
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Stereoselective synthesis of macrocyclic peptides via a dual olefin metathesis and ethenolysis approach†
Shane L.
Mangold
and
Robert H.
Grubbs
*
Arnold and Mabel Beckman Laboratories of Chemical Synthesis, Division of Chemistry and Chemical Engineering, California Institute of Technology, Pasadena, California 91125, USA. E-mail: rhg@caltech.edu; Fax: +1-626-564-9297
First published on 21st May 2015
Abstract
Macrocyclic compounds occupy an important chemical space between small molecules and biologics and are prevalent in many natural products and pharmaceuticals. The growing interest in macrocycles has been fueled, in part, by the design of novel synthetic methods to these compounds. One appealing strategy is ring-closing metathesis (RCM) that seeks to construct macrocycles from acyclic diene precursors using defined transition-metal alkylidene catalysts. Despite its broad utility, RCM generally gives rise to a mixture of E- and Z-olefin isomers that can hinder efforts for the large-scale production and isolation of such complex molecules. To address this issue, we aimed to develop methods that can selectively enrich macrocycles in E- or Z-olefin isomers using an RCM/ethenolysis strategy. The utility of this methodology was demonstrated in the stereoselective formation of macrocyclic peptides, a class of compounds that have gained prominence as therapeutics in drug discovery. Herein, we report an assessment of various factors that promote catalyst-directed RCM and ethenolysis on a variety of peptide substrates by varying the olefin type, peptide sequence, and placement of the olefin in macrocycle formation. These methods allow for control over olefin geometry in peptides, facilitating their isolation and characterization. The studies outlined in this report seek to expand the scope of stereoselective olefin metathesis in general RCM.
Introduction
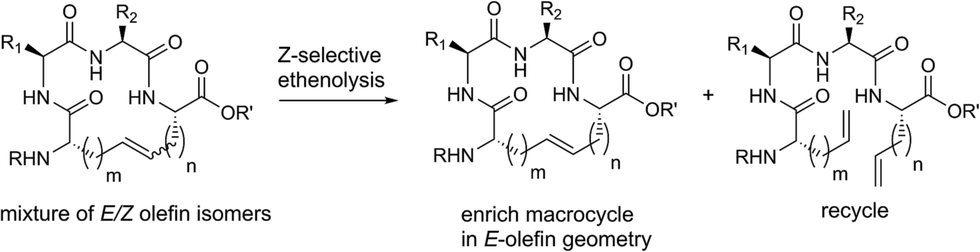
The emergence of chemical strategies for accessing macrocyclic motifs has fostered a renewed interest in their development and macrocycles now fulfill roles in diverse applications from natural products1,2 and therapeutics3,4 to platforms in supramolecular chemistry.5 Contemporary strategies for macrocycle formation often rely on the use of macrolactonization,6–8 macrolactamization,9–11 “click” cyclization,12–14 or transition-metal catalyzed reactions including olefin metathesis15,16 and intramolecular cross coupling.1,17,18 Among these strategies, ring-closing metathesis (RCM) has assumed a prominent role in macrocycle formation, in part, as a consequence of the selectivity and functional group compatibility of select olefin metathesis catalysts.19,20 Such chemoselectivity has offered new strategies for retrosynthetic disconnections in complex molecule synthesis and many active pharmaceuticals have been developed around the use of RCM.21,22 One promising application of RCM involves macrocyclization on peptides, often conferring beneficial properties to these compounds including enhanced activity23–25 and improved proteolytic stability.26,27 While RCM has found utility across many disciplines, an outstanding challenge in this transformation has been the ability to control olefin geometry in the product. Although indirect methods have been developed, including alkyne metathesis followed by partial reduction28–31 or substrate-controlled RCM of vinylsiloxanes followed by desilylation,32,33 the scope of these transformations is limited. We envisioned that a more streamlined route could be devised by modulating the equilibrium of olefin metathesis with control over both RCM and the reverse, ring-opening metathesis (ROM) using ethylene and olefin selective metathesis catalysts (Fig. 1).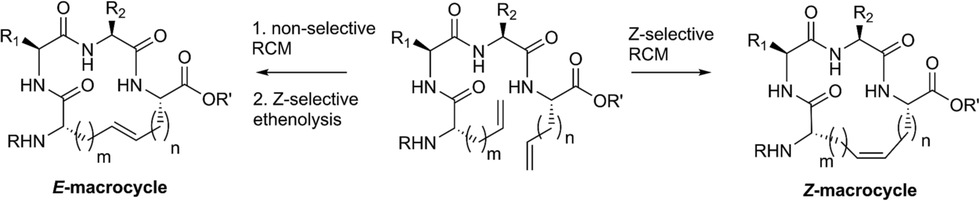 | ||
| Fig. 1 A strategy for controlling olefin geometry in macrocyclic peptides using catalyst-directed RCM and ethenolysis. | ||
We recently explored the use of Z-selective cyclometalated ruthenium catalysts for the derivatization of commodity chemical feedstocks using catalyst-controlled ethenolysis.34,35 These studies led us to consider whether Z-selective ethenolysis could serve as a practical tool for the purification of E-olefins from stereoisomeric mixtures of E- and Z-olefins in complex substrates bearing multiple functionality. Such a strategy could have value in the synthesis and isolation of natural products, peptides, and pharmaceuticals as even small amounts of stereoisomeric impurities can affect their physical or biological properties. As such, we sought to employ a dual RCM/ethenolysis strategy as a means to control olefin geometry in macrocycles. As a rigorous test of our methodology, we focused on the generation of macrocyclic peptides, a class of compounds that are traditionally difficult substrates to synthesize and isolate with defined olefin geometry.1,15 Herein, we provide detailed comparative studies of a variety of ruthenium catalysts in promoting RCM on peptides and assess the role of catalyst structure in controlling the stereoselectivity of RCM. Moreover, through the combined efforts of RCM and catalyst-directed ethenolysis, we offer methods for the selective formation of E- or Z-olefin geometry within macrocyclic peptides.
