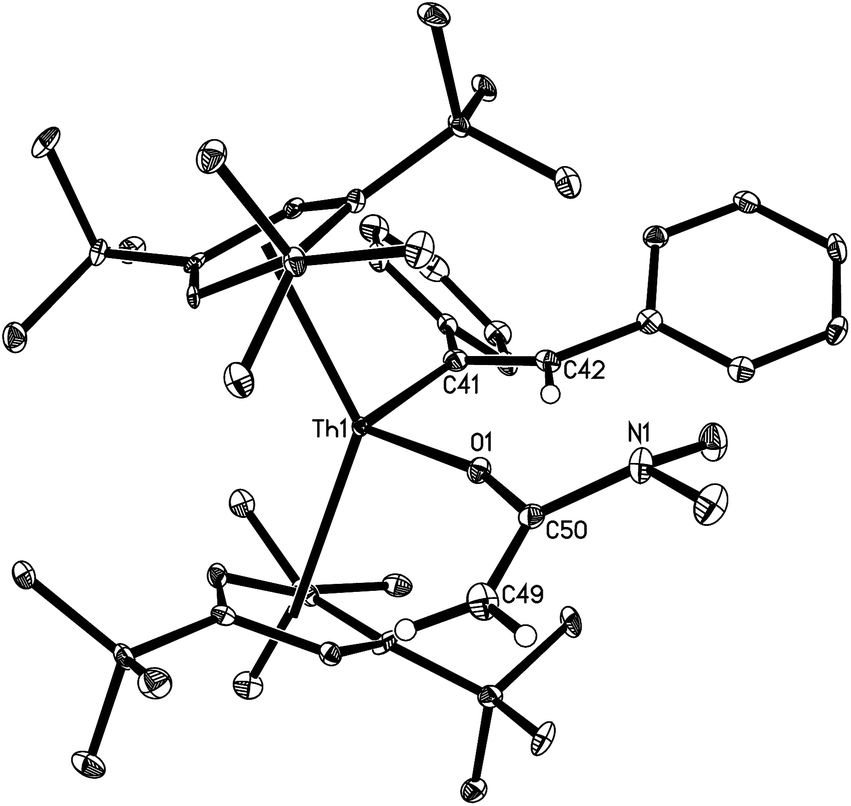 | ||
| Fig. 6 Molecular structure of 13 (thermal ellipsoids drawn at the 35% probability level). | ||
Thorium metallacyclopropenes derived from phenyl(alkyl)acetylenes are very reactive species that are capable to undergo a selective intramolecular C–H bond activation of the cyclopentadienyl ligand 1,2,4-(Me3C)3C5H2. However, while complex 2 derived from diphenylacetylene cannot promote intramolecular C–H bond activations, it activates intermolecularly C–H bonds upon coordination, such as those of pyridine or carbonyl derivatives containing an α-H atom. To further understand these observations, DFT calculations were performed at the B3PW91 level of theory. As a representative example of the phenyl(alkyl)acetylene derivatives complex 5 was chosen. We first compared the energetics of the intramolecular C–H bond activation and its selectivity for complexes 2 and 5 (Fig. 7). These computations revealed several interesting features: (1) The intramolecular C–H bond activation of a methyl group of the 1,2,4-(Me3C)3C5H2 ligand in 2 is energetically unfavorable (ΔG(298 K) = 3.9 kcal mol−1), while that promoted by complex 5 is exergonic (Fig. 7), presumably because of electronic effects. In a simple physical organic picture, an alkyl-group introduces a stronger + I-effect than a phenyl group, which should therefore more strongly destabilize the negative charge on a dianionic [η2-alkenediyl]2− ligand and protonation should occur preferentially at the more basic, alkyl-substituted end. Therefore the thermal stability of the diphenylacetylene derived thorium metallacycloproprene 2 may also reflect the reduced basicity of the diphenyl-substituted [η2-alkenediyl]2− ligand, so that only those metallacyclopropene complexes derived from phenyl(alkyl)acetylenes are thermally converted to the cyclometalated complexes via an intramolecular C–H bond activation of the 1,2,4-(Me3C)3C5H2 ligand. (2) Furthermore, the DFT computations also explain the selectivity of the C–H bond activation: only the RC (R = cyclohexyl) end of phenyl(cyclohexyl)-substituted metallacyclopropene in 5 is capable to undergo σ-bond metathesis (ΔG(298 K) = −4.6 kcal