
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Double-walled pyr topology networks from a novel fluoride-bridged heptanuclear metal cluster†
Kai-Jie
Chen
,
John J.
Perry IV
,
Hayley S.
Scott
,
Qing-Yuan
Yang
and
Michael J.
Zaworotko
*
Department of Chemical & Environmental Sciences, University of Limerick, Limerick, Republic of Ireland. E-mail: Michael.Zaworotko@ul.ie
First published on 22nd May 2015
Abstract
Two isostructural metal–organic materials, Tripp-1-M (Tripp = 2,4,6-tris(4-pyridyl)pyridine; M = Co, Ni), that exhibit binodal 3,6-connected pyr network topology have been prepared and characterized. Tripp-1-M are based upon a novel M7F122+ cluster that possesses 12 connection points but, because of double cross-linking by 3-connected Tripp ligands, it functions as a 6-connected supermolecular building block (SBB).
Introduction
Metal–organic materials (MOMs)1 have attracted rapidly increasing attention from the scientific community in the last two decades thanks to their inherent modularity. This modularity can promote diversity of composition, amenability to systematic design2 and control over certain bulk properties.3 However, not all MOMs are well suited to serve as prototypal platforms for the generation of families of materials with the same topology. Such platforms are important because they enable systematic fine-tuning of both pore size (e.g. organic linkers with different lengths) and pore chemistry (e.g. functional group substitution at the linker or metal substitution at the node). Most platforms are built from single metal ion or small cluster (molecular building block, MBB) nodes and are exemplified by platforms sustained by carboxylate clusters such as 4-connected (4-c) M2(RCOO)4 (e.g.HKUST-1,4 PCN-6,5 MOF-2,6 DMOF-1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 7 and NU-1008), 6-c M3O(RCOO)6 (e.g. MIL-88,9 MIL-10110 and PCN-60011) and 6-c M4O(RCOO)6 (e.g.MOF-512 and MOF-17713). The exploitation of larger, high symmetry clusters offers the possibility of much higher levels of connectivity and even greater control over topology. Such “supermolecular building blocks”, SBBs, are exemplified by 12-connected (12-c) Zr6O4(OH)4(RCOO)1214 and 24-c “nanoball” Cu24(1,3-bdc)24 clusters.15 High connectivity mixed carboxylate/N-donor clusters have also been utilised in this context.16 In addition to affording greater control over topology because of fewer possible topological outcomes, higher connectivity nodes can result in greater robustness.17
7 and NU-1008), 6-c M3O(RCOO)6 (e.g. MIL-88,9 MIL-10110 and PCN-60011) and 6-c M4O(RCOO)6 (e.g.MOF-512 and MOF-17713). The exploitation of larger, high symmetry clusters offers the possibility of much higher levels of connectivity and even greater control over topology. Such “supermolecular building blocks”, SBBs, are exemplified by 12-connected (12-c) Zr6O4(OH)4(RCOO)1214 and 24-c “nanoball” Cu24(1,3-bdc)24 clusters.15 High connectivity mixed carboxylate/N-donor clusters have also been utilised in this context.16 In addition to affording greater control over topology because of fewer possible topological outcomes, higher connectivity nodes can result in greater robustness.17
In this contribution, we introduce a new inorganic SBB of formula M7F122+ (M = Co, Ni, Fig. 1) and demonstrate that it can serve as a 6-c SBB through double cross-linking of its 12 connection points by a facile to prepare 3-c ligand, 2,4,6-tris(4-pyridyl)pyridine, Tripp (Scheme 1). A new type of double-walled pyr topology (Fig. 2) material which exhibits permanent porosity is thereby generated.