Results and discussion
Diastereoselective RCM on macrocyclic peptides bearing i, i + 2 olefin crosslinks
Despite the therapeutic potential of macrocyclic peptides, they represent a relatively underdeveloped class of compounds due, in part, to their complex structures and limited methods for their synthesis.36–38 We sought to apply RCM as a strategy to streamline the synthesis of cyclic peptides and to investigate the influence of olefin type, position, and size of the macrocycle on the efficiency and stereoselectivity of RCM. Moreover, we aimed to provide detailed comparative studies of a variety of ruthenium catalysts in promoting RCM on peptides (Fig. 2).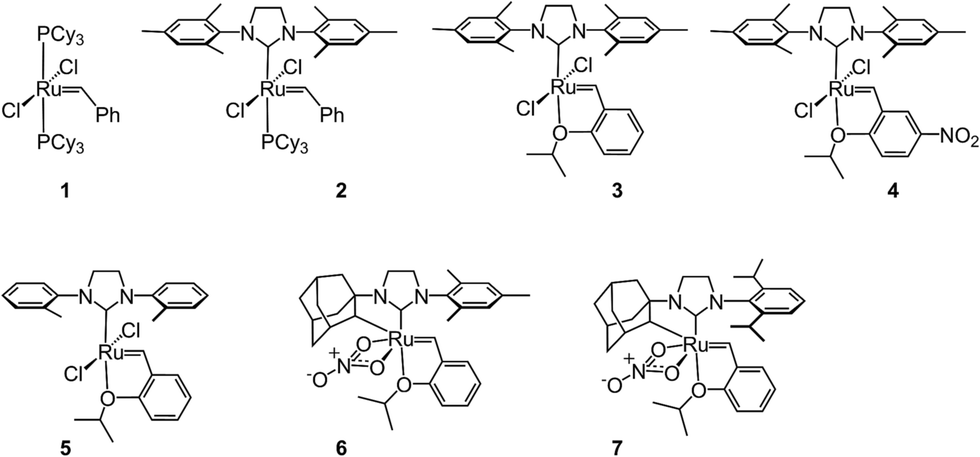 | ||
| Fig. 2 A survey of ruthenium catalysts used to promote RCM on macrocyclic peptides. | ||
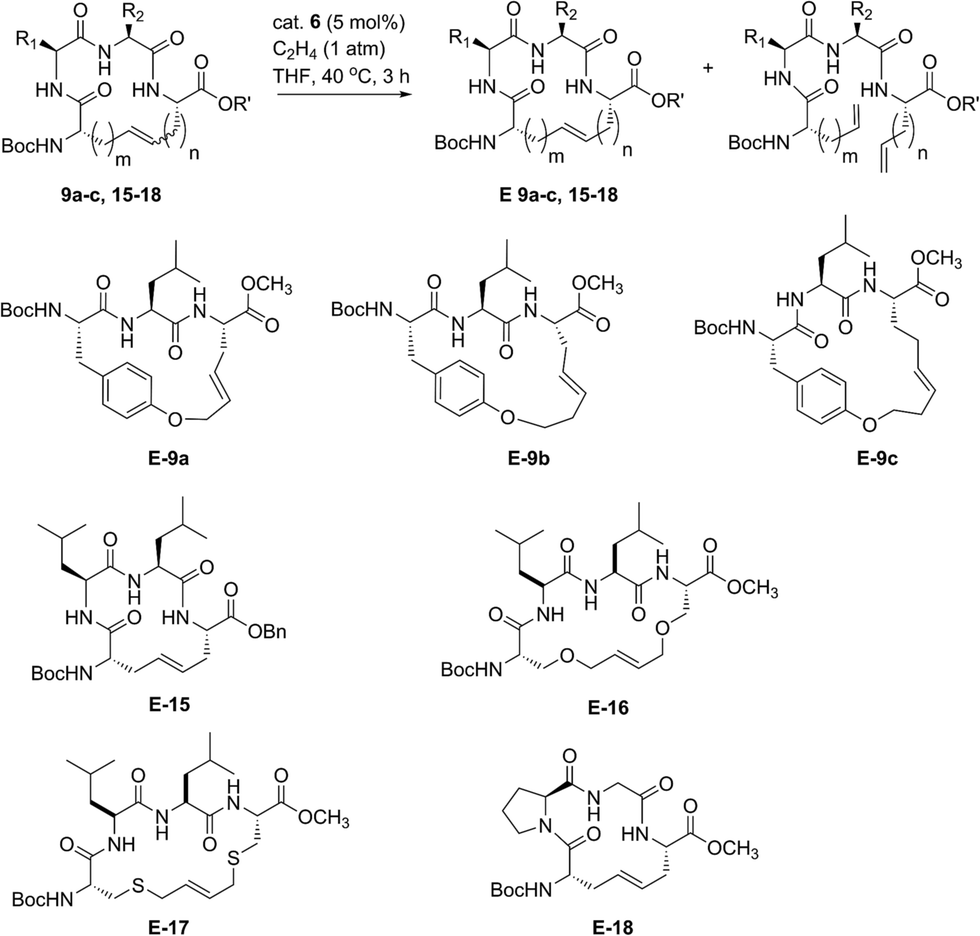
Our initial studies began with the optimization of RCM on dienes 8a–c that contain olefins spanning across i, i + 2 residues using ruthenium catalysts 1–5 and Z-selective cyclometalated catalysts 6 and 7 (Table 1). Using this strategy, we could assess the intrinsic E/Z stereoselectivity of each catalyst in macrocycle formation and gain an understanding of the relative activity of each catalyst to promote RCM. Exposing diene 8a to the first-generation ruthenium catalyst 1 under dilute conditions to promote macrocycle formation afforded the RCM product 9a in 58% yield and with 80% selectivity for the E-olefin isomer. The use of the more active second-generation catalyst 2 afforded 9a in 71% yield and 90% E-selectivity. We next examined the use of chelated isopropoxy catalysts in RCM. Exposing diene 8a to catalyst 3 afforded macrocycle 9a in 63% yield and with 90% E-selectivity. Comparable yields and diastereoselectivities were observed in the presence of the faster initiating catalyst 4, affording 9a in 66% yield and 82% E-selectivity.39 We also explored the use of catalysts bearing less sterically encumbering substituents around the ruthenium center (i.e., tolyl catalyst 5 (ref. 40)) but conversions to 9a were typically low, affording the product in 45% yield.41 The use of Z-selective cyclometalated catalysts 6 and 7 afforded 9a in 47% and 41% yield, respectively. Notably, the olefin selectivity could be reversed to afford macrocycles highly enriched in the Z-olefin isomer.
