
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed direct β-arylation of ketones with diaryliodonium salts: a stoichiometric heavy metal-free and user-friendly approach†
Zhongxing
Huang
,
Quynh P.
Sam
and
Guangbin
Dong
*
The University of Texas at Austin, Department of Chemistry, Austin, TX 78712, USA
First published on 17th June 2015
Abstract
We herein report a new protocol for the Pd-catalyzed β-arylation of ketones without stoichiometric heavy metals. Widely accessible diaryliodonium salts are used as both the oxidant and aryl source. This tandem redox catalysis merges ketone dehydrogenation and conjugate addition without an additional oxidant or reductant. This transformation features the use of a unique bis-N-tosylsulfilimine ligand and the combination of potassium trifluoroacetate/trifluoroacetic acid to maintain an appropriate acidity of the reaction medium. The reaction tolerates both air and moisture, and shows a broad substrate scope. Kinetics studies, along with filtration and poisoning tests, support the involvement of palladium nanoparticles in the catalysis.
Introduction
The transformation and functionalization of carbonyl compounds are of fundamental importance in organic synthesis. While conventional approaches mainly involve the electrophilic ipso carbon and acidic α-C–H bond of carbonyl compounds, recent advances have moved beyond the intrinsic reactivity allowing functionalization of the unactivated β-C–H bonds.1 Among all of these β-functionalization methods, arylation reactions have received particular attention due to the prevalence of β-aryl carbonyl moieties in a large collection of bioactive compounds, including drug candidates, anti-oxidants and pesticides (Fig. 1).2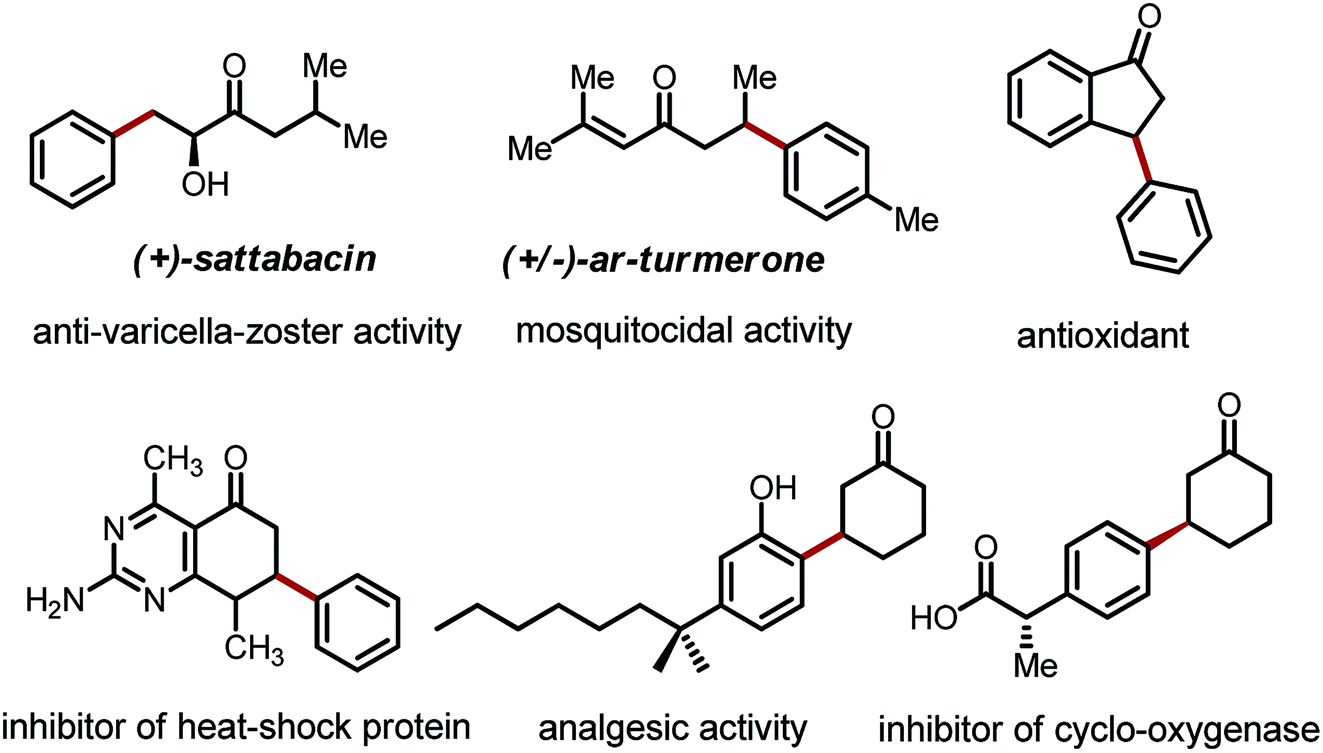 | ||
| Fig. 1 Selected bioactive compounds with the β-aryl moiety. | ||
Traditionally, β-aryl carbonyl compounds are prepared via conjugate addition of aryl nucleophiles to α,β-unsaturated carbonyl compounds.3 During the past decade, several β-arylation strategies that can directly employ saturated carbonyl compounds have emerged. The palladium-catalyzed direct β-arylation of amides was first achieved with a bidentate directing auxiliary.4 This strategy was pioneered by Daugulis,4a Corey4b and Chen,4d and further extended with the use of iron and nickel catalysts.5 Yu and coworkers also reported successful β-arylation reactions of weakly coordinating carboxylic acids and N-aryl amides.6 The β-arylation of esters via a migratory coupling pathway was first discovered by Hartwig,7a and later systematically developed by Baudoin and coworkers.7b,c The β′-arylation of 1,3-dicarbonyl compounds has also been reported using palladium catalysis, albeit with limited substrate scope.8
While carboxylic acid derivatives have been extensively studied as substrates, the direct β-arylation of normal ketones, in contrast, remained an unknown transformation until 2013. Combining photo-redox and enamine catalysis, MacMillan and coworkers disclosed a novel β-arylation of cyclic ketones with electron-deficient arylnitriles (ArCN) as the aryl source (Scheme 1, eqn (1)).9 In the same year, our group devised a palladium tandem catalysis protocol to achieve β-arylation of ketones with aryl iodides (eqn (2)).10 This approach merged palladium-catalyzed ketone dehydrogenation,11 aryl–halogen bond activation, and conjugate addition3a (Scheme 2, catalytic cycle AB′C′D).