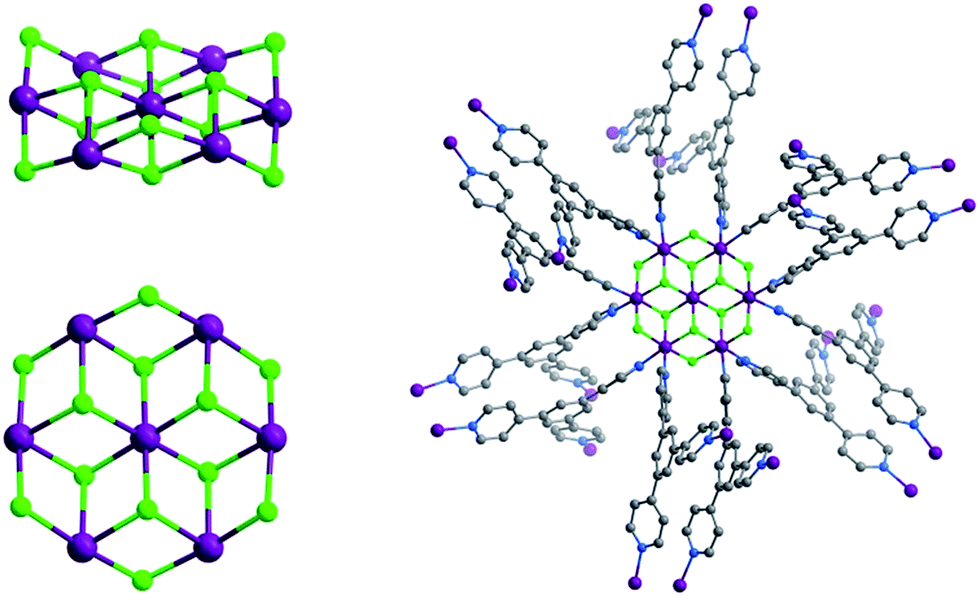 | ||
| Fig. 1 (Left) Perspective and above views of the novel M7F122+ cluster that sustains Tripp-1-M. (right) Illustration of the 12 connection points of the Co7F12 SBB in Tripp-1-Co (Co, F, N and C atoms in purple, green, blue and grey). Solvent molecules, hydrogen atoms and counter-ions are omitted for the sake of clarity. | ||
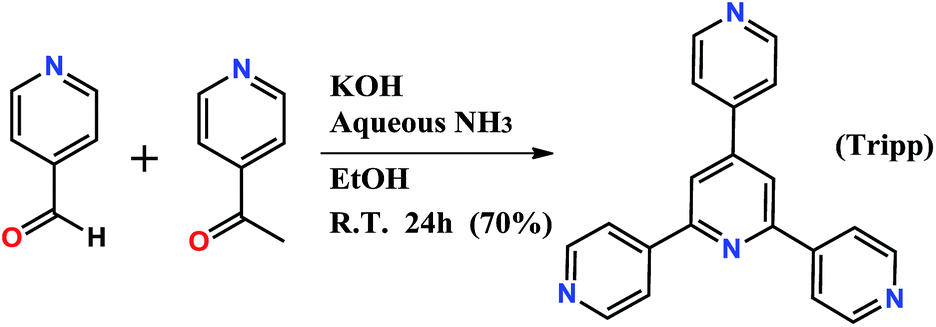 | ||
| Scheme 1 Synthesis of 2,4,6-tris(4-pyridyl)pyridine (Tripp). | ||
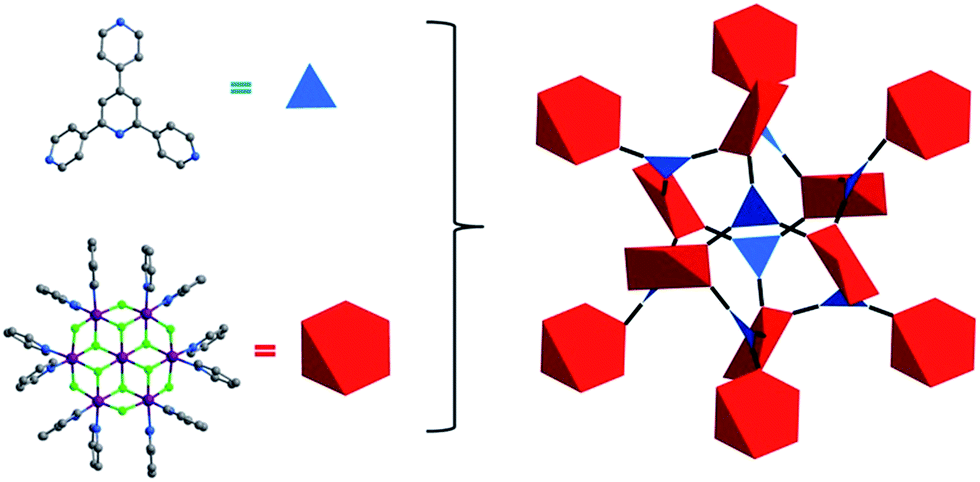 | ||
| Fig. 2 Illustration of the topology of Tripp-1-M. The Tripp ligand is represented by a 3-c triangle (blue) whereas the M7F122+ SBB is reduced to a 6-c octahedron (red). | ||
In previous work, double18,19 or quadruple20 cross-linking of carboxylate21 or oxide22 based SBBs has been shown to represent a suitable approach to build MOMs with well-known23 or hitherto novel24 topology. However, the use of fluoride-bridged MBBs and SBBs as nodes for the construction of three-dimensional MOMs remains rare.25 This is despite the fact that discrete fluoride-bridged metal clusters are known26,27 and that such structures can exhibit interesting magnetic properties.28
Results and discussion
Tripp was prepared by the cyclization reaction of 4-acetylpyridine and 4-pyridinecarbaldehyde (Scheme 1).29 Single crystals of Tripp-1-Co were initially obtained by solvothermal reaction between Co(NO3)2·6H2O, Tripp and (NH4)2SiF6 in DMF (for full details of synthetic procedures see ESI†). Tripp-1-Co crystallizes in the cubic space group Pa![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) . A crystallographic 3-fold axis runs through the centre of the Tripp node and the disordered atoms of the central pyridine ring were therefore refined as 2/3 carbon and 1/3 nitrogen. All atoms of central pyridine ring are presented as carbon atoms for clarity in Fig. 1 and 3. In Tripp-1-Co, every Tripp ligand links three M7F122+ SBBs and every SBB is connected by 12 Tripp ligands. However, the arrangement of the 12 connection points enables double cross-linking by pairs of Tripp ligands (Fig. 1) meaning that the connectivity is effectively reduced to 6. Therefore, each pair of Tripp ligands can be simplified to a single node, the M7F12 SBB can be treated as a 6-c node and the Tripp ligand as a 3-connected node. The outcome of this connectivity is structure that exhibits binodal 3,6-connected pyr topology (Fig. 2), of which there are relatively few examples22b,30 when compared to other types of 3,6 nets such as those with rtl, ant, sit or qom topology.
. A crystallographic 3-fold axis runs through the centre of the Tripp node and the disordered atoms of the central pyridine ring were therefore refined as 2/3 carbon and 1/3 nitrogen. All atoms of central pyridine ring are presented as carbon atoms for clarity in Fig. 1 and 3. In Tripp-1-Co, every Tripp ligand links three M7F122+ SBBs and every SBB is connected by 12 Tripp ligands. However, the arrangement of the 12 connection points enables double cross-linking by pairs of Tripp ligands (Fig. 1) meaning that the connectivity is effectively reduced to 6. Therefore, each pair of Tripp ligands can be simplified to a single node, the M7F12 SBB can be treated as a 6-c node and the Tripp ligand as a 3-connected node. The outcome of this connectivity is structure that exhibits binodal 3,6-connected pyr topology (Fig. 2), of which there are relatively few examples22b,30 when compared to other types of 3,6 nets such as those with rtl, ant, sit or qom topology.