|
|
||||||
|---|---|---|---|---|---|---|
| Catalyst | Yielda (%) | Selectivityb (E![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Z) :Z) |
||||
| 9a | 9b | 9c | 9a | 9b | 9c | |
| m = 1, n = 1 | m = 2, n = 1 | m = 2, n = 2 | m = 1, n = 1 | m = 2, n = 1 | m = 2, n = 2 | |
| a Isolated yields. b Determined by 1H and 13C NMR spectroscopy. | ||||||
| 1 | 58 | 54 | 68 | 81:19 |
72:28 |
66:34 |
| 2 | 71 | 61 | 77 | 91:9 |
88:12 |
82:18 |
| 3 | 63 | 55 | 70 | 90:10 |
85:15 |
83:17 |
| 4 | 66 | 60 | 66 | 82:18 |
85:15 |
78:22 |
| 5 | 45 | 24 | 44 | 81:19 |
n.d. | 81:19 |
| 6 | 47 | 21 | 26 | 13:87 |
n.d. | 15:85 |
| 7 | 41 | 17 | 24 | 7:93 |
n.d. | 5:95 |
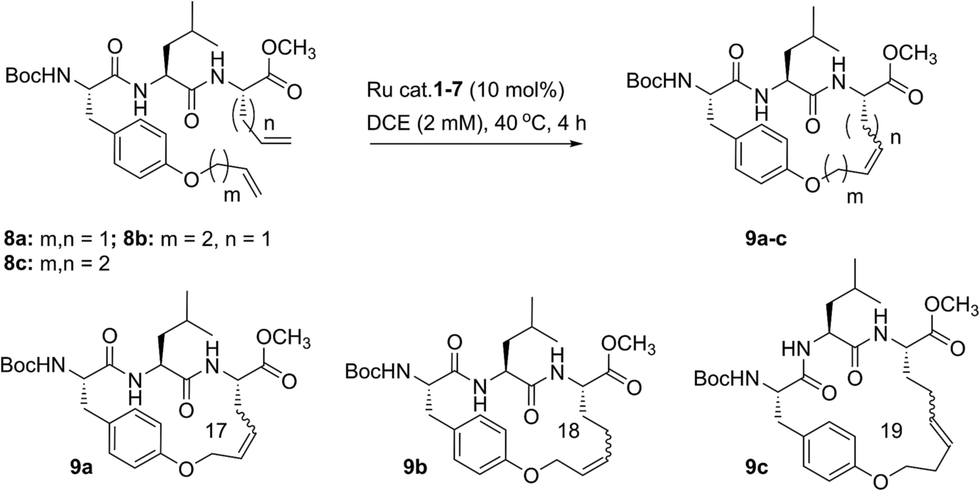
To probe the influence of macrocycle size on the stereoselectivity of RCM, we incorporated an additional methylene unit into one (i.e., peptide 8b) or both (8c) positions of the olefin-bearing amino acids. We anticipated that such modifications might influence the E/Z ratio of olefin geometry in the product due to the varying ring sizes that form upon macrocyclization.42 Moreover, we could determine if the identity of the olefin (i.e., allylic or homoallylic) had any influence on the efficiency of RCM using catalysts 1–7. Exposing substrate 8b to catalysts 1–5 resulted in variable yields and E/Z ratios for the formation of macrocycle 9b, from 54% yield and 70% diastereoselectivity for catalyst 1 to 24% yield and 80% E-selectivity for catalyst 5. The cyclometalated ruthenium catalysts 6 and 7 were less active than catalysts 1–5 in RCM of 8b, with conversions below 25% for the formation of 9b. Interestingly, formation of the 18-membered macrocycle 9b was consistently lower than formation of 9a,43 and this finding prompted us to examine the structurally analogous peptide 8c, bearing an additional methylene that would give rise to the 19-membered macrocycle 9c upon RCM. In general, the conversion of peptide 8c to macrocycle 9c (66–77%) was improved relative to the conversion of 8b to macrocycle 9b (50–60%). In the presence of the first-generation catalyst 1, the selectivity for the E-isomer was lower for 9c (66%) compared to 9a (81%) and 9b (77%) and this trend was consistent with catalyst 2 in RCM. As with macrocycles 9a and 9b, catalysts 3 and 4 were more active than catalyst 5 in RCM, affording the desired macrocycle 9c in 70% yield and 80% E-olefin selectivity as compared to 44% yield and 80% selectivity in the presence of catalyst 5. Interestingly, the propensity of macrocycles 9b and 9c to form with greater Z-selectivity relative to 9a using non-selective catalysts 1–5 did not facilitate RCM in the presence of Z-selective catalysts 6 and 7 as shown by the comparatively low yields of 9b (21%) and 9c (26%) to 9a (41%) with 6 or 7. This finding is consistent with previous reports suggesting that cyclometalated ruthenium catalysts, in some cases, cannot overcome any substrate bias that may favor the formation of Z-olefin geometry during metathesis.44 These studies provide evidence that subtle variations of catalyst structure and macrocycle size can greatly impact the yield and diastereoselectivity of RCM on olefin-bearing peptides. For macrocycles bearing homoallylic olefin tethers (i.e., 9c), the E/Z diastereoselectivity of RCM was generally lower than for dienes consisting of allylic olefin tethers (i.e., 9a). In this regard, macrocyclization of 9c was improved relative to 9a, mostly notably in the presence of phosphine-containing catalysts 1 and 2.
The influence of heteroatoms and peptide sequence in stereoselective RCM on peptides bearing i, i + 3 olefin crosslinks
Our studies regarding the activity of catalysts 1–7 in RCM on substrates 8a–c suggests that the size of the macrocycle can influence E/Z diastereoselectivity. To explore this further, we synthesized peptides bearing an additional amino acid between olefin crosslinks. This would enable access to additional cyclic structures and provide insight into the effect of varying the position of olefin-containing amino acids along the peptide in RCM. Moreover, we could investigate a larger variety of amino acids, including those bearing allylic heteroatoms in the side chain, in macrocyclic ring closure. These studies were motivated by our observation that both the peptide sequence and identity of the olefin can profoundly affect the efficiency of metathesis in homodimerization and cross metathesis on peptides and we wondered if these trends would extend to RCM.44We first evaluated the influence of allylic heteroatoms in facilitating RCM and their influence on E/Z diastereoselectivity. Exposing the allyl-modified peptide 10 to our optimized reaction conditions afforded macrocycle 15 in 70% yield with 74% E-selectivity using catalyst 1 (Table 2). By comparison, the O-allyl serine (11) and S-allyl cysteine (12) modified peptides gave the desired macrocycles 16 and 17 in 73% and 77% yield and with 92% and 90% E-selectivity, respectively.45 These trends were observed in the presence of the second-generation ruthenium catalyst 2; in this instance, the formation of macrocycle 15 could be obtained in 74% yield, as compared to macrocycles 16 (76%) and 17 (78%) in RCM. We next exposed peptides 10–12 to catalysts 3 and 4. The O-allyl modified peptide 11 was converted to macrocycle 16 in 73% yield, compared to 61% yield for the conversion of allyl peptide 10 to 15 using catalyst 3. Similar yields and selectivities were observed with catalyst 4, whereby peptide 11 afforded slightly higher yields of the RCM product 16 (74%) relative to the conversion of 10 to 15 (64%). We next exposed the allyl cysteine-modified peptide 12 to RCM. The formation of macrocycle 17 was generally improved relative to 15 or 16, occurring in 80% yield and with 90% E-selectivity using catalysts 1–3. As observed with substrates 8a–c, the use of tolyl catalyst 5 under the RCM conditions led to lower conversions to macrocycles 15–17 (39–44%). By comparison, the use of Z-selective catalyst 6 and 7 in RCM resulted in yields ranging from 32–40% for formation of 15–17 but with reversal of olefin selectivity. Unlike with the use of isopropoxy catalysts 3 and 4 in RCM, an absence of a pronounced heteroatom effect was observed with cyclometalated catalysts 6 and 7 that may be attributable to their comparatively lower reactivity in RCM. These studies imply that the identity of the olefin can influence the efficiency and diastereoselectivity of RCM.