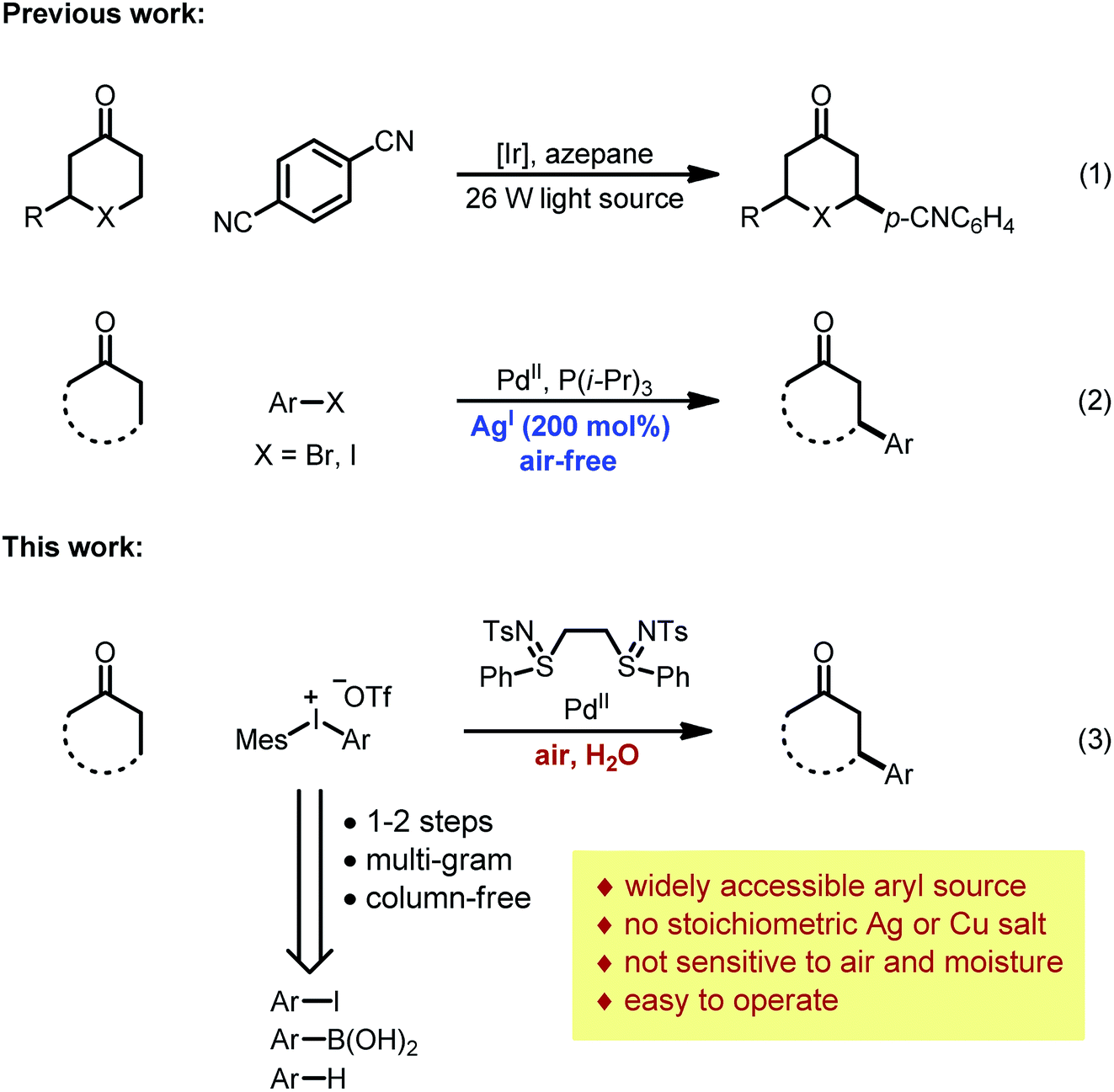 | ||
| Scheme 1 Direct β-arylation of ketones. | ||
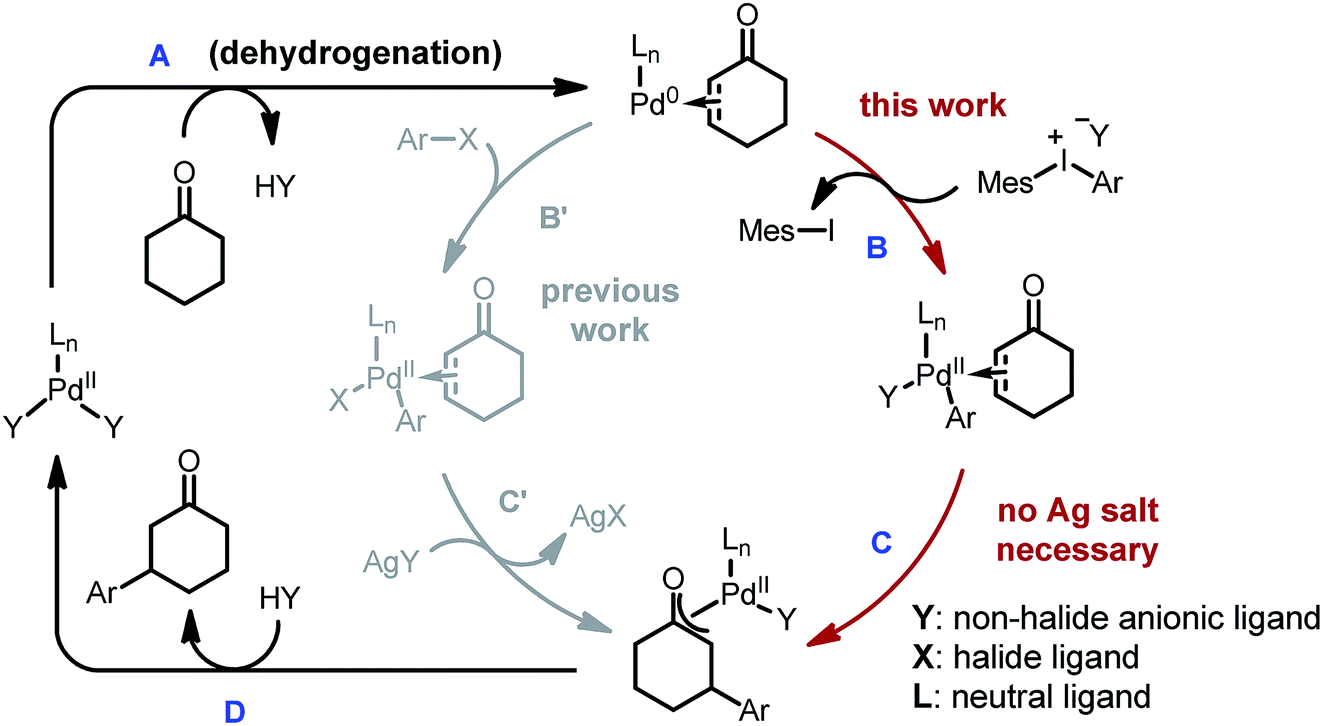 | ||
| Scheme 2 Proposed strategies for the β-arylation of ketones. | ||
However, three major limitations exist with our previous method. Firstly, to extract the halide ligand from the palladium intermediate and restore the active dicarboxylate catalyst (PdLnY2, Scheme 2), a stoichiometric amount of silver salt was required (step C′). Besides the cost and environmental issues, the addition of silver salts also complicates the reaction mechanism due to the possibility of serving as a potential oxidant for the Pd(0) species.12Secondly, the practicality of the reaction conditions is hampered by the use of the air-sensitive P(i-Pr)3 ligand, which necessitates air-free operations and highly purified reagents. Thirdly, α- or β-substituted cyclic substrates were found to be unreactive under the previously reported conditions. To address the aforementioned challenges, we herein describe the detailed development and mechanistic exploration of an alternative approach for β-arylation of simple ketones, in which a Pd(OAc)2/bis-N-tosylsulfilimine combination is used as the precatalyst and diaryliodonium salts are employed as the arylation reagent (Scheme 1, eqn (3)). This new method is free of stoichiometric heavy metals; moreover, it tolerates both air and water, and shows a broader substrate scope.