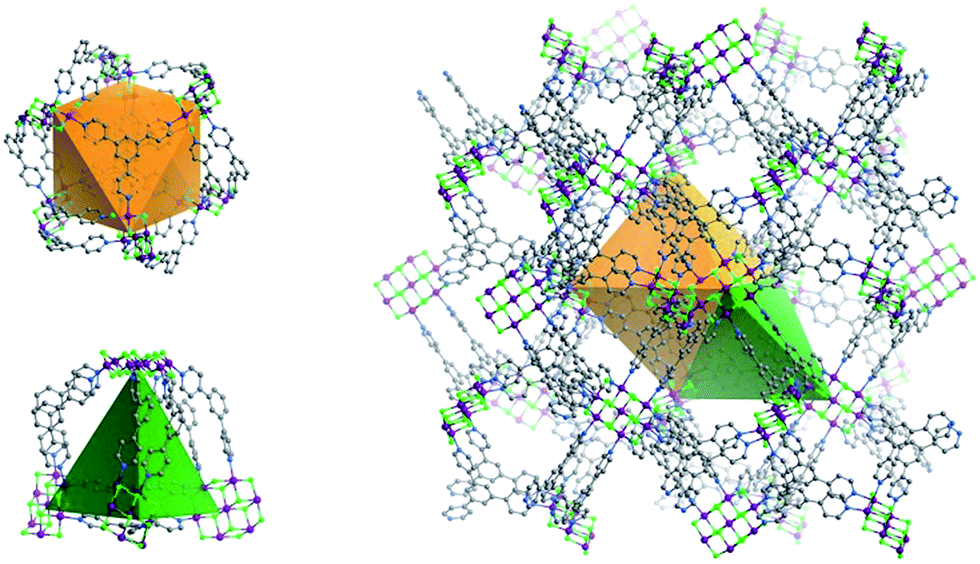 | ||
| Fig. 3 The two types of double-walled cages (OC, left above; TC left, below) found in Tripp-1-M (right). | ||
The structure of Tripp-1-M is comprised of two types of double-walled cages; tetrahedral cages (TCs) and octahedral cages (OCs) with face-sharing configurations (Fig. 3). The M7F122+ SBB is located at the vertex of every TC and OC, while the double-walls are constructed from Tripp ligands. The pore diameter of the TC and OC are ca. 7.7 and 7.6 Å, respectively. Every OC is surrounded by eight adjacent TCs and every TC is surrounded by four OCs. However, there are two faces and one face capped by double walls of Tripp ligands in the OCs and TCs, respectively. Therefore, each OC connects with only six TCs and each TC crosslinks with three OCs, which corresponds to the required 3,6-connectivity of a pyr net. The guest-accessible porosity (considering the presence of counter ions) of Tripp-1-Co is 53.5%, based on Platon software.31 Two other structures with pyr topology using similar tripyridyl-based 3-c ligands, 2,4,6-tri(4-pyridyl)-1,3,5-triazine (TPT) and 1,3,5-benzene tricarboxylic acid tris[N-(4-pyridyl)amide (TPBTC), with 6-c metal ions Hg2+ and Cd2+, were reported by Robson and Kitagawa, respectively.32 These tripyridyl-based ligands can also be utilized as 3-c organic nodes to construct nets with high porosity.33 Further, some discrete cages consisting of 4-c Pd2+ ions linked by TPT and cage-based three-dimensional nets were reported by Fujita and co-workers.34 In contrast, the facile to prepare Tripp ligand has not been as widely studied as TPT and TPBTC.
The double-walled nature of Tripp-1-M is unusual and, to our knowledge, such as structure has not been prepared using a single ligand and a single SBB. However, Bu et al. recently reported two isostructural double-walled cage-based MOMs that were designed using a strategy based upon size-matching between two tritopic ligands (TPT and 2,4,6-tris[1-(3-carboxylphenoxy)-yl-methyl]mesitylene) connected by with same paddle-wheel unit.18
Analysis of crystal structure of Tripp-1-Co revealed that the Co–F distances in heptanuclear cluster lie the range from 2.036 (5) to 2.121 (7) Å, which are consistent with the values found in other fluoride-bridged Co(II) structures, e.g. [Co5F2(tetrazolate)4(H2O)4]35 and [Co12(RCOO)6(PO4)4F4(H2O)6](NO3)2.36 The crystal structure of Tripp-1-Co also revealed that the charge of each heptanuclear cobalt cluster is balanced by one SiF62− anion that exhibits three-fold F⋯F (distance: 2.68 (2) Å) interactions with three bridging F− anions (Fig. S1†). Energy-dispersive X-ray spectroscopy verified the presence of Si and F in crystals of Tripp-1-Co (Fig. 4). There are previous reports concerning the generation of F− by decomposition of PF6− and BF4−,26,37 so we speculated that the source of the bridging fluoride anions in the Co7F122+ SBBs was in situ decomposition of SiF62−. Further, there are two drawbacks to the use of this synthetic method: the relatively low solubility of (NH4)2SiF6 in DMF; the requirement to decompose SiF62− anions before the SBB can form. These drawbacks mean that unreacted (NH4)2SiF6 is isolated in a physical mixture with Tripp-1-Co crystals, mitigating against phase purity and also resulting in low product yield (ca. 10%). Therefore, we tested a different synthetic approach involving reflux of starting materials in DMF/MeOH with NH4F instead of (NH4)2SiF6 as the F− source. This method facilitated an increase in yield to 80%. The composition of Tripp-1-Co was changed since SiF62− counterions are no longer present. Rather, two NO3− anions from the Co(NO3)2 starting material balance charge as indicated by the presence of two diagnostic peaks measured using FT-IR at around 1320 and 1400 cm−1 (Fig. S4†).38 The relatively high solubility of NH4F in MeOH enabled subsequent isolation of pure reaction product. The purity of bulk product was established by powder X-ray diffraction (PXRD) patterns of as-synthesized samples, which are good matches to those calculated from the crystal structure of Tripp-1-Co (Fig. S5†). To further verify the composition of the M7F122+ cluster, X-ray photoelectron spectroscopy (XPS) analysis of a sample prepared from NH4F was conducted, and a molar ratio for F:Co of 1.44 (expected 1.71) was observed (Fig. 4). A series of control experiments conducted without using NH4F as a source of F− revealed that different concentrations of metal and ligand and different reaction temperatures failed to afford the desired Tripp-1-Co product. Nevertheless, these experiments confirm the essential role that F− plays in construction of the M7F122+ SBB and subsequently the overall MOM framework. The isostructural nickel analogue of Tripp-1-Co, Tripp-1-Ni, was obtained via the same modified synthetic protocol with a yield of ca. 70% as verified by PXRD (Fig. S6†).