mol−1), while the reaction at the PhC-position is energetically unfavorable (ΔG(298 K) = 4.7 kcal mol−1) (Fig. 7). Again, this difference in reactivity might be ascribed to the electronic effect as just mentioned above. (3) Moreover, the barrier for the conversion of 5 to 8 is only ΔG‡(298 K) = 17.5 kcal mol−1 and can be overcome under the reaction conditions. The computational results are also consistent with the experimentally observed stability of complex 2 upon heating. The energetic profile for the intermolecular reaction of 2 with pyridine is shown in Fig. 8 and it involves the adduct COM10 and the transition state TS10. In the σ-bond metathesis transition state TS10 the two forming bond distances of Th–C and C–H are 2.687 and 1.513 Å, respectively, ca. 0.22 and 0.42 Å longer than those in product 10. The conversion of COM10 to the product 10 is energetically favorable by ΔG(298 K) = −13.3 kcal mol−1, and proceeds via transition state TS10 with an activation barrier (ΔG‡(298 K)) of 19.2 kcal mol−1, which can be overcome at ambient temperature and therefore is consistent with the experimental observations.
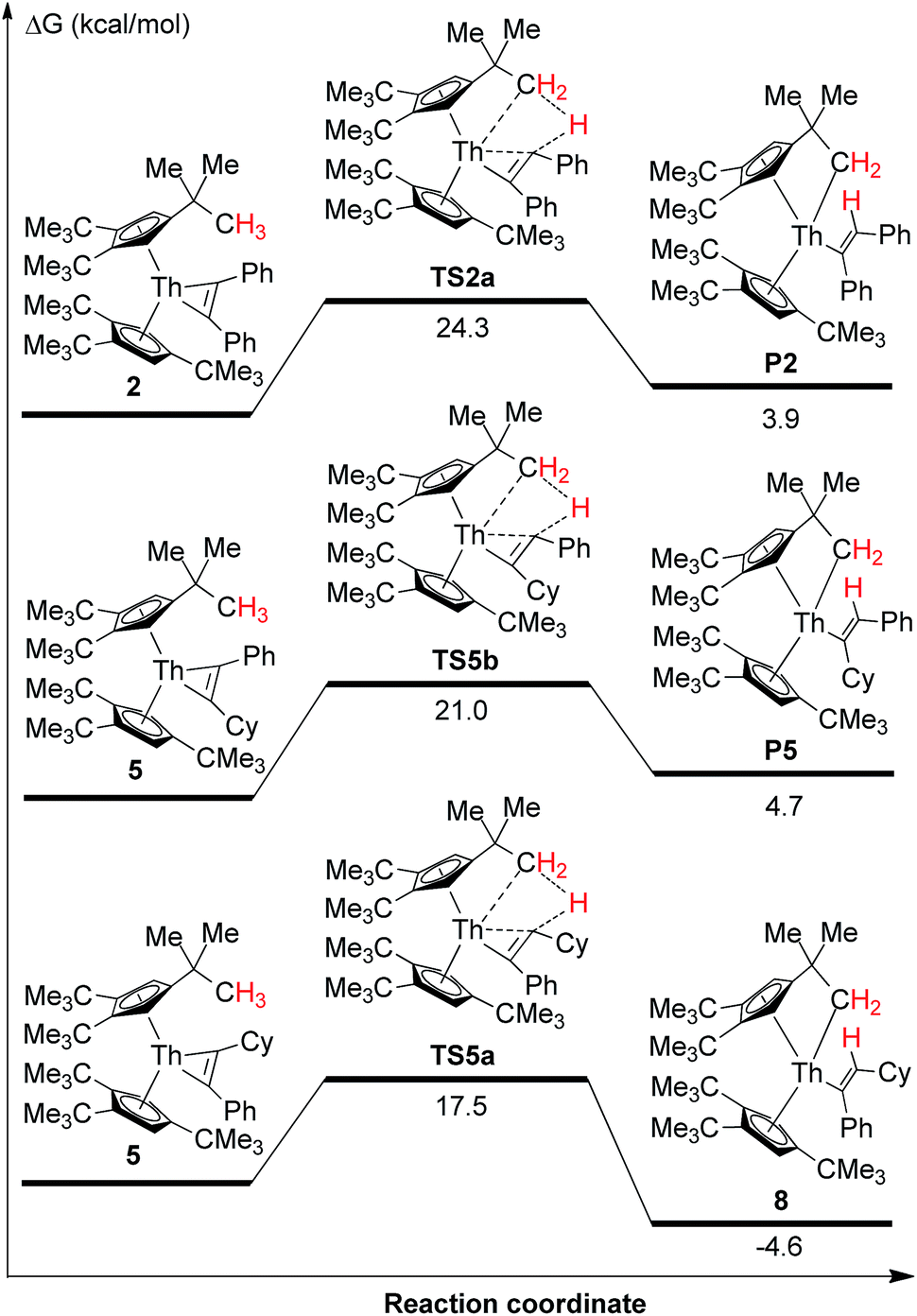 | ||
| Fig. 7 Free energy profile (kcal mol−1) for the conversions of 2 and 5. Cy = cyclohexyl. | ||
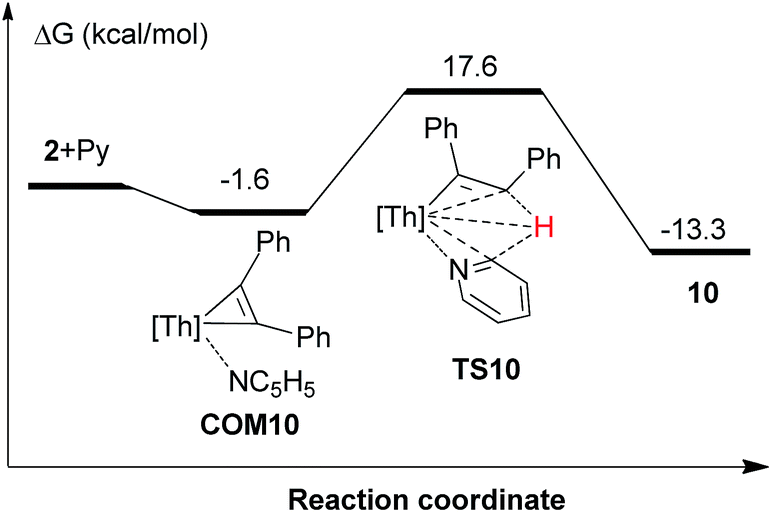 | ||
| Fig. 8 Free energy profile (kcal mol−1) for the reaction of 2 + Py. [Th] = [η5-1,2,4-(Me3C)3C5H2]2Th. | ||
Conclusions
In conclusion, the first examples of inter- and intramolecular C–H bond activations mediated by thorium metallacyclopropenes were comprehensively investigated. When the substitutents on the thorium metallacyclopropene are changed from phenyl to alkyl, a distinctive change in reactivity is observed, which is also illustrated by their relative stabilities. The thorium metallacyclopropenes derived from phenyl(alkyl)acetylenes are very reactive and cannot be isolated, instead, they thermally convert to cyclometalated complexes via an intramolecular C–H bond activation of the 1,2,4-(Me3C)3C5H2 ligand. In contrast, the thorium metallacyclopropene 2 derived from diphenylacetylene is thermally stable. The change in relative stability is also reflected