|
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Yielda (%) | Selectivityb (E:Z) |
||||||||
| 15 | 16 | 17 | 18 | 19 | 15 | 16 | 17 | 18 | 19 | |
| a Isolated yield. b Determined by 1H and 13C NMR spectroscopy. | ||||||||||
| 1 | 70 | 73 | 77 | 37 | 14 | 74:27 |
92:8 |
90:10 |
77:23 |
80:20 |
| 2 | 74 | 76 | 81 | 48 | 18 | 90:10 |
93:7 |
92:8 |
91:9 |
89:11 |
| 3 | 61 | 73 | 79 | 41 | <10 | 86:14 |
90:10 |
90:10 |
80:20 |
n.d. |
| 4 | 64 | 74 | 83 | 36 | 12 | 82:18 |
81:19 |
85:15 |
82:18 |
n.d. |
| 5 | 39 | 44 | 42 | 20 | <5 | 85:15 |
90:10 |
91:9 |
81:19 |
n.d. |
| 6 | 32 | 36 | 40 | 17 | <5 | 12:88 |
16:84 |
13:87 |
n.d. | n.d. |
| 7 | 30 | 34 | 33 | 18 | <5 | 10:90 |
9:91 |
5:95 |
n.d. | n.d. |
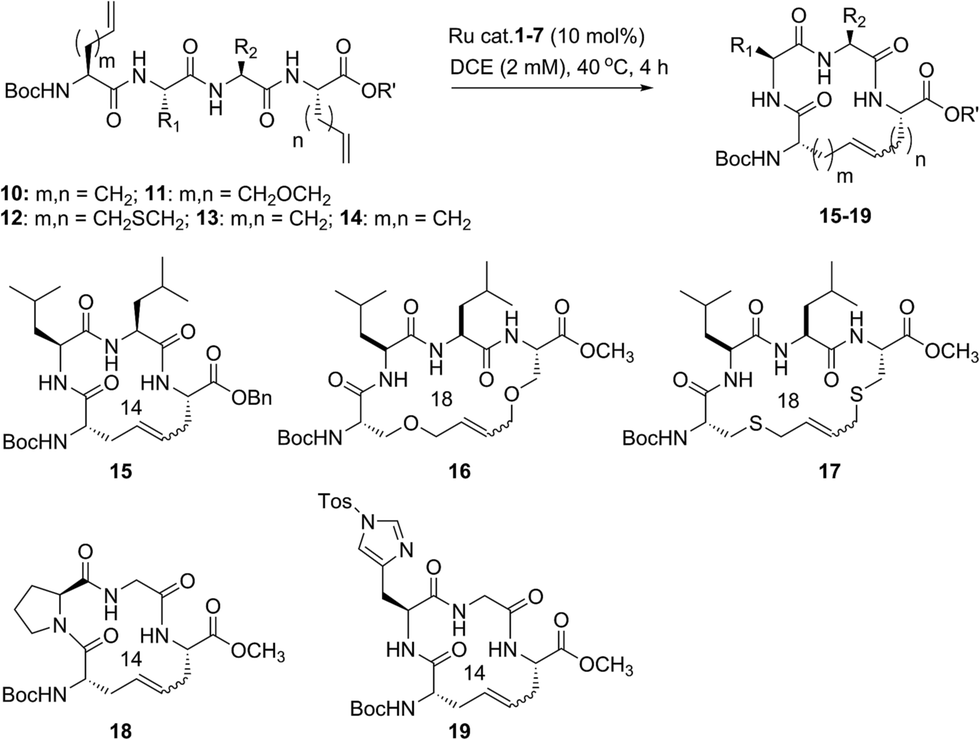
We next examined peptides of varying amino acid sequence in RCM. Our previous work regarding the activity of catalysts 6 and 7 in homodimerization and cross metathesis revealed that a subset of olefin-bearing amino acids had a deactivating effect on olefin metathesis.44 Specifically glycine, proline, and histidine were shown to be unreactive in homodimerization and cross metathesis. We set out to discover if such amino acids generally inhibit metathesis by using a broader range of catalysts and whether incorporation of these amino acids within a larger peptide could override their apparent inactivity. To test this, we generated peptide 13 containing the amino acids proline and glycine at positions along the peptide proximal to the olefin-bearing amino acids. In the presence of catalysts 6 and 7, conversions of 13 to 18 were less than 20%. For comparison, we examined catalysts 1–5 in RCM on diene 13. Yields to the corresponding macrocycle 18 were variable, ranging from 20% in the presence of catalyst 5 to 48% with catalyst 2. For those catalysts that could achieve reasonable yields of 22, the selectivity was above 80% in favor of the E-olefin isomer. As a further test, we synthesized peptide 14 bearing histidine in place of proline and evaluated its activity in RCM. Conversions of 14 to macrocycle 19 were consistently below 25% in the presence of catalysts 1–7. Under these conditions, formation of the 14-membered ring may be hindered by the proximity of histidine,46 which has been shown to have a deactivating effect on metathesis activity.47 Taken together, such results point to the importance of olefin identity, peptide sequence, and catalyst structure in controlling both the efficiency and diastereoselectivity of RCM on macrocyclic peptides. For those peptides bearing i, i + 3 olefin crosslinks, incorporation of allylic heteroatoms into the amino acid side-chain generally favored RCM, most notably in the presence of isopropoxy catalysts 3 and 4 and to a lesser extent with phosphine containing catalysts 1 and 2 and cyclometalated ruthenium catalysts 6 and 7. These observations reflect the importance of directly comparing various catalyst structures in RCM and seek to guide further strategies for optimizing olefin metathesis on peptide-containing substrates.
Z-Selective ethenolysis for the enrichment of macrocyclic peptides in E-olefin geometry
Encouraged by the success of RCM on a variety of peptide substrates, we next evaluated strategies to transform macrocyclic peptides having a mixture of olefin isomers into those bearing a single olefin isomer. Such a strategy could facilitate the isolation and characterization of olefin-containing macrocycles and offer a means to more easily investigate the influence of olefin geometry on the stability, activity, and conformation of this important class of compounds.We envisioned an olefin enrichment strategy using a catalyst-controlled ethenolysis pathway (Fig. 3). This approach capitalizes on the inherent reversibility of olefin metathesis by using ethylene to drive ring-opening metathesis.48,49 By having a catalyst that is selective for the formation of one olefin isomer (e.g., catalysts 6 and 7) it should be possible to selectivity degrade olefin isomers from the corresponding mixtures. We imagined that this strategy could serve as a valuable tool to form cyclic peptides having a single olefin isomer which, to date, has been a synthetic challenge using olefin metathesis.
 | ||
| Fig. 3 Catalyst-controlled ethenolysis as a strategy to selectively enrich olefin geometry within macrocyclic peptides. | ||
For our initial studies, we examined catalyst 6 in Z-selective ethenolysis using macrocycles bearing i, i + 2 or i, i + 3 crosslinks and having variable ratios of olefin isomers. In this way, we could determine if the E/Z ratio in macrocyclic peptides affect the efficiency of ethenolysis. Under our optimized ethenolysis conditions, nearly complete Z-degradation of substrate 9a occurred in the presence of ethylene (1 atm) and catalyst 6 (5 mol%), affording enrichment of 9a in the E-olefin isomer in greater than 98% (entry 1, Table 3).50 More significantly, the ethenolysis conditions were able to transform macrocycle 9b from a 80:20 mixture of isomers to those bearing almost exclusive formation of the E-isomer (entry 2). To test the generality of the method, these conditions were applied to macrocyclic peptides 9c and 15–18. Complete consumption of the Z-isomer was observed for 9c and 15–17, affording the pure E macrocycles with enrichment above 98% (entries 3–6). A notable exception was compound 18 that resulted in comparatively low enrichment (entry 7).51 From these studies, the efficiency of Z-selective ethenolysis does not appear to be greatly influenced by the initial E/Z ratio of olefins in macrocycles 9 and 15–18. For those peptides that underwent efficient ethenolysis, the resulting starting material could be recovered and resubjected to the RCM conditions, providing a method for increasing the overall yield of product through iterative RCM/ethenolysis metathesis events. These results suggest that catalyst-controlled ethenolysis can serve as a practical method for the selective formation of E-olefins in macrocyclic peptides. This strategy, when coupled to Z-selective RCM can afford macrocycles predominantly enriched in E or Z olefin geometry.