Results and discussion
Research hypothesis
Due to their good benchtop stability, low toxicity and easy accessibility from various aromatic compounds,13 diaryliodonium salts have been widely applied in cross-coupling and C–H arylation reactions as an important aryl source.13,14 We envisioned that replacing aryl iodides with diaryliodonium salts would hold a key advantage for the β-arylation reaction, because during the oxidation step the diaryliodonium salts would transfer an aryl and non-halide anionic ligand to the palladium center (Scheme 2, step B). Thus, stoichiometric halide scavengers (e.g. silver salts) would not be necessary to regenerate the active palladium catalyst (catalytic cycle ABCD). Also, due to the enhanced reactivity of diaryliodonium salts, the use of trialkylphosphines could be avoided, and inert substrates might participate in the reaction.Optimization studies
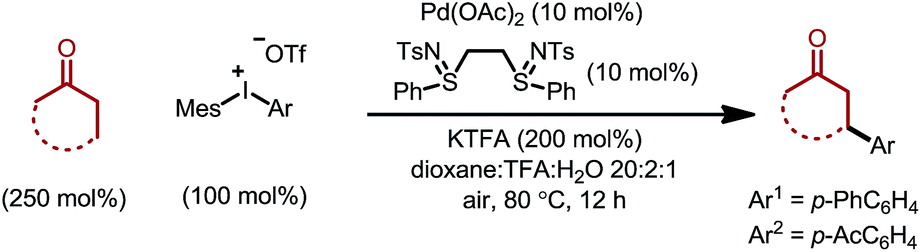
We initiated our study by using cyclohexanone 1a and mesitylphenyliodonium triflate 2a as the standard substrates. The mesitylaryliodonium salt was chosen for two reasons. Firstly, the use of bulky mesityl as one of the aryl groups is known to alleviate chemoselectivity issues, since the less sterically hindered aryl group would be transferred preferably.13f Secondly, iodomesitylene, a byproduct after the oxidative addition (Scheme 2, step B), would barely interfere with the reaction, e.g. competitive reaction with the Pd(0) intermediate, also due to its steric hindrance.After a thorough survey of the reaction parameters (e.g. catalyst, ligand, additive and solvent), the coupling between cyclohexanone 1a and iodonium salt 2a afforded the desired β-arylation product 3a in 70% yield (Table 1). Our optimized reaction conditions utilize Pd(OAc)2 as the catalyst and bis-N-tosylsulfilimine L1 as the ligand. A weak base, potassium trifluoroacetate (KTFA), and an acidic medium consisting of 1,4-dioxane, trifluoroacetic acid (TFA) and water were employed in the reaction. While 2.5 equiv. of ketone 1a was used to ensure fast initiation,11e most of the extra ketone remained intact during the reaction. The over-oxidation was insignificant, and only a trace amount of diarylation was observed. In addition, this new β-arylation reaction proved to be robust and user-friendly: all of the reagents can be added in one batch without glovebox or Schlenk techniques, and no inert atmosphere is necessary.
|
|
|||
|---|---|---|---|
| Entry | Variations from the ‘standard’ conditions | Yield of 3a (%) | 1a (%) |
a Standard conditions: mesitylphenyliodonium salt 2a (0.2 mmol), cyclohexanone 1a (0.5 mmol), Pd(OAc)2 (0.02 mmol), L1 (0.02 mmol, d.r. >20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 racemic/meso), KTFA (0.4 mmol), 1,4-dioxane (1 mL), TFA (100 μL), H2O (50 μL), 80 °C, 12 h.
b All yields were determined via gas chromatography using dodecane as the internal standard. :1 racemic/meso), KTFA (0.4 mmol), 1,4-dioxane (1 mL), TFA (100 μL), H2O (50 μL), 80 °C, 12 h.
b All yields were determined via gas chromatography using dodecane as the internal standard.
|
|||
| 1 | [MesIPh]BF4 instead of 2a | 50 | 158 |
| 2 | [MesIPh]TFA instead of 2a | 56 | 168 |
| 3 | [Ph2I]OTf instead of 2a | 36 | 199 |
| 4 | PhI instead 2a | 5 | 240 |
| 5 | w/o Pd(OAc)2 | 0 | 244 |
| 6 | w/o L1 | 8 | 235 |
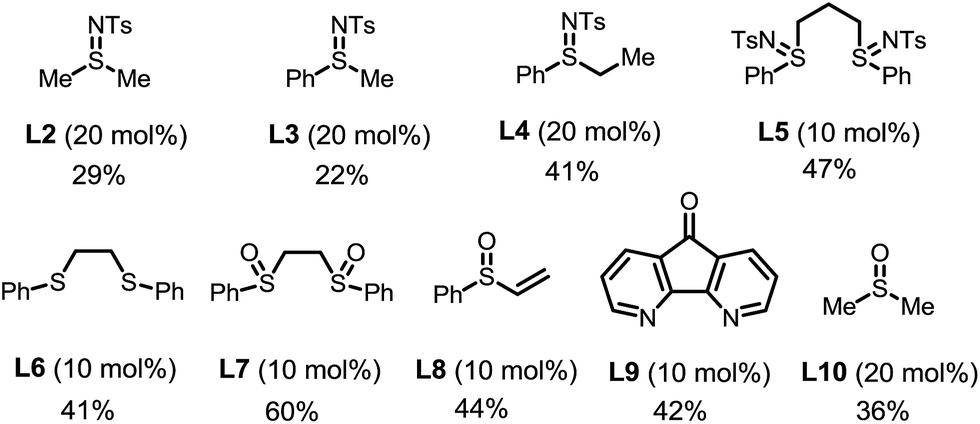
| 7 | L2–L10 instead of L1 | Listed below | — |
| 8 | w/o KTFA and TFA | 13 | 128 |
| 9 | w/o TFA | 36 | 179 |
| 10 | w/o KTFA | 34 | 179 |
| 11 | 100 mol% HOTf instead of TFA | 2 | 142 |
| 12 | KOAc instead of KTFA | 66 | 176 |
| 13 | NaOAc instead of KTFA | 59 | 167 |
| 14 | HOAc instead of TFA | 40 | 181 |
| 15 | HFIP instead of TFA | 43 | 195 |
| 16 | w/o H2O | 47 | 176 |
| 17 | N2 instead of air | 73 | 170 |
| 18 |
1a:2a = 1:1 |
48 | 47 |
|
|||
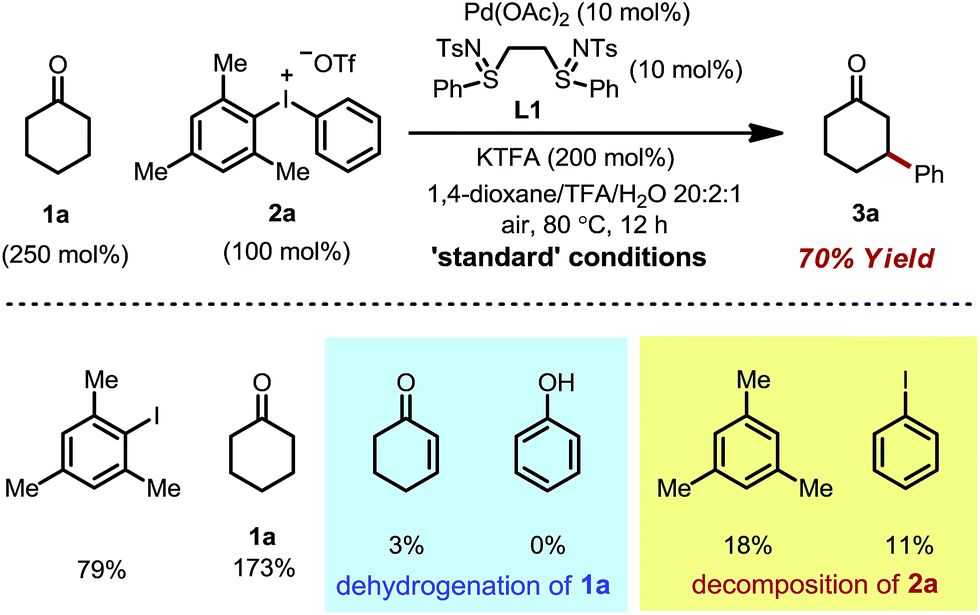
Next, a set of control experiments were performed to gain a deeper understanding of the reaction (Table 1). Common mesitylphenyliodonium salts with other counteranions are also suitable arylation reagents under the reaction conditions (entries 1 and 2). However, the diphenyliodonium salt led to a greatly decreased yield (entry 3). The poor efficiency can be attributed to the iodobenzene (PhI) byproduct released during the reaction, since oxidative addition of iodobenzene to Pd(0) is facile, and the resulting iodide ligand would poison the palladium catalyst. Such a hypothesis was also supported by the marginal yield when 2a was directly replaced by PhI (entry 4). The reaction was completely terminated without the palladium catalyst, indicating its pivotal role in this tandem catalysis (entry 5).