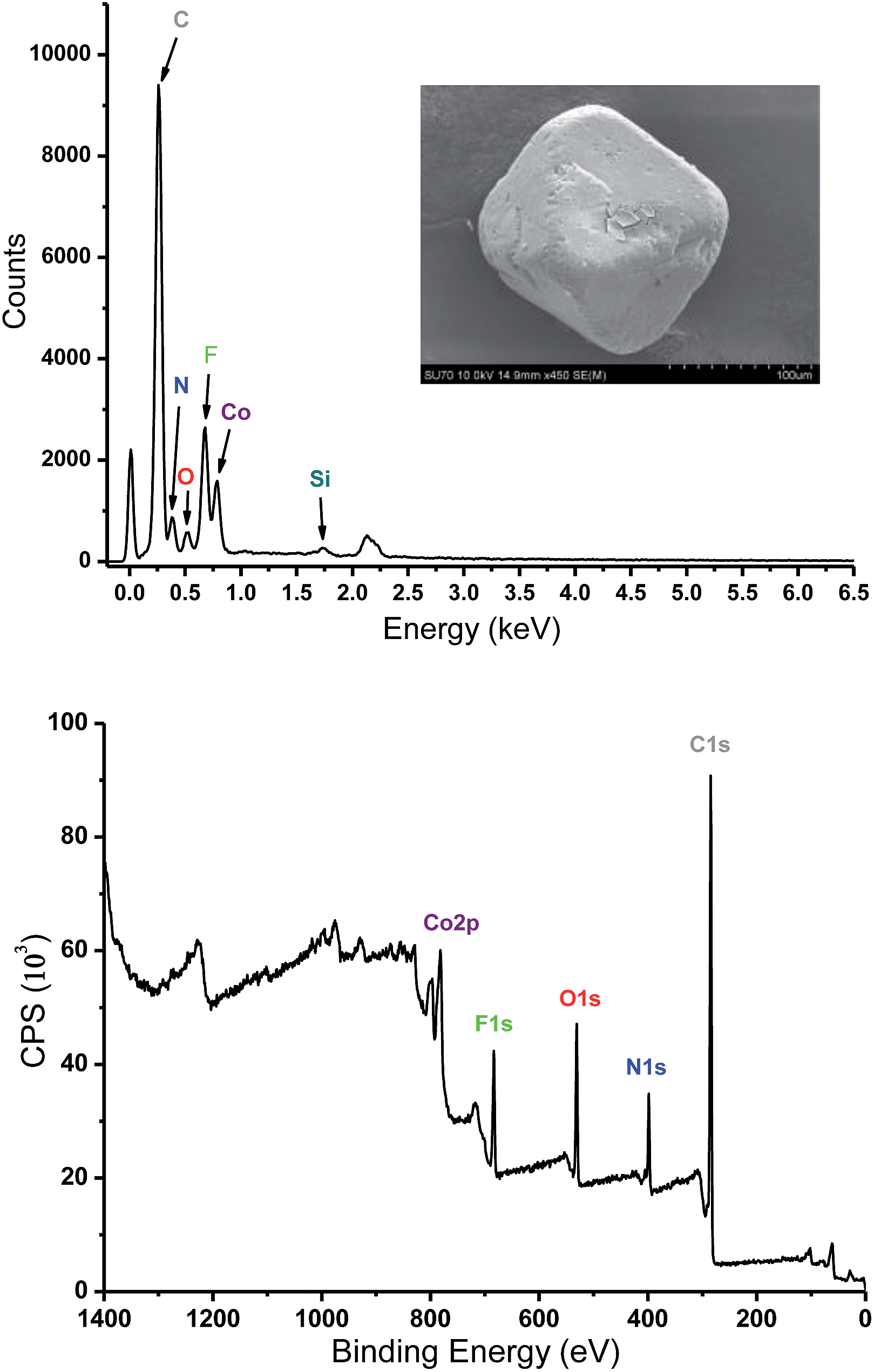 | ||
| Fig. 4 Energy-dispersive X-ray spectroscopy (top) and X-ray photoelectron spectroscopy (bottom) of Tripp-1-Co. | ||
To address the thermal stability of Tripp-1-M, thermo-gravimetric analysis (TGA) was conducted for both as-synthesized and MeOH exchanged samples. The resulting TGA plots reveal that solvent guest molecules in the as-synthesized samples can be fully exchanged with MeOH, which in turn can be removed below 110 °C. No further weight loss until after 300 °C was observed, which we presume is a consequence of framework decomposition (Fig. S8†). Framework integrity was also verified by PXRD experiments conducted after desolvation of MeOH-exchanged samples at 120 °C. Furthermore, samples exposed to the air under ambient conditions for two months, were observed to exhibit PXRD patterns conforming to those calculated from single-crystal data (Fig. S5 and S6†). These results demonstrate that both Tripp-1-Co and Tripp-1-Ni possess good thermal and air/moisture stability, which we attribute to some extent to the high connectivity of the M7F122+ SBB.