in DFT computations, which showed that the intramolecular C–H bond activation of the ligand 1,2,4-(Me3C)3C5H2 induced by 5 is energetically favourable, while that promoted by 2 is not. Nevertheless, in contrast to zirconium metallacyclopropenes,1b complex 2 is capable of promoting the intermolecular C–H bond activations of substrates, such as pyridine or carbonyl derivatives containing α-H atoms upon coordination. This leads to the formation of the corresponding pyridyl alkenyl or enolyl alkenyl complexes. The further development of new actinide metallacyclopropene complexes and the exploration of the thorium cyclometalated complexes and pyridyl alkenyl complexes in organic syntheses are ongoing projects in these laboratories.Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant no. 21472013, 21172022, 21272026, 21373030), the Program for Changjiang Scholars and Innovative Research Team in University, and the Deutsche Forschungsgemeinschaft (DFG) through the Emmy-Noether and Heisenberg program (WA 2513/2 and WA 2513/6, respectively). We thank Dr Xuebin Deng for his help with the crystallography, and Prof. Richard A. Andersen for helpful discussions.Notes and references
- For selected reviews, see: (a) S. L. Buchwald and R. B. Nielsen, Chem. Rev., 1988, 88, 1047–1058 CrossRef CAS; (b) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann and A. Spannenberg, Organometallics, 2003, 22, 884–900 CrossRef CAS; (c) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg and V. B. Shur, Eur. J. Inorg. Chem., 2004, 4739–4749 CrossRef CAS PubMed; (d) U. Rosenthal, Angew. Chem., Int. Ed., 2004, 43, 3882–3887 CrossRef CAS PubMed; (e) U. Rosenthal, V. V. Burlakov, P. Arndt, W. Baumann and A. Spannenberg, Organometallics, 2005, 24, 456–471 CrossRef CAS; (f) U. Rosenthal, V. V. Burlakov, M. A. Bach and T. Beweries, Chem. Soc. Rev., 2007, 36, 719–728 RSC; (g) H. S. La Pierre and K. Meyer, Prog. Inorg. Chem., 2014, 58, 303–415 CAS; (h) K. D. J. Parker and M. D. Fryzuk, Organometallics, 2015, 34, 2037–2047 CrossRef CAS.