The choice of the ligand is crucial. The reaction without any ligand only gave a trace amount of product 3a (entry 6). Among all of the ligands examined, the bis-N-tosylsulfilimine ligand L1, easily prepared in one step from 1,2-bis(phenylthio)ethane and chloramine-T, gave the highest yield. Although sulfur-based ligands (e.g. sulfides and sulfoxides) are widely used,15 to the best of our knowledge, the family of bis-sulfilimines has not been previously employed as ligands for transition-metal catalysis. The reaction was found to be sensitive to the structure of the sulfilimine ligand. Mono-dentate sulfilimine ligands (L2–L4) and the bis-sulfilimine ligand with an elongated backbone (L5) are found to be inferior to L1. The corresponding bis-sulfide and bis-sulfoxide ligands (L6, L7) were effective, albeit giving lower yields. While also providing the product, phenylvinylsulfoxide (L8),16 4,5-diazafluoren-9-one (L9)11b,c and dimethylsulfoxide (L10) proved less efficient than L1.
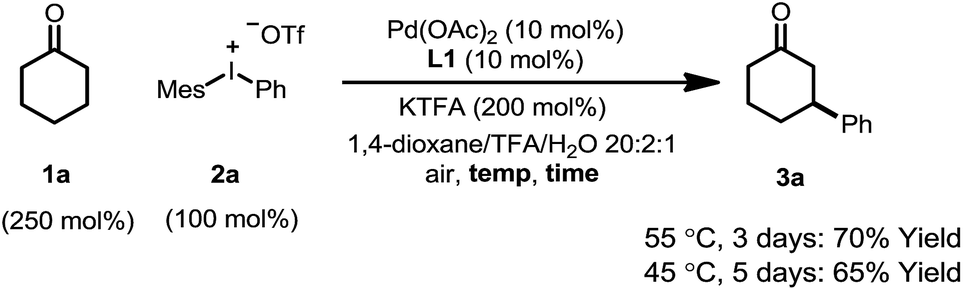
The combination of KTFA/TFA proved to be indispensable: the yield dropped significantly in the absence of one or both of the reagents (Table 1, entries 8–10). It is likely that these two reagents act as a ‘buffer pair’ to control the acidity of the reaction medium. The strong acidity of TFA would facilitate the protonation of the palladium enolate to give the product (Scheme 2, step D). Nevertheless, triflic acid (HOTf) should be produced when 2a was consumed (step A), which proved to be detrimental to the reaction (entry 11). Although TFA and KTFA can be replaced by other salts and acids, the yields dropped variably (entries 12–15). The addition of water was found to be important to promote the reaction, although the exact reason is unclear (entry 16, vide infra). A control reaction that was fully degassed and run under a nitrogen atmosphere gave a similar yield (73%, entry 17), excluding the possibility of oxygen serving as a stoichiometric oxidant. The β-arylation reaction also proceeded smoothly with a high mass balance when an equimolar amount of the two reactants was used (entry 18). Furthermore, we also discovered that the reaction maintained its catalytic activity at lower temperatures, although a prolonged reaction time was required (eqn (4)).
 | (4) |
Substrate scope
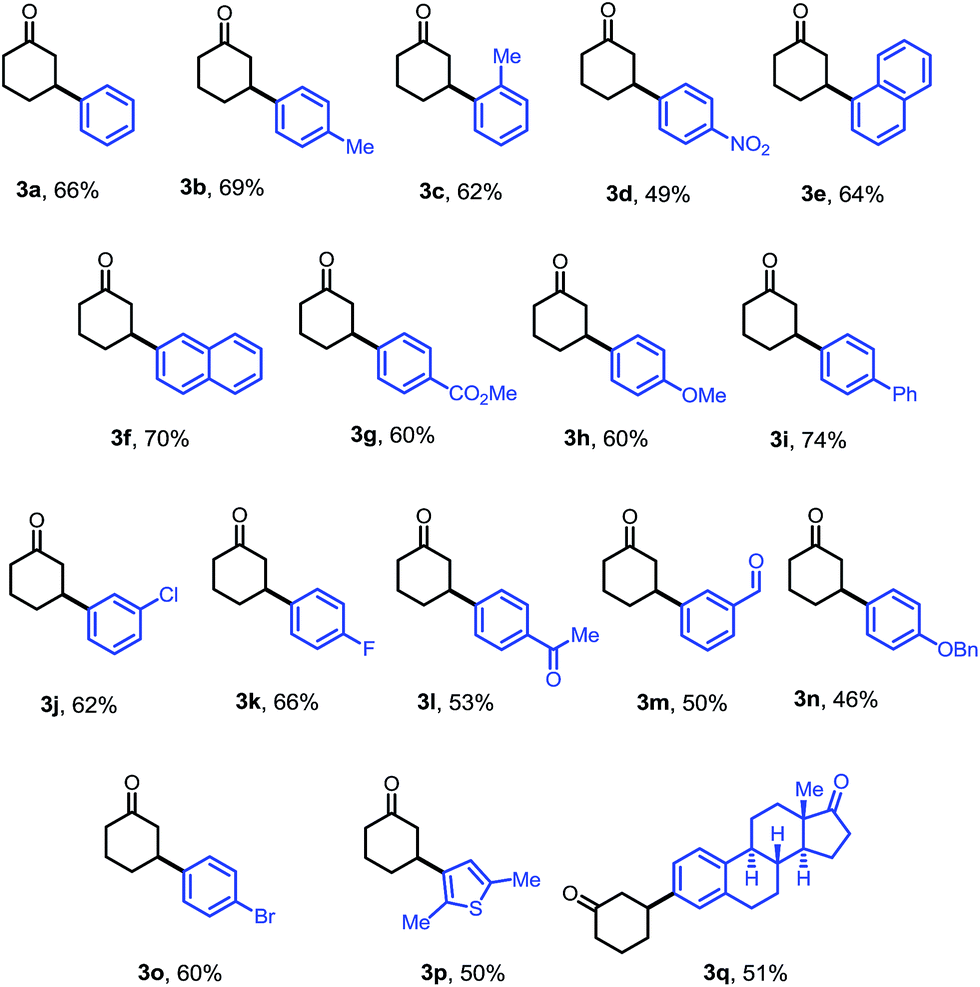
The optimized conditions were then adopted to examine the substrate scope of this β-arylation reaction (Table 2). Aryl groups with a wide span of electronic properties (electron-rich and -deficient) all participated to give the corresponding β-aryl ketones (3a–b, 3d, 3f–i and 3k). The reaction is also compatible with various para-, meta- and ortho- substituents on the arenes (3c, 3e, 3j and 3m). Base- and nucleophile-sensitive functional groups (those that find it hard to survive under traditional conjugate addition conditions), such as methyl ketone (3l) and aldehyde (3m), remained intact in the reaction. Note that aryl bromide (3o), not compatible with our previous system,10 can be tolerated under these conditions, which serves as a handle for further derivatization through cross couplings. It is also encouraging to note that the diaryliodonium salt containing a thiophene moiety also reacted to give the arylation product (3p). A complex estrone-derived iodonium salt smoothly delivered the product (3q). In this case, the cyclohexanone ring was selectively arylated while the cyclopentanone motif of the estrone remained intact.|
|
|---|
|
a Reaction conditions: mesitylaryliodonium salt (0.4 mmol), 1a (1.0 mmol), Pd(OAc)2 (0.04 mmol), L1 (0.04 mmol, d.r. >20:1 racemic/meso), KTFA (0.8 mmol), 1,4-dioxane (2 mL), TFA (200 μL), H2O (100 μL), 80 °C, 12 h.