RCM of resin-bound α-helical peptides bearing i, i + 4 and i, i + 7 crosslinks
The investigation of macrocyclic ring closure on peptides containing i, i + 2 or i, i + 3 olefinic crosslinks revealed that the peptide sequence and olefin identity can influence the efficiency and diastereoselectivity of RCM. Moreover, macrocycles consisting of a mixture of olefin isomers could be enriched in E-olefin geometry using Z-selective ethenolysis and that the initial E/Z ratio did not appear to influence the efficiency of ethenolysis. We sought to further expand these studies to α-helical peptides bearing i, i + 4 or i, i + 7 olefinic tethers. Such compounds, often referred to as stapled peptides,52–54 have gained attention as potential therapeutics in a variety of areas including cancer, infectious disease, and metabolism.25–27,55To date, most strategies for macrocyclization on α-helical peptides rely on RCM and subsequent hydrogenation to generate a fully saturated hydrocarbon tether along the peptide helix.55,56 In this sense, little attention has been focused on examining the role of olefin geometry on the conformation or biological activity of macrocyclic peptides. We wanted to explore if the RCM/ethenolysis reaction manifold could provide a method to synthesize stapled peptides with defined olefin geometry. Moreover, we sought to extend the methodology to peptides on resin for enabling a more streamlined and high-throughput method of peptide synthesis, identification, and purification. In choosing the peptide sequences, we were attracted to those having a variety of amino acids and olefin crosslinks and our studies began with the optimization of RCM on peptides bearing i, i + 4 crosslinks that afford a 21-membered macrocycle (Table 4). We first evaluated RCM on resin-bound peptide 20,55 using the first-generation catalyst 1. After extensive optimization, conversions to the desired cyclic peptide 26 could be achieved in 94% conversion and with 66% E-selectivity. By comparison, the second-generation catalyst 2 afforded 26 in 97% conversion and 80% E-selectivity under the same reaction conditions. Exposing 20 to catalysts 3 and 4 led to conversions of 84% and 88%, respectively. As observed with other olefin-containing peptides, the tolyl catalyst 5 was less active in RCM, with 75% conversion of 20 to 26. Applying the cyclometalated catalysts 6 and 7 to the RCM conditions afforded 26 in 83% and 81% conversion, respectively. In these instances, the selectivity was in favor of the Z-olefin isomer. We also examined peptides containing the amino acids proline44 and histidine57 that were shown to reduce the efficiency of RCM on peptides bearing i, i + 3 olefin crosslinks (i.e., peptides 13 and 14). We reasoned that incorporating olefin tethers at the i, i + 4 positions might facilitate RCM on-resin by serving to preorganize the reactive side chains on the same face of the α-helix. Such preorganization of the olefins, in addition to expanding the size of the macrocycle, might favor RCM over competing deactivation by amino acid side chains. To test this, we synthesized peptides containing proline and histidine at positions distal from the olefin crosslinks (i.e., peptide 21) or proximal to the crosslinks (22) and examined their activity in RCM. Exposing peptide 21 to catalysts 1 and 2 led to nearly full conversion (90%) of 21 to macrocycle 27. The use of catalysts 3 and 4 in RCM of 21 resulted in slightly lower conversion (80%) and with 75% E-selectivity. Exposing 21 to catalyst 5 afforded macrocycle 27 in 70% conversion, comparable to that of catalysts 6 and 7 (75%). To probe further the role of amino acid sequence in RCM of peptides bearing i, i + 4 crosslinks, we exposed the histidine-containing peptide 22 to similar reaction conditions. In the presence of catalysts 1–4 conversions to macrocycle 28 ranged from 80% with catalyst 4 to 90% in the presence of catalyst 1. For these cases, the diastereoselectivity of macrocycle formation ranged from 62% in the presence of 1 to 80% E-selectivity in the presence of catalysts 2–4. A slight decrease in conversion to 28 was seen in the presence of catalysts 6 and 7 but with reversal of olefin selectivity. These results imply that the efficiencies of RCM on resin-bound peptides 26–28 are comparable, even for wide variation in peptide sequence.
|
|
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Catalyst | Conversiona (%) | Selectivitya (E:Z) |
||||||||||
| 26 | 27 | 28 | 29 | 30 | 31 | 26 | 27 | 28 | 29 | 30 | 31 | |
| m, n = 1 | m, n = 1 | m, n = 1 | m, n = 4 | m, n = 4 | m, n = 4 | m, n = 1 | m, n = 1 | m, n = 1 | m, n = 4 | m, n = 4 | m, n = 4 | |
| a Determined by analytical HPLC-MS. | ||||||||||||
| 1 | 94 | 90 | 93 | 88 | 96 | 92 | 66:23 |
64:36 |
70:30 |
58:42 |
62:38 |
65:35 |
| 2 | 97 | 90 | 85 | 94 | 95 | 90 | 80:20 |
75:25 |
83:17 |
80:20 |
75:25 |
79:21 |
| 3 | 84 | 81 | 88 | 80 | 84 | 85 | 80:20 |
75:25 |
74:26 |