- Selected papers on group 4 metallacyclopropenes, see: (a) M. D. Walter, C. D. Sofield and R. A. Andersen, Organometallics, 2008, 27, 2959–2970 CrossRef CAS; (b) T. Beweries, C. Fischer, S. Peitz, V. V. Burlakov, P. Arndt, W. Baumann, A. Spannenberg, D. Heller and U. Rosenthal, J. Am. Chem. Soc., 2009, 131, 4463–4469 CrossRef CAS PubMed; (c) K. Kaleta, M. Ruhmann, O. Theilmann, T. Beweries, S. Roy, P. Arndt, A. Villinger, E. D. Jemmis, A. Schulz and U. Rosenthal, J. Am. Chem. Soc., 2011, 133, 5463–5473 CrossRef CAS PubMed; (d) M. Haehnel, M. Ruhmann, O. Theilmann, S. Roy, T. Beweries, P. Arndt, A. Spannenberg, A. Villinger, E. D. Jemmis, A. Schulz and U. Rosenthal, J. Am. Chem. Soc., 2012, 134, 15979–15991 CrossRef CAS PubMed; (e) M. Haehnel, S. Hansen, K. Schubert, P. Arndt, A. Spannenberg, H. Jiao and U. Rosenthal, J. Am. Chem. Soc., 2013, 135, 17556–17565 CrossRef CAS PubMed; (f) S. K. Podiyanachari, G. Kehr, C. Mück-Lichtenfeld, C. Daniliuc and G. Erker, J. Am. Chem. Soc., 2013, 135, 17444–17456 CrossRef PubMed; (g) L. Becker, P. Arndt, H. Jiao, A. Spannenberg and U. Rosenthal, Angew. Chem., Int. Ed., 2013, 52, 11396–11400 CrossRef CAS PubMed; (h) Ò. Àrias, A. R. Petrov, T. Bannenberg, K. Altenburger, P. Arndt, P. G. Jones, U. Rosenthal and M. Tamm, Organometallics, 2014, 33, 1774–1786 CrossRef; (i) D. J. Mindiola, L. A. Watson, K. Meyer and G. L. Hillhouse, Organometallics, 2014, 33, 2760–2769 CrossRef CAS PubMed; (j) N. Peulecke, A. Ohff, P. Kosse, A. Tillack, A. Spannenberg, R. Kempe, W. Baumann, V. V. Burlakov and U. Rosenthal, Chem.–Eur. J., 1998, 4, 1852–1861 CrossRef CAS; (k) M. Zirngast, C. Marschner and J. Baumgartner, Organometallics, 2008, 27, 2570–2583 CrossRef CAS; (l) U. Rosenthal, A. Ohff, M. Michalik, H. Görls, V. V. Burlakov and V. B. Shur, Angew. Chem., Int. Ed., 1993, 32, 1193–1195 CrossRef PubMed; (m) V. V. Burlakov, A. V. Polyakov, A. I. Yanovsky, Y. T. Struchkov, V. B. Shur, M. E. Vol'pin, U. Rosenthal and H. Görls, J. Organomet. Chem., 1994, 476, 197–206 CrossRef CAS; (n) J. Hiller, U. Thewalt, M. Polášek, L. Petrusová, V. Varga, P. Sedmera and K. Mach, Organometallics, 1996, 15, 3752–3759 CrossRef CAS; (o) D. Thomas, N. Peulecke, V. V. Burlakov, B. Heller, W. Baumann, A. Spanneberg, R. Kempe, U. Rosenthal and R. Beckhaus, Z. Anorg. Allg. Chem., 1998, 624, 919–924 CrossRef CAS.