|
|
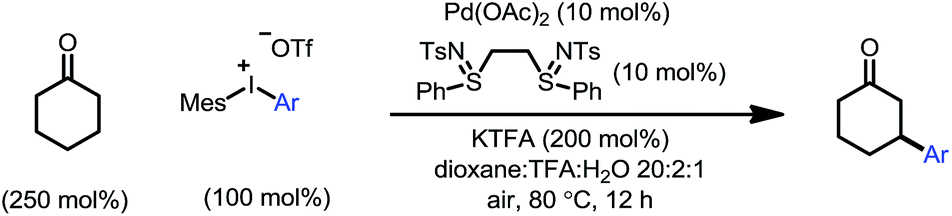
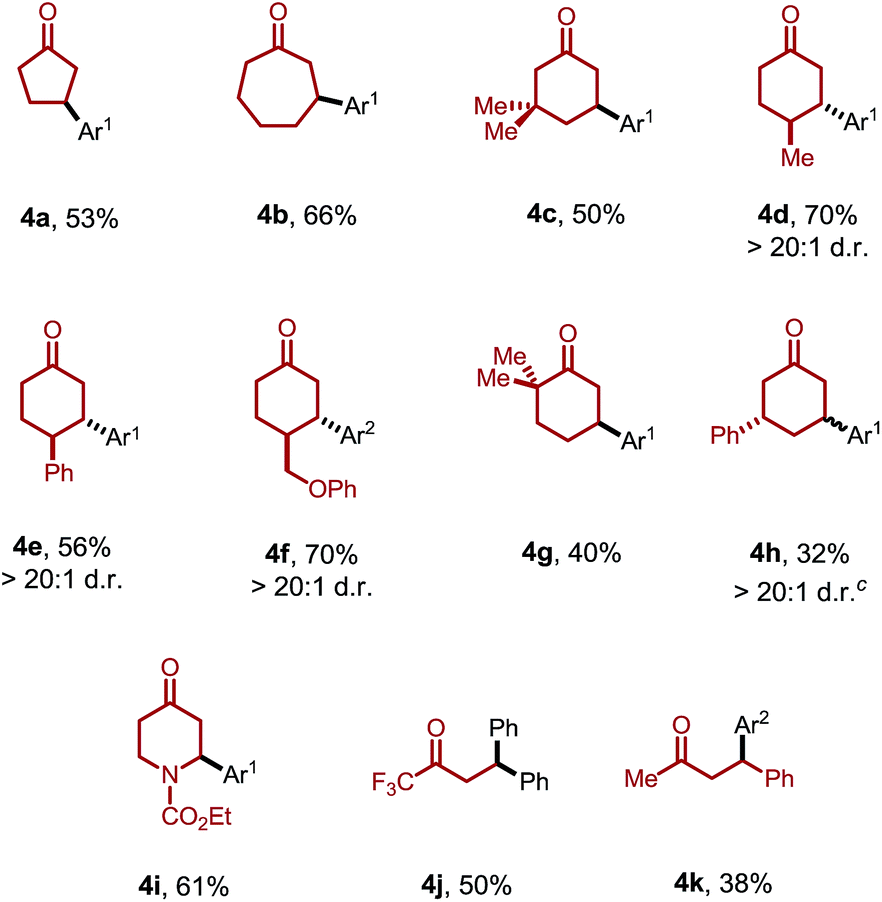
Moreover, the ketone scope was greatly improved with this new catalytic system (Table 3). Cyclic ketones with different ring-sizes were arylated in good yields (4a and 4b). Unlike our previous method, this new catalytic system enabled arylation of cyclohexanones containing α-, β-, or γ-substituents. For example, 3,3-dimethylcyclohexanone proved to be a suitable substrate (4c). Substituents at the C4 position yielded the trans products (4d–f) with excellent diastereoselectivity (>20:1). Sterically hindered 2,2-dimethylcyclohexanones were also compatible. Interestingly, when the product 3a was subjected to the reaction conditions, 3,3′-diarylcyclohexanone 4h was formed with excellent site- and diastereoselectivity. The relatively lower yields with 4g and 4h can be attributed to the competing decomposition of the diaryliodonium salt. 4-Piperidinone derivatives, a class of important pharmaceutical intermediates, can also be β-arylated (4i). While linear ketones are more challenging substrates, they still hold great promise under the new reaction conditions. The use of trifluoromethyl ethyl ketone selectively afforded the diarylation product (4j), indicating that, after the aryl migratory insertion, the Pd(II)-enolate intermediate underwent a faster β-hydrogen elimination instead of protonation. Mono-β-arylation was observed for 4-phenyl-butan-2-one (4k), and a considerable amount of the dehydrogenative β-arylation product was also formed.17
|
|
|---|
|
a Reaction conditions: mesitylaryliodonium salt (0.4 mmol), ketone (1.0 mmol), Pd(OAc)2 (0.04 mmol), L1 (0.04 mmol, d.r. >20:1 racemic/meso), KTFA (0.8 mmol), 1,4-dioxane (2 mL), TFA (200 μL), H2O (100 μL), 80 °C, 12 h.
b Diastereoselectivity was determined using crude NMR spectra.
c While 4h was isolated as a single diastereomer, attempts to determine the relative stereochemistry (cis or trans) were unsuccessful.
|
|
Mechanistic studies
To gain a better understanding of the reaction, a set of kinetics studies was first performed, where gas chromatography was employed to monitor the reaction progress. Under the standard conditions, the β-arylation of cyclohexanone 1a with the mesitylphenyliodonium salt 2a exhibited an induction period that varied from 30 minutes to an hour (Fig. 2). The initiation of the product formation was usually marked by the formation of an opaque dark red solution, while the yield ceased to increase when the diaryliodonium salt 2a was consumed.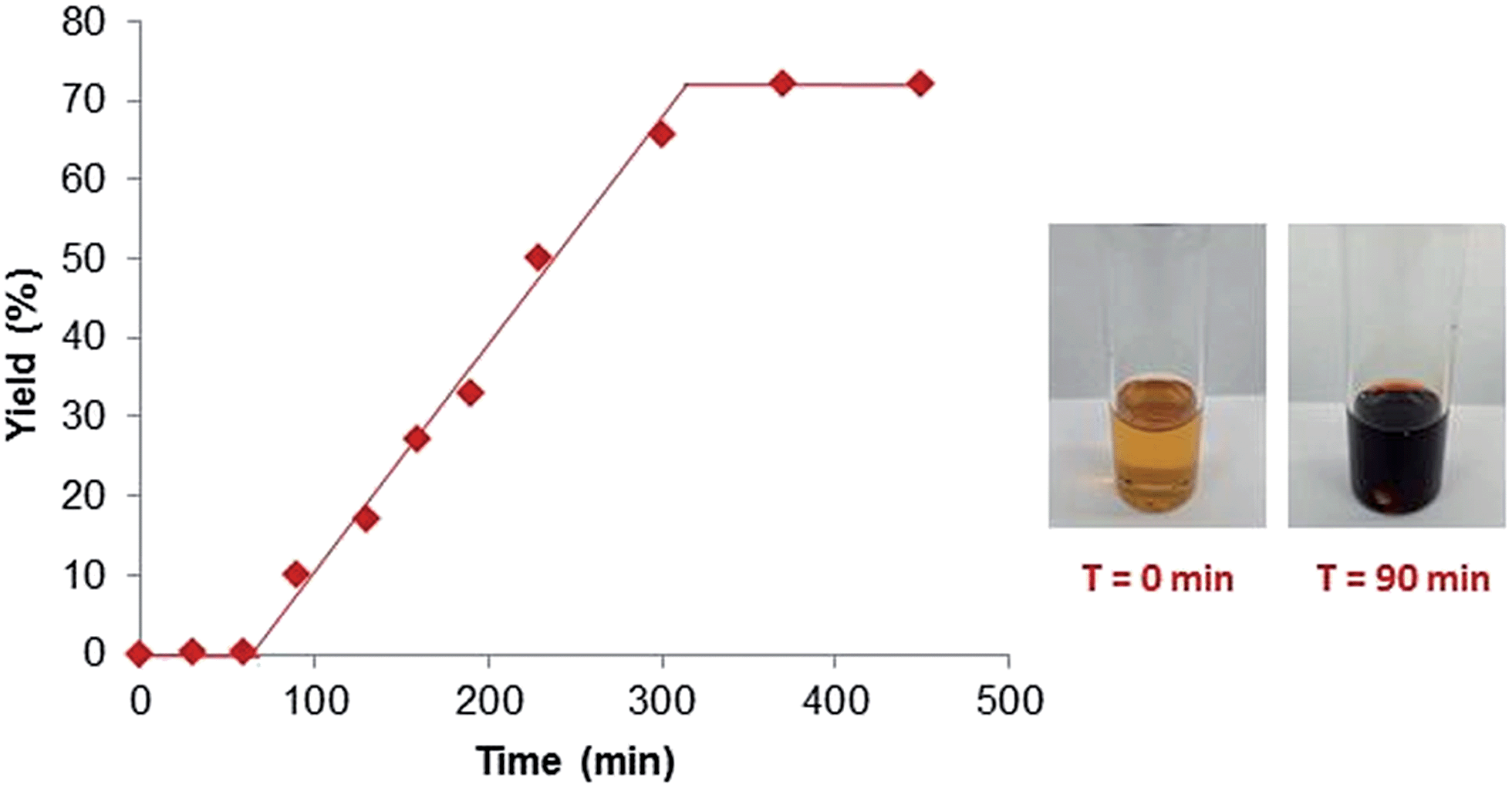 | ||