78:22 |
78:22 |
80:20 |
| 4 | 88 | 80 | 92 | 85 | 88 | 85 | 72:28 |
74:26 |
72:28 |
71:29 |
81:19 |
81:19 |
| 5 | 76 | 70 | 70 | 60 | 84 | 80 | 66:33 |
70:30 |
74:26 |
n.d. | 71:29 |
74:26 |
| 6 | 83 | 75 | 80 | 70 | 80 | 75 | 20:80 |
22:78 |
18:82 |
20:80 |
21:79 |
17:83 |
| 7 | 81 | 75 | 70 | 55 | 75 | 70 | 17:83 |
19:81 |
23:77 |
n.d. | 18:82 |
20:80 |
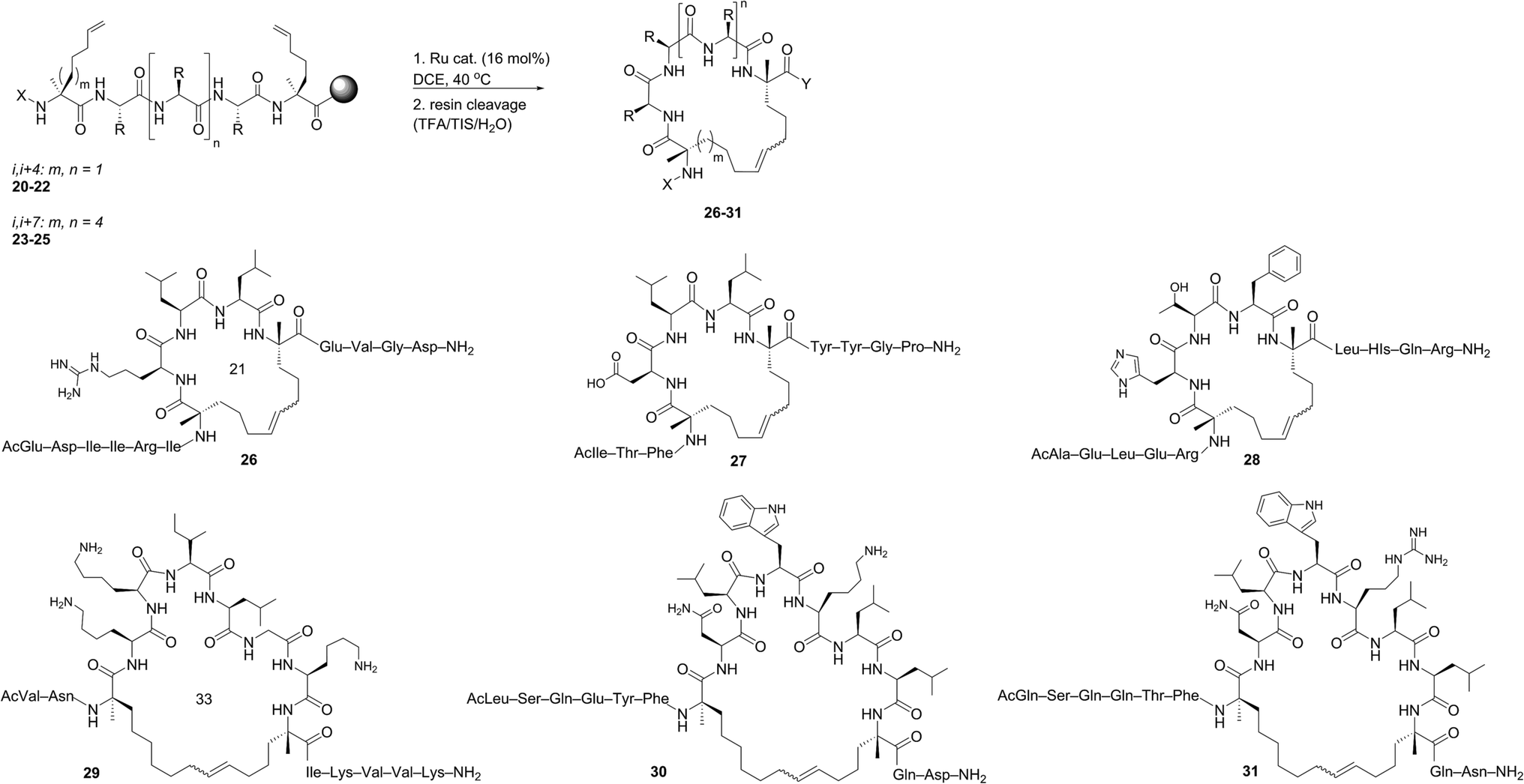
We next evaluated α-helical peptides bearing i, i + 7 olefin crosslinks for the formation of 33-membered macrocycles, as our goal was to compare the influence of macrocycle size on olefin diastereoselectivity in RCM for resin-bound peptides. For our initial studies, we monitored the conversion of peptide 23 to macrocycle 29 (ref. 24) using the first- and second-generation ruthenium catalysts 1 and 2. The conversion to macrocycle 29 was achieved in 88% in the presence of 1 and 94% with the use of catalyst 2, respectively. Under these conditions, the selectivity of the E-olefin was only 58% in the presence of 1 but increased to 80% in the presence of catalyst 2. The use of isopropoxy catalysts 3 and 4 afforded 29 in 3:1 E:Z selectivity at 80% conversion, trends that were similar to peptides bearing i, i + 4 crosslinks. By comparison, the conversions were typically lower in the presence of catalyst 5 (80%), 6 (83%) and 7 (81%). As observed in the formation of macrocycles bearing i, i + 2 or i, i + 4 crosslinks, the ability to form macrocycles with greater Z-olefin content using non-selective catalysts 1–5 did not facilitate RCM in the presence of Z-selective catalysts 6 and 7. As further validation, we examined the use of RCM for formation of macrocycles 30 and 31. Conversions of 24 (ref. 58) to 30 reached a maximum of 95% with catalyst 1 with slightly lower conversions in the presence of 2 (90%), 3 (84%) or 4 (86%). In these cases, the selectivity ranged from 60% with 1 to 80% in favor of the E-isomer with the use of catalyst 4. These trends were observed in the formation of macrocycle 31 using catalysts 1–4, with conversions greater than 85% and comparable diastereoselectivity. The use of catalysts 5–7 in macrocyclization of 25 (ref. 44) afforded the desired cyclic peptide 31 with slightly lower conversions (60–70%) relative to the formation of macrocycle 30 (75–80%). These comparative studies suggest that increasing the macrocycle size and/or preorganizing the olefins on the same face of the α-helix may facilitate RCM even in the presence of amino acids that normally reduce the efficiency of olefin metathesis. Interestingly, such trends seem be consistent in the presence of phosphine-containing catalysts 1 and 2 or isopropoxy catalysts 3–7.
Z-Selective ethenolysis on resin-bound α-helical peptides
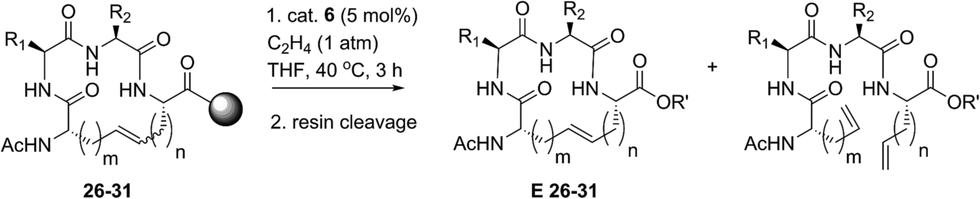
Our results regarding RCM on a variety of olefin-bearing peptides revealed that the diastereoselectivity of macrocyclic ring closure was dictated both by the choice of catalyst and size of the macrocycle. In cases involving peptides bearing i, i + 2 or i, i + 3 olefin crosslinks, RCM generally favored the formation of the E-isomer (∼80% E) in the presence of catalysts 1–5. Alternatively, the use of cyclometalated catalyst 6 or 7 gave rise to macrocycles predominantly of Z-olefin geometry, but at lower yields or conversions as expected for substrates where the E-isomer is normally favored. For macrocycles 8 and 15–18, Z-selective ethenolysis provided a method for further enrichment of E-olefin geometry. While these studies provide a framework for enabling the formation of E or Z olefins in cyclic peptides, we sought to extend our studies of ethenolysis to resin-bound peptides. Such experiments would prove particularly useful as the diastereoselectivity of RCM to form macrocycles 26–31 was typically low. In this sense, the ability to selectively perform ethenolysis on these macrocycles could streamline methods for their identification and purification.Our initial studies began with Z-selective ethenolysis on macrocycle 26 (Table 5). Conversion of 26 to the olefin-enriched macrocycle E-26 occurred in 86%, transforming 26 from an initial ratio of 72% E-olefin to greater than 90% E (entry 1). This trend was observed for the selective ethenolysis of macrocycle 27 which occurred in 65% conversion and transformed an 80% mixture of E/Z isomers of 27 to a macrocycle having greater than 90% selectivity for the E-olefin (entry 2). To probe the general utility of the method, we exposed macrocycles 28–31 containing i, i + 7 crosslinks to the ethenolysis conditions (entries 3–6). Conversions to the enriched macrocycles varied from 78% for 29 to 93% for the formation of 31. As with macrocycles 26 and 27, enrichment of 28–31 to the E-olefin could occur in greater than 90%. These studies, in parallel with ethenolysis on macrocycles 8 and 15–18, point to the utility of RCM and ethenolysis as a practical means of olefin enrichment in cyclic peptides.
Assessing the role of olefin geometry on the conformation of α-helical peptides
Our studies of RCM in tandem with catalyst-directed ethenolysis provided access to macrocyclic peptides enriched in E- or Z-olefin isomers. We sought to examine if changes in olefin geometry induced measureable differences in the overall fold or conformation of macrocyclic peptides. For our initial studies, we examined the α-helical content between non-stapled peptide 21 and the corresponding E or Z macrocycle 27 using circular dichroism (Table 6). The linear peptide 21 was measured to have an α-helical content of 21% (entry 1) which increased upon macrocyclization to 27 affording an α-helicity of 80% for the E-olefin and 71% for the Z-olefin, respectively (entries 2 and 3). These results are in agreement with the observation that macrocyclization by RCM generally induces greater α-helicity within stapled peptides.25,26,59,60 To further expand our studies, we next examined the role of olefin-geometry on the α-helical content within a larger macrocycle and chose peptide 23 containing olefins at i, i + 7 positions. For this peptide, the helical content was 7.5% for the non-cyclized peptide (entry 4) but upon macrocyclization to 29, the α-helicity increased to 21% and 23% for the E- and Z-olefin isomers, respectively (entries 5 and 6). As observed with macrocycle 27, the difference in the helicity between the Z- and E-olefin isomers in 29 was minimal, suggesting that the olefin geometry in 27 and 29 does not contribute substantially to the overall secondary structure of the macrocycles bearing olefin tethers of such lengths. These studies seek to inform further explorations into examining the role of olefin geometry on the stability or biological activity of macrocyclic compounds.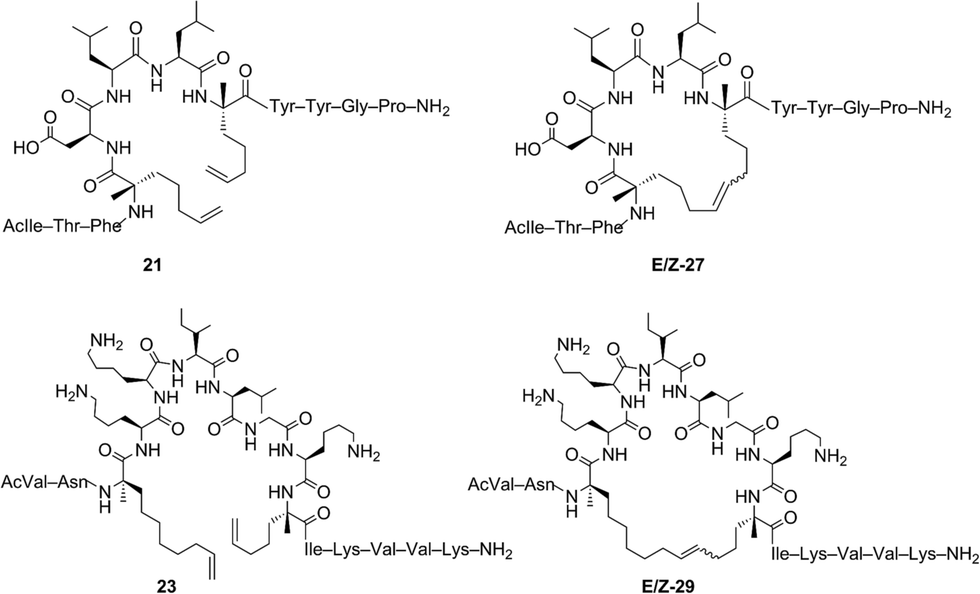
Conclusions
In summary, we report a method for the stereoselective synthesis of macrocyclic peptides using RCM in tandem with catalyst-controlled ethenolysis. The utility of the method was demonstrated on a variety of peptide sequences and olefin crosslinks to enrich macrocycles in E or Z olefin geometry. The strategies outlined herein can facilitate the synthesis and isolation of macrocylic peptides and this approach allowed for the examination of olefin geometry on the conformation of α-helical peptide secondary structures. Notably, these studies provide a comprehensive evaluation of a variety of ruthenium catalysts in facilitating RCM on peptides and highlight the use of cyclometalated ruthenium catalysts to control diastereoselectivity in macrocycle synthesis. It is envisioned that these studies will enable strategies for accessing novel macrocyclic architectures and help elucidate the role of olefin geometry on the stability or biological activity of cyclic peptides. Progress in the design of catalysts that provide such dual capabilities will continue to broaden the scope and applications of olefin metathesis in areas from chemical biology to natural product synthesis and pharmaceutical development.Acknowledgements
This work was financially supported by the NIH (NIH R01-GM031332). NMR spectra were obtained by instruments supported by the NIH (RR027690). Materia, Inc. is acknowledged for the generous donation of catalysts 1–3 and 6–7. The authors would like to thank Scott Virgil (Caltech Center for Catalysis and Chemical Synthesis) and Vanessa Marx for helpful discussions.Notes and references
- K. C. Nicolaou, P. G. Bulger and D. Sarlah, Angew. Chem., Int. Ed., 2005, 44, 4490 CrossRef CAS PubMed.
- X. Yu and D. Sun, Molecules, 2013, 18, 6230 CrossRef CAS PubMed.
- E. M. Driggers, S. P. Hale, J. Lee and N. K. Terrett, Nat. Rev. Drug Discovery, 2008, 7, 608 CrossRef CAS PubMed.
- J. Mallinson and I. Collins, Future Med. Chem., 2012, 4, 1409 CrossRef CAS PubMed.
- F. Diederich, P. J. Stang and R. R. Tykwinski, Modern Supramolecular Chemistry: Strategies for Macrocycle Synthesis, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed.
- A. Parenty, X. Moreau and J. M. Campagne, Chem. Rev., 2006, 106, 911 CrossRef CAS PubMed.
- K. C. Swamy, N. N. Kumar, E. Balaraman and K. V. Kumar, Chem. Rev., 2009, 109, 2551 CrossRef CAS PubMed.
- X.-F. Wu, H. Neumann and M. Beller, Chem. Rev., 2013, 113, 1 CrossRef CAS PubMed.
- S. Wen, G. Packham and A. Ganesan, J. Org. Chem., 2008, 73, 9353 CrossRef CAS PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509 CrossRef CAS PubMed.
- Z. J. Song, D. M. Tellers, M. Journet, J. T. Kuethe, D. Lieberman, G. Humphrey, F. Zhang, Z. Peng, M. S. Waters, D. Zewge, A. Nolting, D. Zhao, R. A. Reamer, P. G. Dormer, K. M. Belyk, I. W. Davies, P. N. Devine and D. M. Tschaen, J. Org. Chem., 2011, 76, 7804 CrossRef CAS PubMed.
- R. A. Turner, A. G. Oliver and R. S. Lokey, Org. Lett., 2007, 9, 5011 CrossRef CAS PubMed.
- G. Chouhan and K. James, Org. Lett., 2011, 13, 2754 CrossRef CAS PubMed.
- D. Pasini, Molecules, 2013, 18, 9512 CrossRef CAS PubMed.
- A. Gradillas and J. Perez-Castells, Angew. Chem., Int. Ed., 2006, 45, 6086 CrossRef CAS PubMed.
- A. H. Hoveyda and A. R. Zhugralin, Nature, 2007, 450, 243 CrossRef CAS PubMed.
- S. R. Chemler, D. Trauner and S. J. Danishefsky, Angew. Chem., Int. Ed., 2001, 40, 4544 CrossRef CAS.
- G. Evano, N. Blanchard and M. Toumi, Chem. Rev., 2008, 108, 3054 CrossRef CAS PubMed.
- M. E. Maier, Angew. Chem., Int. Ed., 2000, 39, 2073 CrossRef CAS.
- G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746 CrossRef CAS PubMed.
- X. Wei, C. Shu, N. Haddad, X. Zeng, N. D. Patel, Z. Tan, J. Liu, H. Lee, S. Shen, S. Campbell, R. J. Varsolona, C. A. Busacca, A. Hossain, N. K. Yee and C. H. Senanayake, Org. Lett., 2013, 15, 1016 CrossRef CAS PubMed.
- W. Erb and J. Zhu, Nat. Prod. Rep., 2013, 30, 161 RSC.
- L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 1466 CrossRef CAS PubMed.
- R. E. Moellering, M. Cornejo, T. N. Davis, C. Del Bianco, J. C. Aster, S. C. Blacklow, A. L. Kung, D. G. Gilliland, G. L. Verdine and J. E. Bradner, Nature, 2009, 462, 182 CrossRef CAS PubMed.
- L. D. Walensky and G. H. Bird, J. Med. Chem., 2014, 57, 6275 CrossRef CAS PubMed.
- G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3 CAS.
- G. H. Bird, E. Gavathiotis, J. L. Labelle, S. G. Katz and L. D. Walensky, ACS Chem. Biol., 2014, 9, 831 CrossRef CAS PubMed.
- A. Furstner, O. Guth, A. Rumbo and G. Seidel, J. Am. Chem. Soc., 1999, 121, 11108 CrossRef.
- A. Furstner and P. W. Davies, Chem. Commun., 2005, 2307 RSC.
- W. Zhang and J. S. Moore, Adv. Synth. Catal., 2007, 349, 93 CrossRef CAS.
- A. Furstner, Angew. Chem., Int. Ed., 2013, 52, 2794 CrossRef PubMed.
- Y. Wang, M. Jimenez, A. S. Hansen, E. A. Raiber, S. L. Schreiber and D. W. Young, J. Am. Chem. Soc., 2011, 133, 9196 CrossRef CAS PubMed.
- D. Gallenkamp and A. Furstner, J. Am. Chem. Soc., 2011, 133, 9232 CrossRef CAS PubMed.
- V. M. Marx, M. B. Herbert, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 94 CrossRef CAS PubMed.
- V. M. Marx, A. H. Sullivan, M. Melaimi, S. C. Virgil, B. K. Keitz, D. S. Weinberger, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 1919 CrossRef CAS PubMed.
- J. S. Davies, J. Pept. Sci., 2003, 9, 471 CrossRef CAS PubMed.
- M. Katsara, T. Tselios, S. Deraos, G. Deraos, M. T. Matsoukas, E. Lazoura, J. Matsoukas and V. Apostolopoulos, Curr. Med. Chem., 2006, 13, 2221 CrossRef CAS PubMed.
- S. Jiang, Z. Li, K. Ding and P. P. Roller, Curr. Org. Chem., 2008, 12, 1502 CrossRef CAS.
- In comparing the relative activity of catalysts 1–7 in RCM, we performed the reactions under dilute conditions and at 40 degrees Celsius. Conducting RCM at higher temperatures led to catalyst decomposition of catalysts 6 and 7. The chelated isopropoxy catalysts 3 and 4 have been shown, in many cases, to be more active at higher temperatures.
- I. C. Stewart, C. J. Douglas and R. H. Grubbs, Org. Lett., 2008, 10, 441 CrossRef CAS PubMed.
- The lower activity of catalyst 5 in RCM may result from a greater incidence of non-productive olefin metathesis which has been observed with less hindered ruthenium catalysts (see ref. 40).
- A. D. Abell, N. A. Alexander, S. G. Aitken, H. Chen, J. M. Coxon, M. A. Jones, S. B. McNabb and A. Muscroft-Taylor, J. Org. Chem., 2009, 74, 4354 CrossRef CAS PubMed.
- Competing dimerization of 8b (ca. ∼ 15%) was observed, which may account for the lower yield of the desired RCM product.
- S. L. Mangold, D. J. O'leary and R. H. Grubbs, J. Am. Chem. Soc., 2014, 136, 12469 CrossRef CAS PubMed.
- The higher yields of macrocycles 16 and 17 relative to 15 could result from the formation of a larger ring size (18-membered ring in 16 and 17) relative to 15 (14-membered ring) in addition to enhancing RCM through an allylic heteroatom effect.
- We observed dimerization (ca. ∼ 10%) in addition to a small percentage of the RCM product for peptide 14.
- R. N. Chapman and P. S. Arora, Org. Lett., 2006, 8, 5825 CrossRef CAS PubMed.
- S. C. Marinescu, D. S. Levine, Y. Zhao, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2011, 133, 11512 CrossRef CAS PubMed.
- H. Miyazaki, M. B. Herbert, P. Liu, X. Dong, X. Xu, B. K. Keitz, T. Ung, G. Mkrtumyan, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 5848 CrossRef CAS PubMed.
- Some E-degradation (ca. ∼ 12%) occurred after prolonged exposure to the ethenolysis conditions.
- The relatively low percentage of E-olefin enrichment for compound 18 may be partially attributed to its lower reactivity in RCM.
- S. J. Miller, H. E. Blackwell and R. H. Grubbs, J. Am. Chem. Soc., 1996, 118, 9606 CrossRef CAS.
- H. B. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281 CrossRef CAS.
- C. E. Schafmeister, J. Po and G. L. Verdine, J. Am. Chem. Soc., 2000, 122, 5891 CrossRef CAS.
- Y. W. Kim, T. N. Grossmann and G. L. Verdine, Nat. Protoc., 2011, 6, 761 CrossRef CAS PubMed.
- G. H. Bird, W. C. Crannell and L. D. Walensky, Curr. Protoc. Chem. Biol., 2011, 3, 99 Search PubMed.
- H. Zhang, Q. Zhao, S. Bhattacharya, A. A. Waheed, X. Tong, A. Hong, S. Heck, F. Curreli, M. Goger, D. Cowburn, E. O. Freed and A. K. Debnath, J. Mol. Biol., 2008, 378, 565 CrossRef CAS PubMed.
- H. Chapuis, J. Slaninova, L. Bednarova, L. Monincova, M. Budesinsky and V. Cerovsky, Amino Acids, 2012, 43, 2047 CrossRef CAS PubMed.
- K. Estieu-Gionnet and G. Guichard, Expert Opin. Drug Discovery, 2011, 6, 937 CrossRef CAS PubMed.
- F. Bernal and S. G. Katz, Methods Mol. Biol., 2014, 1176, 107 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc01507c |
| This journal is © The Royal Society of Chemistry 2015 |