- For selected reviews, see: (a) E.-I. Negishi and T. Takahashi, Acc. Chem. Res., 1994, 27, 124–130 CrossRef CAS; (b) T. Takahashi and Y. Li, Zirconacyclopentadienes in Organic Synthesis, in Titanium and Zirconium in Organic Synthesis, Wiley-VCH, Weinheim, Germany, 2002, pp. 50–85 Search PubMed; (c) J. A. Varela and C. Saá, Chem. Rev., 2003, 103, 3787–3802 CrossRef CAS PubMed; (d) H. Braunschweig and T. Kupfer, Chem. Commun., 2011, 47, 10903–10914 RSC; (e) S. Roy, U. Rosenthal and E. D. Jemmis, Acc. Chem. Res., 2014, 47, 2917–2930 CrossRef CAS PubMed.
- (a) L. Andrews, G. P. Kushto and C. J. Marsden, Chem.–Eur. J., 2006, 12, 8324–8335 CrossRef CAS PubMed; (b) M. Foyentin, G. Folcher and M. Ephritikhine, J. Chem. Soc., Chem. Commun., 1987, 494–495 RSC.
- B. Fang, W. Ren, G. Hou, G. Zi, D.-C. Fang, L. Maron and M. D. Walter, J. Am. Chem. Soc., 2014, 136, 17249–17261 CrossRef CAS PubMed.
- Selected papers about the bonding of organoactinide complexes, see: (a) T. Cantat, C. R. Graves, K. C. Jantunen, C. J. Burns, B. L. Scott, E. J. Schelter, D. E. Morris, P. J. Hay and J. L. Kiplinger, J. Am. Chem. Soc., 2008, 130, 17537–17551 CrossRef CAS PubMed; (b) N. Barros, D. Maynau, L. Maron, O. Eisenstein, G. Zi and R. A. Andersen, Organometallics, 2007, 26, 5059–5065 CrossRef CAS; (c) A. Yahia and L. Maron, Organometallics, 2009, 28, 672–679 CrossRef CAS; (d) W. Ren, X. Deng, G. Zi and D.-C. Fang, Dalton Trans., 2011, 9662–9664 RSC; (e) E. Ciliberto, S. D. Bella, A. Gulino, I. Fragala, J. L. Petersen and T. J. Marks, Organometallics, 1992, 11, 1727–1737 CrossRef CAS; (f) K. Tatsumi and A. Nakamura, J. Am. Chem. Soc., 1987, 109, 3195–3206 CrossRef CAS; (g) J. R. Walensky, R. L. Martin, J. W. Ziller and W. J. Evans, Inorg. Chem., 2010, 49, 10007–10012 CrossRef CAS PubMed; (h) B. M. Gardner, P. A. Cleaves, C. E. Kefalidis, J. Fang, L. Maron, W. Lewis, A. J. Blake and S. T. Liddle, Chem. Sci., 2014, 5, 2489–2497 RSC.