| Fig. 2 Kinetics profile. | ||
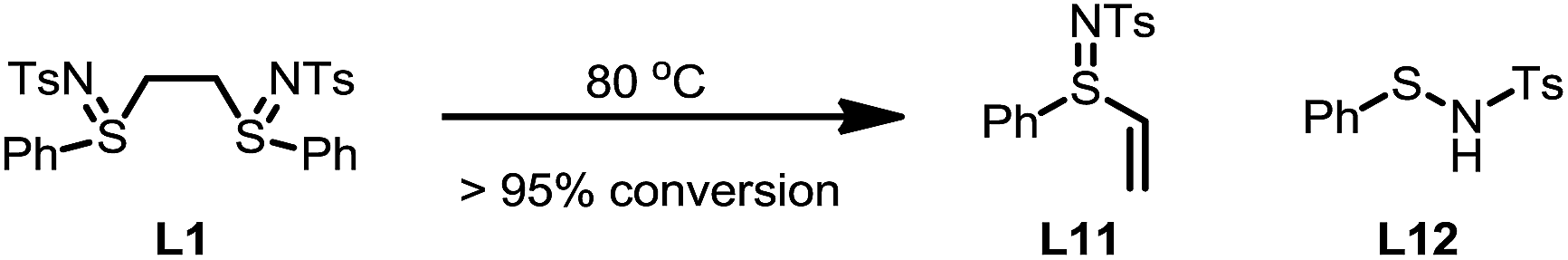
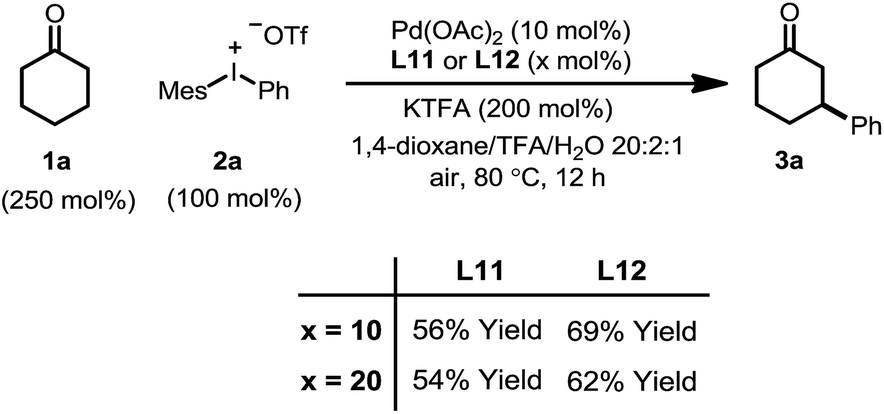
It was also observed that the induction period was accompanied with the elimination of the ligand L1 to an equimolar amount of phenyl vinyl sulfilimine L11 and N-phenylsulfanyl tosylamine L12 in more than 95% conversion (eqn (5)).16,18 In addition, when used independently as the ligand, both ligands proved to be effective for the β-arylation reaction (eqn (6)). However, the direct use of L11 or L12 from the decomposition did not eliminate the induction period.
 | (5) |
 | (6) |
We hypothesized that the induction period and the formation of the dark red opaque solution were likely attributed to the transformation of molecular palladium complexes into active catalysts in the form of clusters. It is known that sulfur-based ligands, acids (e.g. TFA) and solvents with a high dielectric constant (e.g. water) can generate and/or stabilize palladium nanoparticles.19 Recently, Stahl and coworkers also presented evidence for the role of Pd-nanoparticles in the dehydrogenation of cyclohexanones and cyclohexenones,11e although tandem transformations that can be catalyzed by nanoparticles are rare.19
In order to distinguish between a soluble nanoparticle and a heterogeneous catalyst, hot filtration tests20 were first applied to the reaction mixture when the product formation had initiated (Fig. 3). It was discovered that the resulting filtrates sustained the catalytic activity and gave comparable yields to the standard conditions upon heating. However, the filtrand collected was catalytically inactive and failed to deliver any desired product when heated together with new substrates, additives and solvents, suggesting that heterogeneous species were not responsible for the catalysis. The formation of nanoparticle species was also evidenced by dynamic light scattering (DLS) experiments,21 which showed the presence of particles with an average size of 0.9 and 204 nm at 90 min (Fig. S4, ESI†).
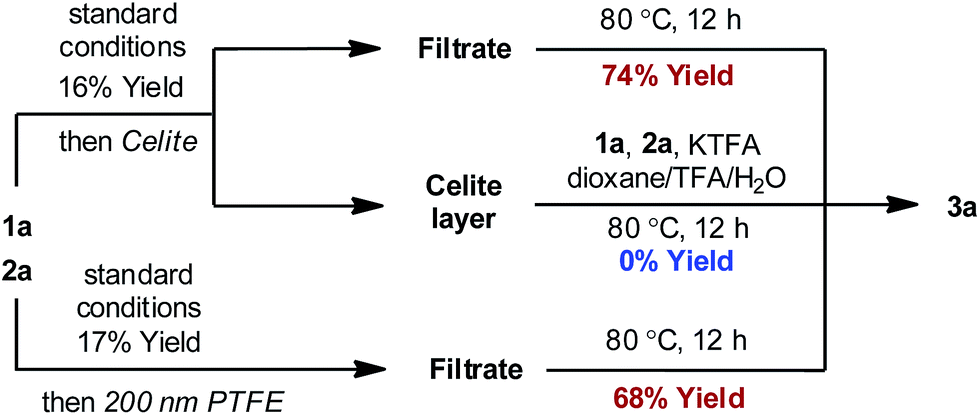 | ||
| Fig. 3 Hot filtration tests. | ||
A supplementary mercury poisoning test22 was also executed to support the presence of palladium nanoparticles. Molecular mercury is known to inhibit noble metal-nanoparticle-catalyzed reactions through amalgamation. Under our reaction conditions, when excess mercury was added during the middle of the reaction, complete inhibition was observed (Fig. 4).
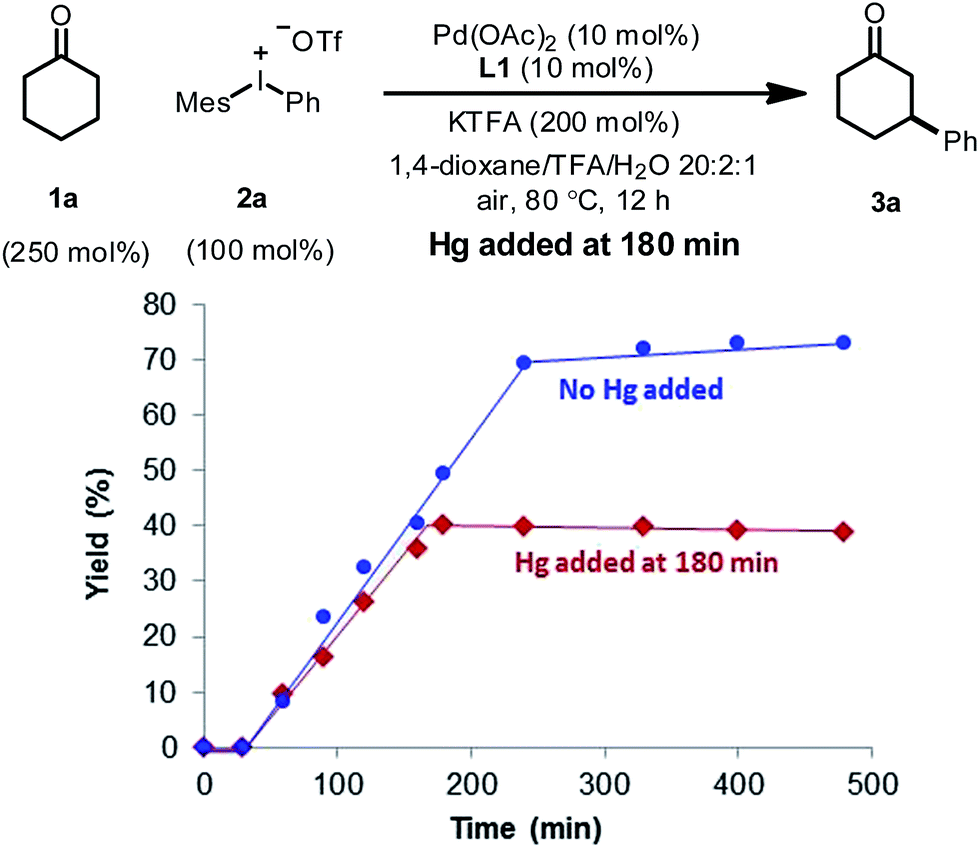 | ||
| Fig. 4 Mercury poisoning test. | ||
Conclusion
In summary, we have developed a distinct catalytic system for the direct β-arylation of ketones with widely accessible diaryliodonium salts. Compared with our previous method using aryl iodides, this new protocol holds several advantages. Firstly, it avoids the use of stoichiometric silver or copper promoters. Secondly, the conditions are more user-friendly: both moisture and air can be tolerated. Thirdly, the substrate scope is also extended to cyclic ketones with α-, β-, or γ-substituents and even aryl bromides can be tolerated. Finally, the catalytic system can sustain the reactivity at lower temperatures. Efforts towards expanding the reaction scope to other β-functionalization reactions and developing enantioselective transformations are ongoing.Abbreviations
| Mes | Mesityl |
| OTf | Trifluoromethanesulfonate |
| HFIP | 1,1,1,3,3,3-Hexafluoroisopropanol |
| P(i-Pr)3 | Triisopropylphosphine |
Acknowledgements
We thank CPRIT (R-1118) and the Welch Foundation (F-1781) for funding. G.D. is a Searle Scholar and a Sloan Fellow. Dr Michael Young and Dr Vincent Lynch are acknowledged for X-ray crystallography. Johnson Matthey is thanked for a donation of Pd salts. Dr Michael Yong and Mr John Thompson are thanked for proofreading the manuscript.References
- For recent reviews on β-C–H functionalization, see: (a) G. Rouquet and N. Chatani, Angew. Chem., Int. Ed., 2013, 52, 11726 CrossRef CAS PubMed; (b) Z. Huang and G. Dong, Tetrahedron Lett., 2014, 55, 5869 CrossRef CAS PubMed; (c) G. Qiu and J. Wu, Org. Chem. Front., 2015, 2, 169 RSC; (d) B. Zhang, H.-X. Guan, B. Liu and B.-F. Shi, Chin. J. Org. Chem., 2014, 34, 1487 CrossRef CAS.
- For representative examples, see: (a) G. N. Roth, A. Chandra and M. G. Nair, J. Nat. Prod., 1998, 61, 542 CrossRef CAS PubMed; (b) L. S. Melvin, M. R. Johnson, C. A. Harbert, G. M. Milne and A. Weissman, J. Med. Chem., 1984, 27, 67 CrossRef CAS.
- (a) A. Gutnov, Eur. J. Org. Chem., 2008, 4547 CrossRef CAS PubMed; (b) T. Hayashi and K. Yamasaki, Chem. Rev., 2003, 103, 2829 CrossRef CAS PubMed.
- For selected examples, see: (a) V. G. Zaitsev, D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2005, 127, 13154 CrossRef CAS PubMed; (b) B. V. S. Reddy, L. R. Reddy and E. J. Corey, Org. Lett., 2006, 8, 3391 CrossRef CAS PubMed; (c) D. Shabashov and O. Daugulis, J. Am. Chem. Soc., 2010, 132, 3965 CrossRef CAS PubMed; (d) Y. Feng, Y. Wang, B. Landgraf, S. Liu and G. Chen, Org. Lett., 2010, 12, 3414 CrossRef CAS PubMed; (e) L. D. Tran and O. Daugulis, Angew. Chem., Int. Ed., 2012, 51, 5188 CrossRef CAS PubMed; (f) Q. Zhang, K. Chen, W. Rao, Y. Zhang, F.-J. Chen and B.-F. Shi, Angew. Chem., Int. Ed., 2013, 52, 13588 CrossRef CAS PubMed; (g) F. Pan, P.-X. Shen, L.-S. Zhang, X. Wang and Z.-J. Shi, Org. Lett., 2013, 15, 4758 CrossRef CAS PubMed; (h) Y. Wei, H. Tang, X. Cong, B. Rao, C. Wu and X. Zeng, Org. Lett., 2014, 16, 2248 CrossRef CAS PubMed; (i) Q. Zhang, X.-S. Yin, S. Zhao, S.-L. Fang and B.-F. Shi, Chem. Commun., 2014, 50, 8353 RSC.
- (a) R. Shang, L. Ilies, A. Matsumoto and E. Nakamura, J. Am. Chem. Soc., 2013, 135, 6030 CrossRef CAS PubMed; (b) Q. Gu, H. H. Al Mamari, K. Graczyk, E. Diers and L. Ackermann, Angew. Chem., Int. Ed., 2014, 53, 3868 CrossRef CAS PubMed; (c) Y. Aihara and N. Chatani, J. Am. Chem. Soc., 2014, 136, 901 Search PubMed; (d) M. Li, J. Dong, X. Huang, K. Li, Q. Wu, F. Song and J. You, Chem. Commun., 2014, 50, 3944 RSC; (e) M. Iyanaga, Y. Aihara and N. Chatani, J. Org. Chem., 2014, 79, 11933 CrossRef CAS PubMed.
- For recent examples, see: (a) R. Giri, N. Maugel, J.-J. Li, D.-H. Wang, S.-P. Breazzano, L.-B. Saunders and J.-Q. Yu, J. Am. Chem. Soc., 2007, 129, 3510 CrossRef CAS PubMed; (b) D.-H. Wang, M. Wasa, R. Giri and J.-Q. Yu, J. Am. Chem. Soc., 2008, 130, 7190 CrossRef CAS PubMed; (c) M. Wasa, K. M. Engle and J.-Q. Yu, J. Am. Chem. Soc., 2009, 131, 9886 CrossRef CAS PubMed; (d) M. Wasa, K. S. L. Chan, X.-G. Zhang, J. He, M. Miura and J.-Q. Yu, J. Am. Chem. Soc., 2012, 134, 18570 CrossRef CAS PubMed; (e) J. He, S. Li, Y. Deng, H. Fu, B. N. Laforteza, J. E. Spangler, A. Homs and J.-Q. Yu, Science, 2014, 343, 1216 CrossRef CAS PubMed; (f) K.-J. Xiao, D. W. Lin, M. Miura, R.-Y. Zhu, W. Gong, M. Wasa and J.-Q. Yu, J. Am. Chem. Soc., 2014, 136, 8138 CrossRef CAS PubMed.
- For examples of β-arylation of esters using migratory coupling, see: (a) M. Jørgensen, S. Lee, X. Liu, J. P. Wolkowski and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 12557 CrossRef PubMed; (b) A. Renaudat, L. Jean-Gérard, R. Jazzar, C. E. Kefalidis, E. Clot and O. Baudoin, Angew. Chem., Int. Ed., 2010, 49, 7261 CrossRef CAS PubMed; (c) S. Aspin, A.-S. Goutierre, P. Larini, R. Jazzar and O. Baudoin, Angew. Chem., Int. Ed., 2012, 51, 10808 CrossRef CAS PubMed.
- (a) R. Zuber, G. Carlens, R. Haag and A. de Meijere, Synlett, 1996, 542 CrossRef CAS PubMed; (b) M. V. Leskinen, K.-T. Yip, A. Valkonen and P. M. Pihko, J. Am. Chem. Soc., 2012, 134, 5750 CrossRef CAS PubMed; (c) M. V. Leskinen, Á. Madarász, K.-T. Yip, A. Vuorinen, I. Pápai, A. J. Neuvonen and P. M. Pihko, J. Am. Chem. Soc., 2014, 136, 6453 CrossRef CAS PubMed.
- M. T. Pirnot, D. A. Rankic, D. B. C. Martin and D. W. C. MacMillan, Science, 2013, 339, 1593 CrossRef CAS PubMed.
- Z. Huang and G. Dong, J. Am. Chem. Soc., 2013, 135, 17747 CrossRef CAS PubMed.
- For recent examples of Pd-catalyzed dehydrogenation of ketones, see: (a) T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 14566 CrossRef CAS PubMed; (b) T. Diao, T. J. Wadzinski and S. S. Stahl, Chem. Sci., 2012, 3, 887 RSC; (c) W. Gao, Z. He, Y. Qian, J. Zhao and Y. Huang, Chem. Sci., 2012, 3, 883 RSC; (d) T. Diao, D. Pun and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8205 CrossRef CAS PubMed; (e) D. Pun, T. Diao and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 8213 CrossRef CAS PubMed.
- An alternative mechanism consisting of two independent catalytic cycles, i.e. an oxidative dehydrogenation and a reductive Heck reaction, cannot be ruled out.
- For seminal works and recent reviews on the synthesis and application of diaryliodonium salts, see: (a) T. Dohi, M. Ito, K. Morimoto, Y. Minamitsuji, N. Takenaga and Y. Kita, Chem. Commun., 2007, 4152 RSC; (b) M. Bielawski and B. Olofsson, Chem. Commun., 2007, 2521 RSC; (c) M. Bielawski, M. Zhu and B. Olofsson, Adv. Synth. Catal., 2007, 349, 2610 CrossRef CAS PubMed; (d) N. R. Deprez and M. S. Sanford, Inorg. Chem., 2007, 46, 1924 CrossRef CAS PubMed; (e) V. V. Zhdankin and P. J. Stang, Chem. Rev., 2008, 108, 5299 CrossRef CAS PubMed; (f) E. A. Merritt and B. Olofsson, Angew. Chem., Int. Ed., 2009, 48, 9052 CrossRef CAS PubMed; (g) M. Bielawski and B. Olofsson, Org. Synth., 2009, 86, 308 CrossRef CAS; (h) R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172 CrossRef CAS PubMed.
- For recent examples of using diaryliodonium salts in C–H arylation reactions, see: (a) B. Xiao, Y. Fu, J. Xu, T.-J. Gong, J.-J. Dai, J. Yi and L. Liu, J. Am. Chem. Soc., 2010, 132, 468 CrossRef CAS PubMed; (b) C.-L. Ciana, R. J. Phipps, J. R. Brandt, F.-M. Meyer and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 458 CrossRef CAS PubMed; (c) H. A. Duong, R. E. Gilligan, M. L. Cooke, R. J. Phipps and M. J. Gaunt, Angew. Chem., Int. Ed., 2011, 50, 463 CrossRef CAS PubMed; (d) A. Bigot, A. E. Williamson and M. J. Gaunt, J. Am. Chem. Soc., 2011, 133, 13778 CrossRef CAS PubMed; (e) J. S. Harvey, S. P. Simonovich, C. R. Jamison and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 13782 CrossRef CAS PubMed; (f) A. E. Allen and D. W. C. MacMillan, J. Am. Chem. Soc., 2011, 133, 4260 CrossRef CAS PubMed; (g) R. J. Phipps, L. McMurray, S. Ritter, H. A. Duong and M. J. Gaunt, J. Am. Chem. Soc., 2012, 134, 10773 CrossRef CAS PubMed; (h) Q. Y. Toh, A. McNally, S. Vera, N. Erdmann and M. J. Gaunt, J. Am. Chem. Soc., 2013, 135, 3772 CrossRef CAS PubMed; (i) M. E. Kieffer, K. V. Chuang and S. E. Reisman, J. Am. Chem. Soc., 2013, 135, 5557 CrossRef CAS PubMed; (j) A. M. Wagner, A. J. Hickman and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 15710 CrossRef CAS PubMed; (k) F. Zhang, S. Das, A. J. Walkinshaw, A. Casitas, M. Taylor, M. G. Suero and M. J. Gaunt, J. Am. Chem. Soc., 2014, 136, 8851 CrossRef CAS PubMed; (l) S. G. Modha and M. F. Greaney, J. Am. Chem. Soc., 2015, 137, 1416 CrossRef CAS PubMed.
- For a recent review on sulfur-based ligands, see: M. Mellah, A. Voituriez and E. Schulz, Chem. Rev., 2007, 107, 5133 CrossRef CAS PubMed.
- It is noteworthy that partial decomposition of the bis-sulfoxide ligands was reported by White and coworkers, see: M. S. Chen, N. Prabagaran, N. A. Labenz and M. C. White, J. Am. Chem. Soc., 2005, 127, 6970 CrossRef CAS PubMed.
- For a recent example of dehydrogenative β-arylation, see: P. Gandeepan, P. Rajamalli and C.-H. Cheng, ACS Catal., 2014, 4, 4485 CrossRef CAS.
- For a selected review on elimination of alkyl bis-N-tosylsulfilimines, see: S. Oae and N. Furukawa, Tetrahedron, 1977, 33, 2359 CrossRef CAS.
- For recent reviews on Pd nanoparticles, see: (a) J. D. Aiken III and R. G. Finke, J. Mol. Catal. A: Chem., 1999, 145, 1 CrossRef; (b) A. Roucoux, J. Schulz and H. Patin, Chem. Rev., 2002, 102, 3757 CrossRef CAS PubMed; (c) D. Astruc, Inorg. Chem., 2007, 46, 1884 CrossRef CAS PubMed; (d) J. A. Widegren and R. G. Finke, J. Mol. Catal. A: Chem., 2003, 198, 317 CrossRef CAS; (e) R. H. Crabtree, Chem. Rev., 2011, 112, 1536 CrossRef PubMed.
- J. E. Hamlin, K. Hirai, A. Millan and P. M. Maitlis, J. Mol. Catal., 1980, 7, 543 CAS.
- B. J. Berne and R. Pecora, Dynamic Light Scattering (With Applications to Chemistry, Biology, and Physics), Dover, Mineola, NY, 2nd edn, 2000 Search PubMed.
- For recent examples of the mercury test, see: (a) E. E. Finney and R. G. Finke, Inorg. Chim. Acta, 2006, 359, 2879 CrossRef CAS PubMed; (b) M. Weck and C. W. Jones, Inorg. Chem., 2007, 46, 1865 CrossRef CAS PubMed; (c) C. W. Jones, Top. Catal., 2010, 53, 942 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1058344–1058346. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01636c |
| This journal is © The Royal Society of Chemistry 2015 |