The permanent porosities of Tripp-1-Co and Tripp-1-Ni were established by measuring CO2 sorption isotherms at 195 K (Fig. 5). The apparent BET surface area was calculated to be 822 and 1149 m2 g−1 for Tripp-1-Co and Tripp-1-Ni, respectively. Pore volumes of 0.358 and 0.516 cm3 g−1 for Tripp-1-Co and Tripp-1-Ni were calculated by assuming liquid filling of CO2 at saturated state, which are close to the value of 0.587 cm3 g−1 estimated from the crystal data for Tripp-1-Co. The relatively lower uptake of Tripp-1-Co might be attributed to partial collapse of the framework during activation, which does not appear to occur for Tripp-1-Ni. CO2 and N2 sorption isotherms of Tripp-1-M at 273, 283 and 293 K were also measured. As shown in Fig. 6, CO2 uptakes of 76.7, 61.4 and 49.2 cm3 g−1 at 273, 283 and 293 K, respectively, are much higher than the values of 3.7, 3.2 and 2.5 cm3 g−1 observed for N2 in Tripp-1-Co. Tripp-1-Ni exhibits higher CO2 uptakes of 99.3, 79.8 and 64.3 cm3 g−1 at 273, 283 and 293 K, respectively, than those observed for Tripp-1-Co. Meanwhile, N2 uptakes at 273, 283 and 293 K are only 5.2, 4.3 and 3.5 cm3 g−1 for Tripp-1-Ni. The high CO2 uptakes of Tripp-1-M could be attributed to high pore volume and polarized pore surface originating from the heptanuclear cluster and central pyridine ring of the Tripp ligand. These results indicate that both Tripp-1-M variants exhibit good selectivity for CO2 over N2. Preliminary CO2/N2 selectivity for Tripp-1-Co and Tripp-1-Ni, calculated from the uptakes of CO2 at 0.15 bar and N2 at 0.85 bar, are 41.4 and 40.8, 36.8 and 36.7, and 36.2 and 32.9 at 273, 283 and 293 K. These uptakes and selectivities are comparable to many well-known MOMs containing polar functional groups and/or open metal sites, both of which are absent in Tripp-1-M materials.39
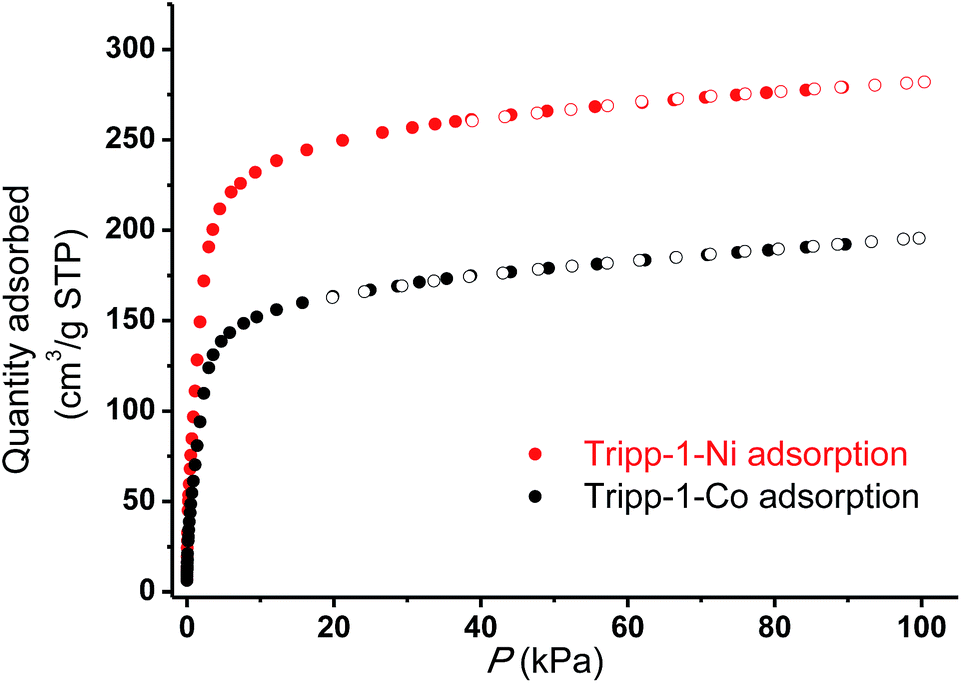 | ||
| Fig. 5 CO2 adsorption isotherms (filled symbols) and desorption (empty symbols) for Tripp-1-Co (black) and Tripp-1-Ni (red) conducted at 195 K. | ||
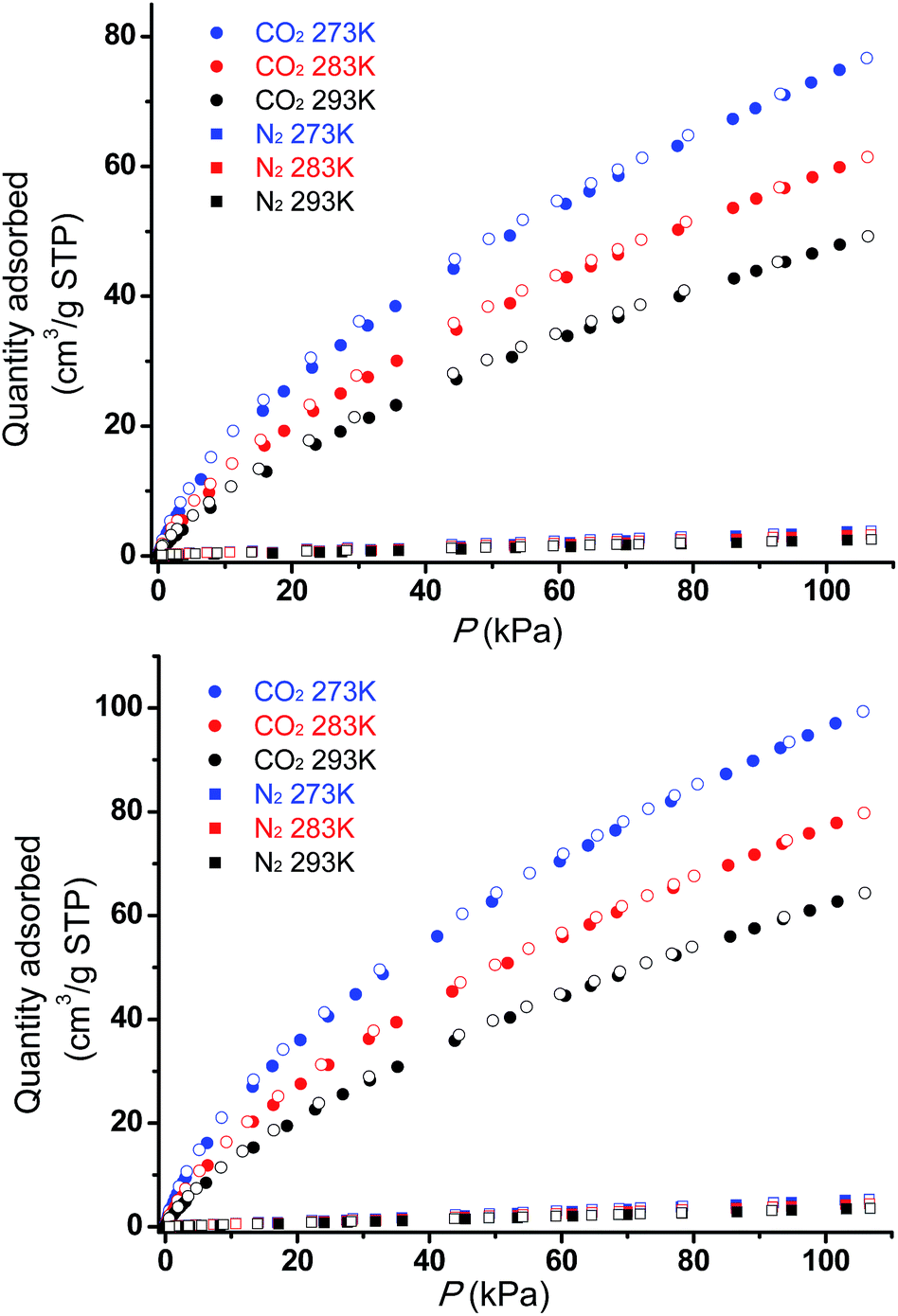 | ||
| Fig. 6 CO2 and N2 adsorption (filled symbols) and desorption (empty symbols) isotherms for Tripp-1-Co (top) and Tripp-1-Ni (bottom) at three temperatures, 273 K, 283 K and 293 K. | ||
To assess the strength of interaction between CO2 and framework, the CO2 isotherms measured at 273, 283 and 293 K were fitted using the virial equation (Fig. S9†), and the isosteric heat of adsorption (Qst) was calculated using the Clausius–Clapeyron equation. The enthalpies at zero loading for Tripp-1-Co and Tripp-1-Ni are 25.6 and 26.3 kJ mol−1, respectively (Fig. S10†). These values are also consistent with those observed in other classes of MOMs such as MOF-5 (34 kJ mol−1),40HKUST-1 (35 kJ mol−1),41MAF-25 (26 kJ mol−1),42InOF-1 (29 kJ mol−1),43NOTT-140 (25 kJ mol−1).44
Conclusions
In summary, we report a novel fluoride-bridged heptanuclear metal cluster-based SBB which has not been previously observed as a discrete structure. This cluster has 12-connection points, but 3-c Tripp ligands doubly cross-link to adjacent SBBs in order to form Tripp-1-M, two isostructural MOMs with binodal 3,6-connected pyr network topology. Good thermal and air/moisture stabilities were observed and gas sorption experiments demonstrate that both Tripp-1-Co and Tripp-1-Ni exhibit permanent porosity. The novel M7F122+ SBB reported herein has the potential to serve as an SBB for a wider range of MOMs with tailored pore sizes and surface chemistries. Follow-on studies on this platform will address properties related to gas sorption, catalysis and magnetism and are currently underway.Acknowledgements
The authors thank the Science Foundation Ireland for funding of this research (SFI Award 13/RP/B2549). We also thank Dr Yina Guo and Dr Fathima laffir for help with Energy-dispersive X-ray spectroscopy and X-ray photoelectron spectroscopy measurements, respectively.Notes and references
- J. J. Perry IV, J. A. Perman and M. J. Zaworotko, Chem. Soc. Rev., 2009, 38, 1400–1417 RSC.
- (a) L. R. MacGillivray, Metal-Organic Frameworks: Design and Application, Wiley&Sons, 2010 Search PubMed; (b) S. R. Batten, S. M. Neville and D. R. Turner, Coordination Polymers: Design, Analysis and Application, RSC publishing, 2009 Search PubMed; (c) A. J. Blake, N. R. Champness, P. Hubberstey, W.-S. Li, M. A. Withersby and M. Schröder, Coord. Chem. Rev., 1999, 183, 117–138 CrossRef CAS; (d) M. W. Hosseini, Acc. Chem. Res., 2005, 38, 313–323 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS.
- S. Ma, D. Sun, M. Ambrogio, J. A. Fillinger, S. Parkin and H.-C. Zhou, J. Am. Chem. Soc., 2007, 129, 1858–1859 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571–8572 CrossRef CAS.
- D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033–5036 CrossRef CAS PubMed.
- O. K. Farha, A. Özgür Yazaydın, I. Eryazici, C. D. Malliakas, B. G. Hauser, M. G. Kanatzidis, S. T. Nguyen, R. Q. Snurr and J. T. Hupp, Nat. Chem., 2010, 2, 944–948 CrossRef CAS PubMed.
- C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828–1831 CrossRef CAS PubMed.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040–2042 CrossRef PubMed.
- K. Wang, D. Feng, T.-F. Liu, J. Su, S. Yuan, Y.-P. Chen, M. Bosch, X. Zou and H.-C. Zhou, J. Am. Chem. Soc., 2014, 136, 13983–13986 CrossRef CAS PubMed.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- H. K. Chae, D. Y. Siberio-Perez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature, 2004, 427, 523–527 CrossRef CAS PubMed.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850–13851 CrossRef PubMed.
- G. J. McManus, Z. Wang and M. J. Zaworotko, Cryst. Growth Des., 2004, 4, 11–13 CAS.
- Y.-S. Wei, K.-J. Chen, P.-Q. Liao, B.-Y. Zhu, R.-B. Lin, H.-L. Zhou, B.-Y. Wang, W. Xue, J.-P. Zhang and X.-M. Chen, Chem. Sci., 2013, 4, 1539–1546 RSC.
- F. Nouar, J. F. Eubank, T. Bousquet, L. Wojtas, M. J. Zaworotko and M. Eddaoudi, J. Am. Chem. Soc., 2008, 130, 1833–1835 CrossRef CAS PubMed.
- D. Tian, Q. Chen, Y. Li, Y.-H. Zhang, Z. Chang and X.-H. Bu, Angew. Chem., Int. Ed., 2014, 53, 837–841 CrossRef CAS PubMed.
- (a) Z.-Z. Lu, R. Zhang, Y.-Z. Li, Z.-J. Guo and H.-G. Zheng, J. Am. Chem. Soc., 2011, 133, 4172–4174 CrossRef CAS PubMed; (b) M.-H. Zeng, Q.-X. Wang, Y.-X. Tan, S. Hu, H.-X. Zhao, L.-S. Long and M. Kurmoo, J. Am. Chem. Soc., 2010, 132, 2561–2563 CrossRef CAS PubMed.
- J. J. Perry IV, V. C. Kravtsov, G. J. McManus and M. J. Zaworotko, J. Am. Chem. Soc., 2007, 129, 10076–10077 CrossRef PubMed.
- S. Y. Yang, L. S. Long, Y. B. Jiang, R. B. Huang and L. S. Zheng, Chem. Mater., 2002, 14, 3229–3231 CrossRef CAS.
- (a) A. Schoedel, L. Wojtas, S. P. Kelley, R. D. Rogers, M. Eddaoudi and M. J. Zaworotko, Angew. Chem., Int. Ed., 2011, 50, 11421–11424 CrossRef CAS PubMed; (b) E. Y. Lee, S. Y. Jang and M. P. Suh, J. Am. Chem. Soc., 2005, 127, 6374–6381 CrossRef CAS PubMed.
- A. Schaate, P. Roy, T. Preuße, S. J. Lohmeier, A. Godt and P. Behrens, Chem.–Eur. J., 2011, 17, 9320–9325 CrossRef CAS PubMed.
- (a) M. Zhang, Y.-P. Chen, M. Bosch, T. Gentle, K. Wang, D. Feng, Z. U. Wang and H.-C. Zhou, Angew. Chem., Int. Ed., 2014, 53, 815–818 CrossRef CAS PubMed; (b) W. Morris, B. Volosskiy, S. Demir, F. Gándara, P. L. McGrier, H. Furukawa, D. Cascio, J. F. Stoddart and O. M. Yaghi, Inorg. Chem., 2012, 51, 6443–6445 CrossRef CAS PubMed; (c) J. E. Mondloch, W. Bury, D. Fairen-Jimenez, S. Kwon, E. J. DeMarco, M. H. Weston, A. A. Sarjeant, S. T. Nguyen, P. C. Stair, R. Q. Snurr, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 10294–10297 CrossRef CAS PubMed.
- K. M. Ok and D. O'Hare, Dalton Trans., 2008, 5560–5562 RSC.
- J. C. Jansen, H. Van Koningsveld and J. Reedijk, Nature, 1977, 269, 318–319 CrossRef CAS PubMed.
- M. Romanelli, G. A. Kumar, T. J. Emge, R. E. Riman and J. G. Brennan, Angew. Chem., Int. Ed., 2008, 47, 6049–6051 CrossRef CAS PubMed.
- (a) T. Birk, K. S. Pedersen, C. A. Thuesen, T. Weyhermüller, M. Schau-Magnussen, S. Piligkos, H. Weihe, S. Mossin, M. Evangelisti and J. Bendix, Inorg. Chem., 2012, 51, 5435–5443 CrossRef CAS PubMed; (b) G. A. Timco, S. Carretta, F. Troiani, F. Tuna, R. J. Pritchard, C. A. Muryn, E. J. L. McInnes, A. Ghirri, A. Candini, P. Santini, G. Amoretti, M. Affronte and R. E. P. Winpenny, Nat. Nanotechnol., 2009, 4, 173–178 CrossRef CAS PubMed.
- C. B. Smith, C. L. Raston and A. N. Sobolev, Green Chem., 2005, 7, 650–654 RSC.
- (a) X.-J. Li, F.-L. Jiang, M.-Y. Wu, L. Chen, J.-J. Qian, K. Zhou, D.-Q. Yuan and M.-C. Hong, Inorg. Chem., 2014, 53, 1032–1038 CrossRef CAS PubMed; (b) J. Lieffrig, O. Jeannin and M. Fourmigué, J. Am. Chem. Soc., 2013, 135, 6200–6210 CrossRef CAS PubMed; (c) J. J. Henkelis, S. A. Barnett, L. P. Harding and M. J. Hardie, Inorg. Chem., 2012, 51, 10657–10674 CrossRef CAS PubMed; (d) H. K. Chae, J. Kim, O. D. Friedrichs, M. O'Keeffe and O. M. Yaghi, Angew. Chem., Int. Ed., 2003, 42, 3907–3909 CrossRef CAS PubMed; (e) L. Xu, Y.-U. Kwon, B. de Castro and L. Cunha-Silva, Cryst. Growth Des., 2013, 13, 1260–1266 CrossRef CAS; (f) M. J. Manos, M. S. Markoulides, C. D. Malliakas, G. S. Papaefstathiou, N. Chronakis, M. G. Kanatzidis, P. N. Trikalitis and A. J. Tasiopoulos, Inorg. Chem., 2011, 50, 11297–11299 CrossRef CAS PubMed; (g) D. B. Cordes and L. R. Hanton, Inorg. Chem., 2007, 46, 1634–1644 CrossRef CAS PubMed; (h) B. Gao, S.-X. Liu, C.-D. Zhang, L.-H. Xie, C.-Y. Sun and M. Yu, J. Coord. Chem., 2007, 60, 911–918 CrossRef CAS PubMed; (i) M. Du, Z.-H. Zhang, L.-F. Tang, X.-G. Wang, X.-J. Zhao and S. R. Batten, Chem.–Eur. J., 2007, 13, 2578–2586 CrossRef CAS PubMed; (j) E. Lefebvre, F. Conan, N. Cosquer, J.-M. Kerbaol, M. Marchivie, J. Sala-Pala, M. M. Kubicki, E. Vigier and C. J. Gomez Garcia, New J. Chem., 2006, 30, 1197–1206 RSC; (k) B. Gao, S.-X. Liu, L.-H. Xie, M. Yu, C.-D. Zhang, C.-Y. Sun and H.-Y. Cheng, J. Solid State Chem., 2006, 179, 1681–1689 CrossRef CAS PubMed; (l) A. Mondal, G. Mostafa, A. Ghosh, I. Rahaman Laskar and N. Ray Chaudhuri, J. Chem. Soc., Dalton Trans., 1999, 9–10 RSC.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- (a) S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS PubMed; (b) S. R. Batten, B. F. Hoskins and R. Robson, Angew. Chem., Int. Ed., 1995, 34, 820–822 CrossRef PubMed.
- (a) X. Zhao, X. Bu, Q.-G. Zhai, H. Tran and P. Feng, J. Am. Chem. Soc., 2015, 137, 1396–1399 CrossRef CAS PubMed; (b) F.-Y. Yi, J. Zhang, H.-X. Zhang and Z.-M. Sun, Chem. Commun., 2012, 48, 10419–10421 RSC.
- (a) T. Osuga, T. Murase and M. Fujita, Angew. Chem., Int. Ed., 2012, 51, 12199–12201 CrossRef CAS PubMed; (b) Y. Inokuma, S. Yoshioka, J. Ariyoshi, T. Arai, Y. Hitora, K. Takada, S. Matsunaga, K. Rissanen and M. Fujita, Nature, 2013, 495, 461–466 CrossRef CAS PubMed; (c) S. Horiuchi, T. Murase and M. Fujita, Chem.–Asian J., 2011, 6, 1839–1847 CrossRef CAS PubMed.
- W. Ouellette, K. Darling, A. Prosvirin, K. Whitenack, K. R. Dunbar and J. Zubieta, Dalton Trans., 2011, 12288–12300 RSC.
- S. A. Kumalah Robinson, M.-V. L. Mempin, A. J. Cairns and K. T. Holman, J. Am. Chem. Soc., 2011, 133, 1634–1637 CrossRef CAS PubMed.
- S. C. Lee and R. H. Holm, Inorg. Chem., 1993, 32, 4745–4753 CrossRef CAS.
- (a) K. B. Yatsimirskii, Pure Appl. Chem., 1977, 49, 115 CrossRef CAS; (b) Y.-Q. Chen, G.-R. Li, Z. Chang, Y.-K. Qu, Y.-H. Zhang and X.-H. Bu, Chem. Sci., 2013, 4, 3678–3682 RSC.
- (a) M. H. Mohamed, S. K. Elsaidi, L. Wojtas, T. Pham, K. A. Forrest, B. Tudor, B. Space and M. J. Zaworotko, J. Am. Chem. Soc., 2012, 134, 19556–19559 CrossRef CAS PubMed; (b) P. S. Nugent, V. L. Rhodus, T. Pham, K. Forrest, L. Wojtas, B. Space and M. J. Zaworotko, J. Am. Chem. Soc., 2013, 135, 10950–10953 CrossRef CAS PubMed; (c) B. Arstad, H. Fjellvåg, K. Kongshaug, O. Swang and R. Blom, Adsorption, 2008, 14, 755–762 CrossRef CAS; (d) J. Kim, S.-T. Yang, S. B. Choi, J. Sim, J. Kim and W.-S. Ahn, J. Mater. Chem., 2011, 21, 3070–3076 RSC; (e) R. Vaidhyanathan, S. S. Iremonger, K. W. Dawson and G. K. H. Shimizu, Chem. Commun., 2009, 5230–5232 RSC; (f) K.-J. Chen, R.-B. Lin, P.-Q. Liao, C.-T. He, J.-B. Lin, W. Xue, Y.-B. Zhang, J.-P. Zhang and X.-M. Chen, Cryst. Growth Des., 2013, 13, 2118–2123 CrossRef CAS.
- Z. Zhao, Z. Li and Y. S. Lin, Ind. Eng. Chem. Res., 2009, 48, 10015–10020 CrossRef CAS.
- Q.-M. Wang, D. Shen, M. Bülow, M. Ling Lau, S. Deng, F. R. Fitch, N. O. Lemcoff and J. Semanscin, Microporous Mesoporous Mater., 2002, 55, 217–230 CrossRef CAS.
- J.-B. Lin, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2010, 132, 6654–6656 CrossRef CAS PubMed.
- J. Qian, F. Jiang, D. Yuan, M. Wu, S. Zhang, L. Zhang and M. Hong, Chem. Commun., 2012, 48, 9696–9698 RSC.
- C. Tan, S. Yang, N. R. Champness, X. Lin, A. J. Blake, W. Lewis and M. Schröder, Chem. Commun., 2011, 47, 4487–4489 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, single-crystal XRD data, PXRD patterns, TGA curves, sorption data fit and Qst plot. CCDC 1060661. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01515d |
| This journal is © The Royal Society of Chemistry 2015 |