- Selected reviews, see: (a) K. R. D. Johnson and P. G. Hayes, Chem. Soc. Rev., 2013, 42, 1947–1960 RSC; (b) P. L. Arnold, M. W. McMullon, J. Rieb and F. E. Kühn, Angew. Chem., Int. Ed., 2015, 54, 82–100 CrossRef CAS PubMed.
- M. A. Schwindt, T. Lejon and L. S. Hegedus, Organometallics, 1990, 9, 2814–2819 CrossRef CAS.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, Chem.–Eur. J., 2011, 17, 12669–12682 CrossRef CAS PubMed.
- G. M. Sheldrick, SADABS, Program for Empirical Absorption Correction of Area Detector Data, University of Göttingen, Göttingen, Germany, 1996 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- SQUEEZE: P. V. D. Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.02, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- W. Küchle, M. Dolg, H. Stoll and H. Preuss, Mol. Phys., 1991, 74, 1245–1263 CrossRef PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Selected actinide metallacyclopentadienes, see: (a) P. J. Fagan, J. M. Manriquez, T. J. Marks, C. S. Day, S. H. Vollmer and V. W. Day, Organometallics, 1982, 1, 170–180 CrossRef CAS; (b) P. J. Fagan, J. M. Manriquez, E. A. Maatta, A. M. Seyam and T. J. Marks, J. Am. Chem. Soc., 1981, 103, 6650–6667 CrossRef CAS; (c) J. M. Manriquez, P. J. Fagan, T. J. Marks, S. H. Vollmer, C. S. Day and V. W. Day, J. Am. Chem. Soc., 1979, 101, 5075–5078 CrossRef CAS; (d) W. J. Evans, S. A. Kozimor and J. W. Ziller, Chem. Commun., 2005, 4681–4683 RSC; (e) B. Fang, G. Hou, G. Zi, D.-C. Fang and M. D. Walter, Dalton Trans., 2015, 44, 7927–7934 RSC.
- (a) L. A. Seaman, E. A. Pedrick, T. Tsuchiya, G. Wu, E. Jakubikova and T. W. Hayton, Angew. Chem., Int. Ed., 2013, 52, 10589–10592 CrossRef CAS PubMed; (b) I. Korobkov, B. Vidjayacoumar, S. I. Gorelsky, P. Billone and S. Gambarotta, Organometallics, 2010, 29, 692–702 CrossRef CAS; (c) I. Korobkov, A. Arunachalampillai and S. Gambarotta, Organometallics, 2004, 23, 6248–6252 CrossRef CAS; (d) W. Ren, E. Zhou, B. Fang, G. Zi, D.-C. Fang and M. D. Walter, Chem. Sci., 2014, 5, 3165–3172 RSC.
- T. J. Marks and W. A. Wachter, J. Am. Chem. Soc., 1976, 98, 703–710 CrossRef CAS.
- W. Ren, N. Zhao, L. Chen and G. Zi, Inorg. Chem. Commun., 2013, 30, 26–28 CrossRef CAS PubMed.
- C. N. Carlson, T. P. Hanusa and W. W. Brennessell, J. Am. Chem. Soc., 2004, 126, 10550–10551 CrossRef CAS PubMed.
- J. A. Pool, B. L. Scott and J. L. Kiplinger, J. Am. Chem. Soc., 2005, 127, 1338–1339 CrossRef CAS PubMed.
- (a) A. Haskel, T. Straub and M. S. Eisen, Organometallics, 1996, 15, 3773–3775 CrossRef CAS; (b) M. A. Edelman, P. B. Hitchcock, J. Hu and M. F. Lappert, New J. Chem., 1995, 19, 481–489 CAS.
- R. E. Marsh, M. Kapon, S. Hu and F. H. Herbstein, Acta Crystallogr., Sect. B: Struct. Sci., 2002, 58, 62–77 Search PubMed.
- W. Ren, G. Zi, D.-C. Fang and M. D. Walter, J. Am. Chem. Soc., 2011, 133, 13183–13196 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Cartesian coordinates of all stationary points optimized at B3PW91-PCM level. CCDC 1058993–1058998. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01684c |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |