
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe boron–boron triple bond in NHC→B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e002.gif) B←NHC†‡
B←NHC†‡
Nicole
Holzmann
a,
Markus
Hermann
a and
Gernot
Frenking
*ab
aFachbereich Chemie, Philipps-Universität Marburg, Hans-Meerwein-Str. 2, 35032 Marburg, Germany
bDonostia International Physics Center (DIPC), P.K. 1072, 20080 Donostia, Spain. E-mail: frenking@chemie.uni-marburg.de
First published on 1st June 2015
Abstract
Quantum chemical calculations of the compound B2(NHCMe)2 and a thorough examination of the electronic structure with an energy decomposition analysis provide strong evidence for the appearance of boron–boron triple bond character. This holds for the model compound and for the isolated diboryne B2(NHCR)2 of Braunschweig which has an even slightly shorter B–B bond. The bonding situation in the molecule is best described in terms of NHCMe→B2←NHCMe donor–acceptor interactions and concomitant π-backdonation NHCMe←B2→NHCMe which weakens the B–B bond, but the essential features of a triple bond are preserved. An appropriate formula which depicts both interactions is the sketch NHCMe⇄B![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) B⇄NHCMe. Calculations of the stretching force constants FBB which take molecules that have genuine single, double and triple bonds as references suggest that the effective bond order of B2(NHCMe)2 has the value of 2.34. The suggestion by Köppe and Schnöckel that the strength of the boron–boron bond in B2(NHCH)2 is only between a single and a double bond is repudiated. It misleadingly takes the force constant FBB of OBBO as the reference value for a B–B single bond which ignores π bonding contributions. The alleged similarity between the B–O bonds in OBBO and the B–C bonds in B2(NHCMe)2 is a mistaken application of the principle of isolable relationship.
B⇄NHCMe. Calculations of the stretching force constants FBB which take molecules that have genuine single, double and triple bonds as references suggest that the effective bond order of B2(NHCMe)2 has the value of 2.34. The suggestion by Köppe and Schnöckel that the strength of the boron–boron bond in B2(NHCH)2 is only between a single and a double bond is repudiated. It misleadingly takes the force constant FBB of OBBO as the reference value for a B–B single bond which ignores π bonding contributions. The alleged similarity between the B–O bonds in OBBO and the B–C bonds in B2(NHCMe)2 is a mistaken application of the principle of isolable relationship.
Introduction
In 2012, Braunschweig and co-workers reported the synthesis of the diboryne compound B2(NHCR)2 where diatomic B2 binds two N-heterocyclic carbenes (NHC) which carry bulky groups R at nitrogen.1 The X-ray structure analysis revealed a linear arrangement of the NHCR ligands to the central B2 moiety and a very short B–B bond of 1.45 Å which agrees with the standard value for a triple bond (1.46 Å).2 An earlier theoretical study that was published in 2011 had predicted that the molecule has very strong bonds which can be interpreted in terms of donor–acceptor interactions NHCR→B2←NHCR where diatomic B2 is in the highly excited (3)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1Σg+ singlet state rather than in the X3Σg− ground state (Fig. 1a and b).3
1Σg+ singlet state rather than in the X3Σg− ground state (Fig. 1a and b).3
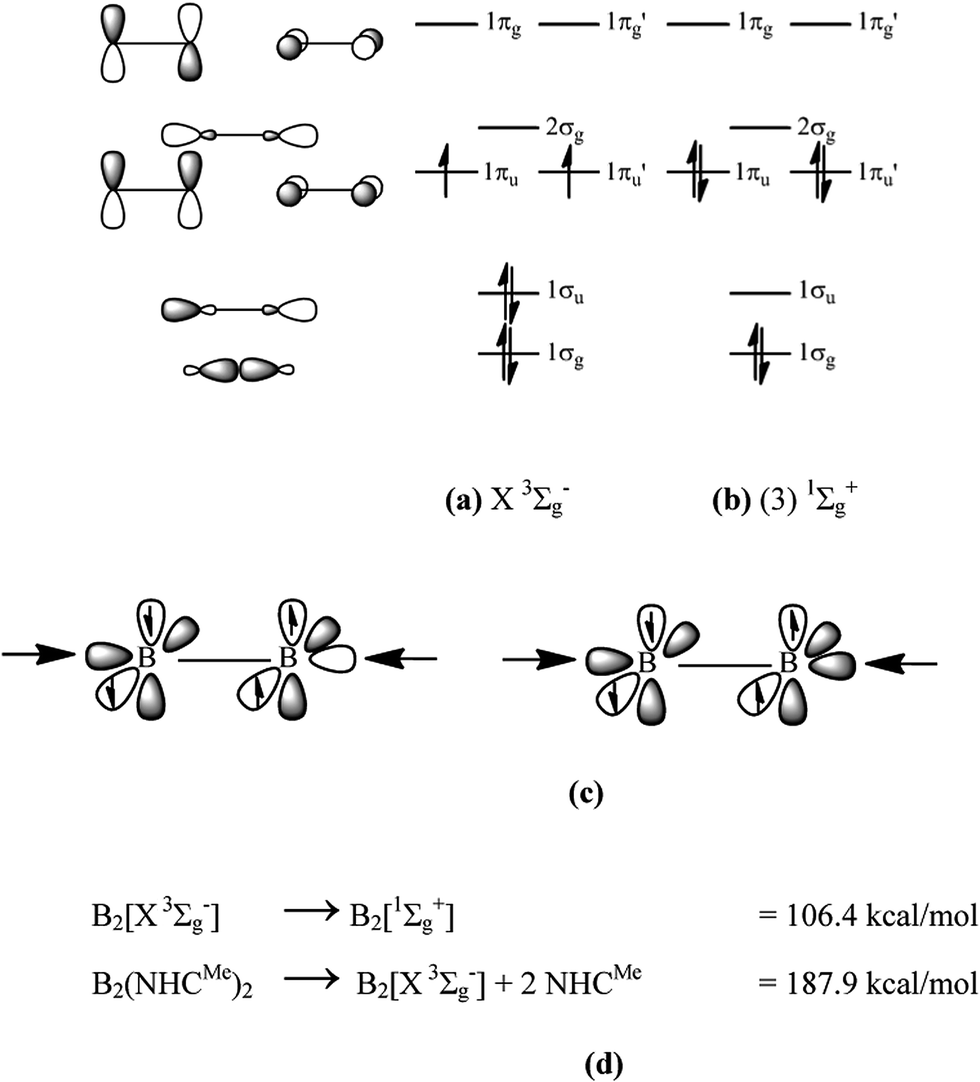 | ||
| Fig. 1 Schematic diagram of the valence orbitals and orbital occupation of B2 in (a) X3Σg− ground state and (b) (3)1Σg+ excited state. (c) Schematic representation of the charge donation from the out-of-phase (+,−) and in-phase (+,+) combinations of the ligand lone-pair σ orbitals into the vacant orbitals of B2 in the 1Σg+ excited state. (d) Calculated energies of the excitation energy of B2 from the ground state to the reference state and bond dissociation energy De of B2(NHCMe)2 at BP86/def2-TZVPP. | ||
Charge donation from the ligands takes place into the empty 1σu MO (out-of-phase +,− donation) and the 2σg MO (in-phase +,+ donation) which is schematically shown in Fig. 1c. In addition to the σ-donation NHCR→B2←NHCR, π-backdonation NHCR←B2→NHCR may occur from the occupied 1πu and 1πu′ orbitals which would weaken the BB triple bond. An energy decomposition analysis was carried out in 2013 which suggests that 2/3 of the total orbital interactions comes from NHCR→B2←NHCR σ-donation and 1/3 comes from NHCR←B2→NHCR π-backdonation.4 Thus, the bond order for the B–B can be expected between 2 and 3 while the triple bond character is retained in the diboryne whose bonding situation is properly sketched with the formula NHCR⇄BB⇄NHCR.
Very recently, Köppe and Schnöckel (KS) published in this journal a reinterpretation of the boron–boron bond in NHCR→B2←NHCR where they question the assignment of a triple bond.5 Using thermodynamic arguments and a correlation of force constants, KS suggest that the strength of the boron–boron bond in NHC→B2←NHC is only between a single and a double bond and that the molecule should better become described with traditional resonance structures. The conclusion of KS was made on the basis of selected experimental and calculated results. The authors write that their results ‘…generates a new interpretation which is in contrast to the triple bond donor–acceptor description visualized by arrows and which casts a critical light on the interpretation of any NHC “stabilized” molecule’. The statement was made without reference to the previous quantum chemical studies of B2(NHCR)23,4,6 nor to any other theoretical work about NHC stabilized molecules.7
We studied the paper by KS5 very carefully and noticed several flaws in their arguments which cast severe doubts on their conclusions. In the following we critically discuss the approach and the conclusions of the authors. Our counter arguments are presented in the same order as in the paper by KS.
Thermodynamic view
KS begin their discussion with a lengthy description of the energy which is required for the formation of B2 in the excited (3)1Σg+ state (which is denoted as B2* in their paper) from solid boron in a gas-phase reaction which is irrelevant for the discussion of the bonding situation and for the formation and stability of the compound. The compound NHCR→B2←NHCR was prepared by Braunschweig by reacting NHCR→(B2Br4)←NHCR with sodium naphthalenide in THF solution which gives the product molecule via debromination reaction.1 It is well known that molecules which have a large positive heat of formation can be isolated as stable compounds in straightforward reactions if the electronic structure is favourable.8 KS arrive at the conclusion: “Therefore, nobody would conclude that B2* is stabilized in solid boron!”. But nobody ever claimed that solid boron would do that! What has been claimed is the significant stabilization of B2* through strong donor–acceptor interactions with NHC ligands.3,4,6
KS make the statement that the arrows in the formula NHC→B2←NHC “suggest only a weak donor (NHC)–acceptor (B2*) interaction in which the bonding of the educt is still visible”. But it was already shown in 20133 that the donor–acceptor interactions NHC→B2←NHC are very strong, which should be expected in light of the low-lying vacant orbitals of B2* (Fig. 1). The strong interactions do not lead to major changes in the structures of the donor and acceptor species which is easy to understand. The two vacant σ orbitals in B2* are bonding and antibonding, respectively, which roughly cancel the impact of the donor–acceptor interactions on the B–B bond length. The slightly longer B–B distance in NHCR→B2←NHCR (1.45 Å) than in free B2* (1.40 Å) is caused by π-backdonation from B2 to the NHC ligands. The geometry changes of the NHC ligands in NHCR→B2←NHCR are likewise rather small, because they involve mainly the carbene lone-pair orbital. Thus, the structure and stability of NHC→B2←NHC are in full accord with the bonding situation of a BB moiety which is stabilized by strong σ-donation of and π-backdonation to the NHC ligands.
Determination and discussion of the force constants in NHC→B2←NHC
The main argument in the paper by KS against a triple bond character in NHCR→B2←NHCR rests on the force constants of the B–B stretching mode. The authors first discuss several molecules which may be used as reference for a B–B single bond. They choose OBBO as candidate, because it would “have a BB bonding situation similar to 1 with a linear X–B–B–X moiety” where 1 stands for NHCR→B2←NHCR. In order to underpin the connection between OBBO and 1 the authors refer to the isolobal relationship O CH2. Then they write that “the Lewis formula O
CH2. Then they write that “the Lewis formula O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/i_char_e001.gif) B–BO is in accordance with the values of the BB and BC force constants”.
B–BO is in accordance with the values of the BB and BC force constants”.
There are two major flaws in the above arguments. One flaw concerns the suggestion that the boron–boron bond in OBBO is a single bond.9 There are two orthogonal π components in the BO moieties which conjugate over the BB fragment. Using OBBO as template for a B–B single bond ignores the contribution of B–B π-bonding. Note that the authors suggest a Lewis structure O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) B–BO for the molecule which has an electron sextet at boron. The second flaw is the suggestion that the isolobal relationship O
B–BO for the molecule which has an electron sextet at boron. The second flaw is the suggestion that the isolobal relationship O CH2 may be used to indicate a similar bonding situation in OBBO and NHCR→BB←NHCR. CH2 has an electronic (3B1) triplet ground state which is 9.0 kcal mol−1 below the (1A1) singlet state.10 Methylene is isolobal with oxygen atom, because both species have a triplet state and a similar shape and energy.11 In contrast, NHC has a singlet ground state which is 85 kcal mol−1 below the triplet state.12 It means that the electronic structure and the chemical reactivity of the carbene carbon atom of NHC are very different from CH2, which is common knowledge in chemistry.13 A pertinent example which demonstrates the large difference between the two carbenes is given by the compounds where they bind to a single carbon atom. C(CH2)2 is the parent allene H2CCCH2 which has a linear structure with CC double bonds and perpendicular CH2 planes. C(NHC)2 is a carbone CL2 which features two donor–acceptor bonds L→C←L to a carbon atom which retains two electron lone pairs.14 The carbodicarbene C(NHCMe)2 has a bond angle (NHCMe)–C–(NHCMe) of 132°15 which is similar to the isolobal carbodiphosphorane C(PPh3)2 which has a bond angle of 135°.16 The first carbodicarbenes C(NHCR)2 were recently synthesized by Bertrand and by Fürstner.17 It is obvious that the isolobal relationship O
CH2 may be used to indicate a similar bonding situation in OBBO and NHCR→BB←NHCR. CH2 has an electronic (3B1) triplet ground state which is 9.0 kcal mol−1 below the (1A1) singlet state.10 Methylene is isolobal with oxygen atom, because both species have a triplet state and a similar shape and energy.11 In contrast, NHC has a singlet ground state which is 85 kcal mol−1 below the triplet state.12 It means that the electronic structure and the chemical reactivity of the carbene carbon atom of NHC are very different from CH2, which is common knowledge in chemistry.13 A pertinent example which demonstrates the large difference between the two carbenes is given by the compounds where they bind to a single carbon atom. C(CH2)2 is the parent allene H2CCCH2 which has a linear structure with CC double bonds and perpendicular CH2 planes. C(NHC)2 is a carbone CL2 which features two donor–acceptor bonds L→C←L to a carbon atom which retains two electron lone pairs.14 The carbodicarbene C(NHCMe)2 has a bond angle (NHCMe)–C–(NHCMe) of 132°15 which is similar to the isolobal carbodiphosphorane C(PPh3)2 which has a bond angle of 135°.16 The first carbodicarbenes C(NHCR)2 were recently synthesized by Bertrand and by Fürstner.17 It is obvious that the isolobal relationship O CH2 does not hold for NHC.
CH2 does not hold for NHC.
We searched for a molecule which possesses a genuine B–B single bond. The vast experimental information which has been accumulated about boron compounds clearly shows that boron atoms in stable molecules have at least partially filled four valence AOs which makes the search for an experimental value of species with a B–B single bond futile. We therefore looked for a model compound with a genuine B–B single bond that can be calculated with an accuracy that rivals experimental values. A suitable species is planar H2B–BH2 where the p(π) orbitals of boron are vacant. The planar (D2h) structure is a transition state for the rotation about the B–B bond of B2H4 which has a perpendicular (D2d) equilibrium structure. The B–B bond in the D2d energy minimum structure has been calculated to be much shorter (1.623 Å) than in the planar form (1.752 Å) which is due to significant hyperconjugation that was estimated from the energy difference between the D2d and D2h structures (17.3 kcal mol−1).18 The reported B–B bond length of 1.65 Å for OBBO which is much shorter than the single bond in planar B2H4 (1.752 Å) indicates a substantial degree of B–B π-bonding which makes the molecule unsuitable as reference for a single bond.
We calculated the force constant for the B–B stretching mode in planar H2B–BH2 and took the value of FBB = 2.49 mdyn Å−1 as reference value for a B–B single bond. A related molecule with a genuine BB double bond is HBBH in the (3Σg−) triplet state which has a B–B σ bond and two singly occupied B–B π orbitals. The calculated force constant for the B–B stretching mode is FBB = 5.38 mdyn Å−1 which is taken as reference value for a BB double bond. The reference value for a BB triple bond comes from the calculated force constant for the (3)1Σg+ state of B2 which has the value of FBB = 7.62 mdyn Å−1. The B–B force constants in mdyn Å−1 of the hydroboranes for a single bond (2.49; planar B2H4), double bond (5.38, triplet B2H2) and triple bond (7.62; (3)1Σg+ B2) exhibit the same ratio 1:2:3 as the force constants FCC for the C–C bonds in the analogous hydrocarbons which were given by KS as 4.4 (C2H6), 9.1 (C2H4) and 15.6 (C2H2).5 The regular increase of the force constant is at first sight a bit surprising, since the additional components of the multiple bonds are π bonds which are weaker than σ bonds. Along with the addition of π bonding, there is a concomitant change in the hybridisation of the σ bond which enhances the overall bonding and leads to the approximate relation 1:2:3.19
Fig. 2 shows a correlation diagram between the force constants and the bond orders of the reference compounds H2B–BH2 (planar), HBBH (3Σg−) and B2 ((3)1Σg+) which exhibits a linear correlation coefficient of R2 = 0.995. The calculated force constant FBB = 3.5 for OBBO suggests a bond order of 1.36, which indicates a significant contribution of π bonding. The force constant FBB = 6.0 which was calculated by KS for NHC→B2←NHC now indicates an effective bond order for the B–B bond of 2.34. This is in agreement with the classification as a molecule which has a BB triple bond that is weakened by π-backdonation. The assignment of the much lower bond order of 1.5 for the complex which was suggested by KS is misleading, because it arbitrarily uses a bond order of 1.0 for OBBO, which neglects the π contribution to the B–B bond.
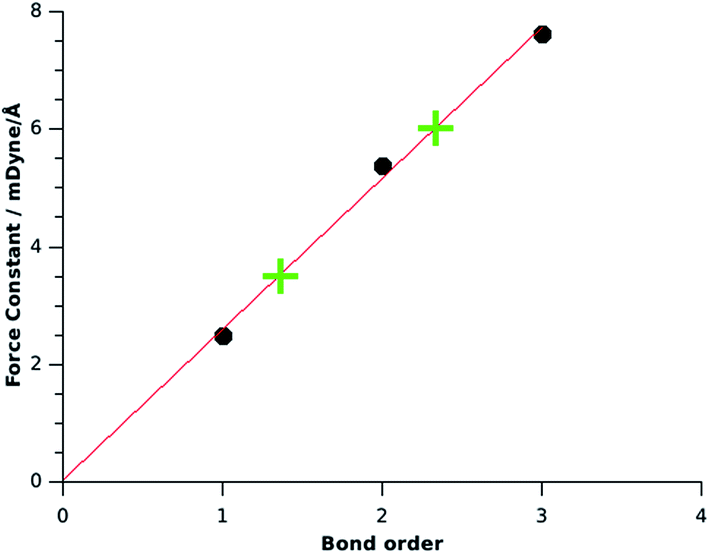 | ||
| Fig. 2 Correlation between the force constants FBB for boron–boron single, double and triple bonds (•) and the values for OBBO and B2(NHCMe)2 (+). | ||
Bonding analysis of NHC→B2←NHC
The above discussion provides information about the relative strength of the B–B bond in the complex NHCR→B2←NHCR compared with reference compounds which were chosen by KS (OBBO) and by us (planar H2BBH2, triplet HBBH and (3)1Σg+ B2) which, however, does not give insight into the nature of the bonding. Quantitative information is available form EDA-NOCV20 (Energy Decomposition Analysis21 combined with Natural Orbitals for Chemical Valence22) calculations. The advantage of the recently developed EDA-NOCV method over the EDA procedure which we used in our earlier bonding analysis4 of NHCMe→B2←NHCMe is, that the orbital interactions are broken down to a sum of pairwise contributions which provides detailed information about the strength of specific pairs of orbitals. The results give a complete picture of the bonding between the interacting fragments which can be connected with classical bonding models of chemistry. A helpful feature of the EDA-NOCV method is, that the results of the calculations can not only numerically be expressed but that the impact of the pairwise orbital interactions on the electronic structure can also graphically be visualized. Details of the calculations are given in ESI.‡ For further information about the EDA-NOCV method23 and recent examples we refer to the literature.24
Table 1 gives the numerical data of the EDA-NOCV calculations of the complex NHCMe→B2←NHCMe for the interactions between the acceptor (3)1Σg+ B2 (for the choice of the latter excited state of B2 see ref. 4) and the donor (NHCMe)2 at the geometry of the complex. The instantaneous interaction energy ΔEint = −307.5 kcal mol−1 is very large and overcompensates the electronic excitation energy of B2 of 106.4 kcal mol−1. The interaction energy ΔEint comprises the Pauli repulsion ΔEPauli (259.0 kcal mol−1) and the binding interactions which comes from the Coulombic attraction ΔEelstat (−252.3 kcal mol−1) and the covalent (orbital) term ΔEorb (−314.2 kcal mol−1). The EDA-NOCV calculations suggest that the donor–acceptor bonds NHCMe→B2←NHCMe have a slightly more covalent (55.5%) character than electrostatic character (44.5%).
1Σg+] and (NHCMe)2 as interacting species. All energy values in kcal mol−1. Calculated NBO partial charge of B2 in e
| Interacting fragments | B2(NHCMe)2 |
|---|---|
| B2[(3)1Σg+] and (NHCMe)2 |
|
| a The value in parenthesis gives the percentage contribution to the total attractive interactions ΔEelstat + ΔEorb. b The value in parenthesis gives the percentage contribution to the total orbital interactions ΔEorb. | |
| ΔEint | −307.5 |
| ΔEPauli | 259.0 |
| ΔEelstata | −252.3 (44.5%) |
| ΔEorba | −314.2 (55.5%) |
| ΔEσ1 L→(B2)←L (+,+) donationb | −112.7 (35.9%) |
| ΔEσ2 L→(B2)←L (+,−) donationb | −86.9 (27.7%) |
| ΔEπ1 L←(B2)→L π-backdonationb | −48.0 (15.3%) |
| ΔEπ2 L←(B2)→L π′-backdonationb | −42.4 (13.5%) |
| ΔErest | −24.2 (7.7%) |
| q(B2) | −0.36 |
The breakdown of the orbital term ΔEorb into pairwise contributions between occupied and vacant MOs of the fragments provide quantitative insight into the σ-donation NHCMe→B2←NHCMe and π-backdonation NHCMe←B2→NHCMe. The associated charge flow can be visualized through the deformation densities Δρ1 − Δρ4 that are shown in Fig. 3, left column. The colour code indicates the direction of the charge flow red→blue. The occupied and vacant orbitals which are connected to the deformation densities are displayed in the three columns at the right side of Fig. 3. There are strongly stabilizing contributions from the in-phase (+,+) NHCMe→B2←NHCMe σ-donation into the LUMO + 1 of (3)1Σg+ B2 (ΔEσ1 = −112.7 kcal mol−1) while the out-of-phase (+,−) donation into the energetically lower-lying LUMO is a bit smaller (ΔEσ2 = −86.9 kcal mol−1) which is due to the better overlap of the former (+,+) donation. There two contributions of the NHCMe←B2→NHCMe π-backdonation are clearly smaller (ΔEπ1 = −48.0 kcal mol−1 and ΔEπ2 = −42.4 kcal mol−1) than the σ-donation. Since both components, σ-donation and π-backdonation significantly contribute to the bonding, a more appropriate notation for the bonding interactions is the formula NHCMe⇄BB⇄NHCMe. Since the π-backdonation weakens the boron–boron bond, a smaller bond order below 3 but above 2 is expected, which is in agreement with the effective bond order of 2.34 that was derived from the force constants.
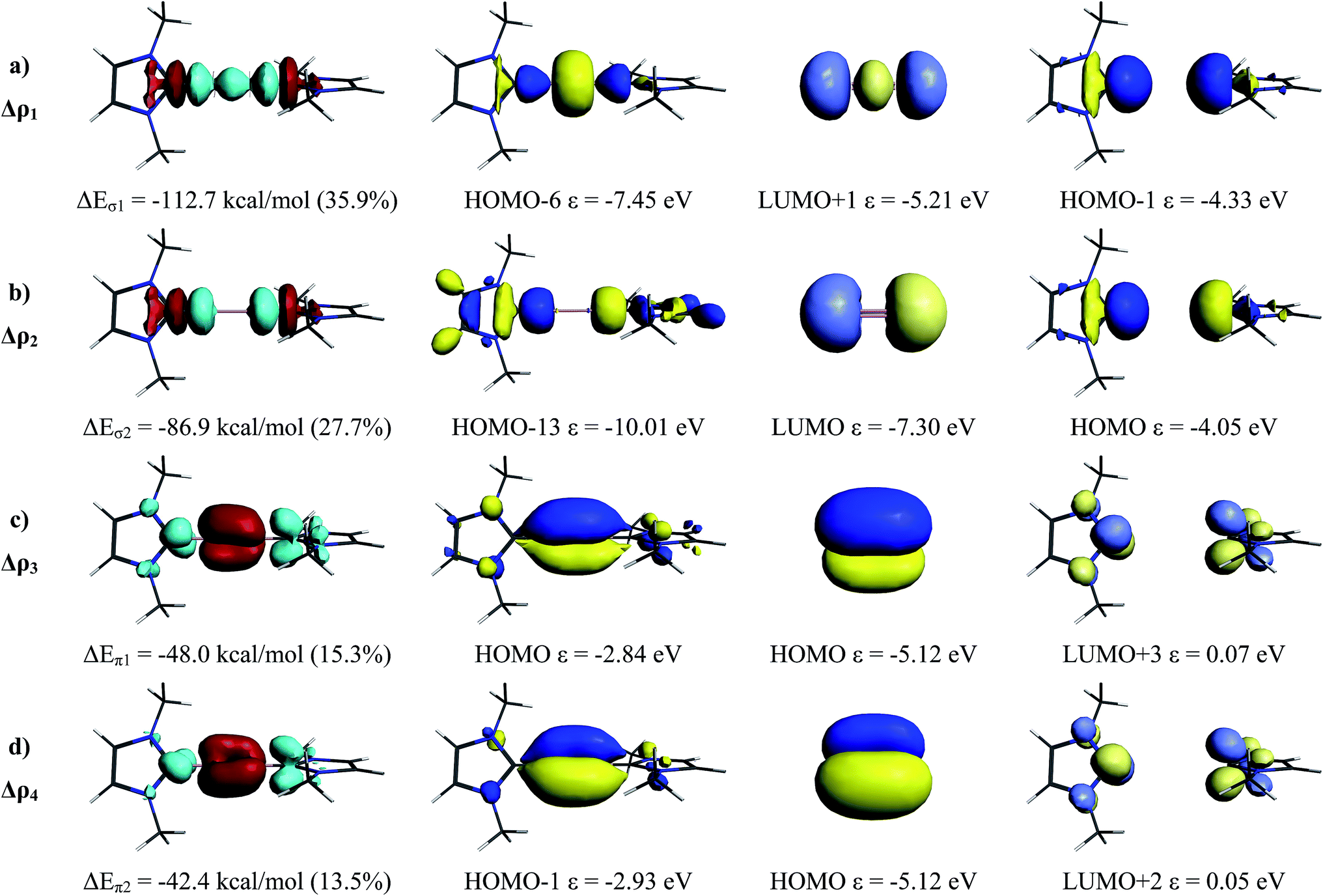 | ||
| Fig. 3 Plot of the interacting donor and acceptor orbitals and calculated eigenvalues ε of (NHCMe)2 and (1Σg+) B2 (right two columns) and matching MOs of the complex NHCMe→BB←NHCMe (second column from the left). Plot of the deformation densities Δρ with connected stabilization energies ΔE of the four most important orbital interactions in B2(NHCMe)2 which indicate the associated charge flow red→blue. | ||
The numerical results of the EDA-NOCV calculations and the graphical display of the deformation densities together with the molecular orbitals provide a bridge between heuristic bonding models of chemistry and a quantum chemical analysis of the electronic structure of the investigated complex. The calculated energy values are not observable data, but they arise from an unambiguously defined partitioning scheme which is not very sensitive to the level of theory. The value of the EDA-NOCV method lies in the fact that it establishes a quantitative classification scheme where comparisons can be made with related compounds. Explanations for experimental findings and predictions for new compounds can be made for different ligands L and atoms E in complexes L→E2←L as well as for compounds L→E←L by considering the electronic states of E2 or E and the donor strength of L. This has been shown for numerous complexes of atoms E of group 13,3,4,6,25 group 1414,15,26 and group 15.8c,27 The same approach has recently been used to predict stable beryllium compounds L→Be2←L.28
We would like to make a comment on the use of the different notations which were used by KS and by us for describing the bonding situation in B2(NHCR)2 (see Scheme 1). The two formulas are not just different writing styles but they refer to different bonding situations between B2 and the NHCR moieties. The notation with arrows NHCMe⇄BB⇄NHCMe indicates heterolytic cleavage of dative bonds while the notation with dashed lines as suggested by KS suggests conjugation over electron-sharing bonds which cleave homolytically. The latter description is appropriate for molecules such as 1,2-butadiene where rupture of the C–C bonds yields radicals. Cleavage of the B–NHCR bonds gives closed-shell fragments. Recognizing the difference between the two types of bonding has led to the discovery of carbones CL2 as a peculiar class of organic compounds and related systems which exhibit unusual geometries and reactivities and to the prediction of novel compounds.29
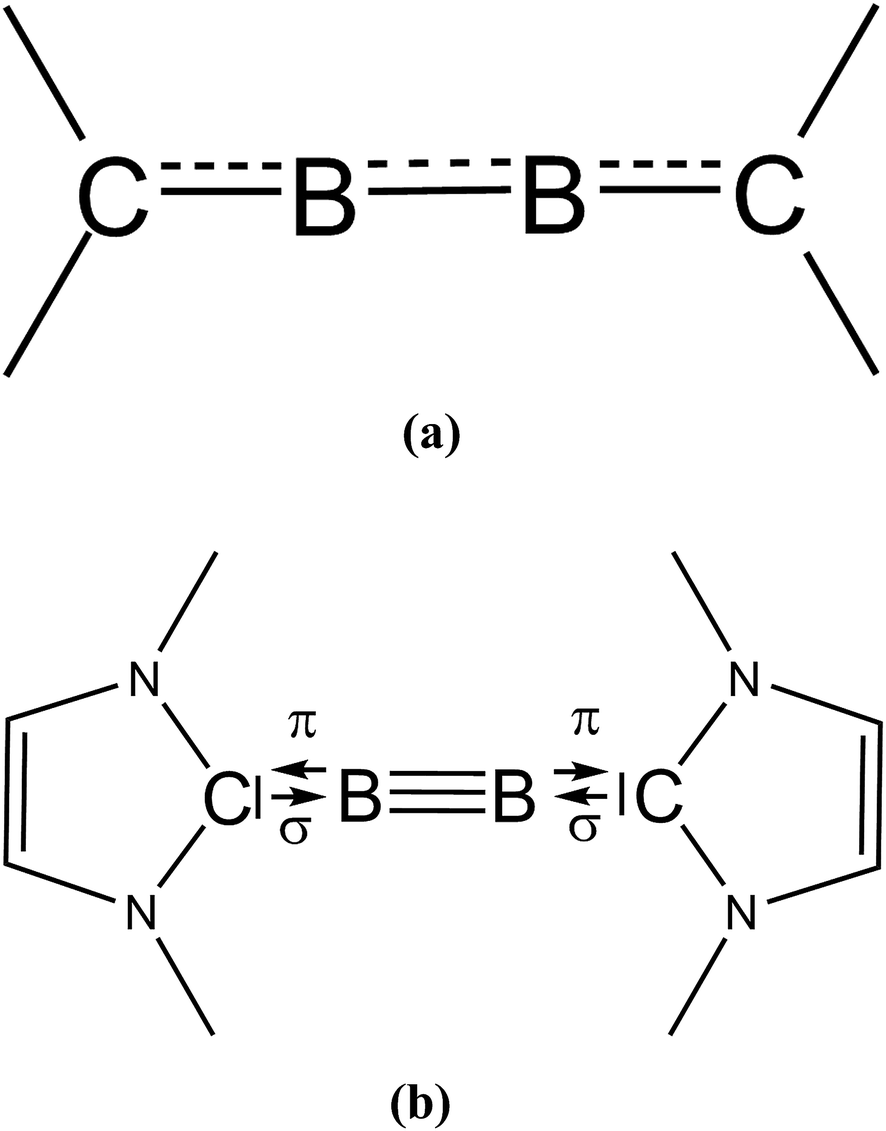 | ||
| Scheme 1 Bonding situation in B2(NHC)2 which were suggested (a) by Köppe and Schnöckel and (b) by us. | ||
Finally, we want to point out that two recent papers by Braunschweig just appeared where the strength of the B–B triple bond in the original diboryne B2(NHCR)2 and in the slightly changed species B2(NHCR′)2 with slightly less bulky substituents R′ than R has experimentally been assessed.30 One paper shows that the observed B–B stretching modes of the Raman spectra nicely fit into the established trend of the CC and NN stretching frequencies of alkynes and dinitrogen.30a The second paper reports 11B–11B spin–spin (J) coupling constants which indicate that the bonding description in terms of a triple bond is justified.30b Thus, theory and experiment agree that B2(NHCR)2 contains has significant boron–boron triple bond character.
Summary and conclusion
The results of this work can be summarized as follows. The calculated data and the thorough bonding analysis of the compound B2(NHCMe)2 provide convincing evidence for the appearance of significant boron–boron triple bond character in the model compound and in the isolated diboryne B2(NHCR)2 of Braunschweig which has an even slightly shorter B–B bond. The bonding situation is best described in terms of NHCMe→B2←NHCMe donor–acceptor interactions and concomitant π-backdonation NHCMe←B2→NHCMe which weakens the B–B bond, but the essential features of a triple bond are preserved. An appropriate formula which depicts both interactions is the sketch NHCMe⇄BB⇄NHCMe.
Calculations of the stretching force FBB constants which take molecules that have genuine single, double and triple bonds suggest that the effective bond order of B2(NHCMe)2 has the value of 2.34. The suggestion by Köppe and Schnöckel that the strength of the boron–boron bond in B2(NHCH)2 is only between a single and a double bond which should be described with a Lewis structure that has a σ bond and a delocalized π bond is repudiated. It misleadingly takes the force constant FBB of OBBO as reference value for a B–B single bond which ignores π bonding contributions. The alleged similarity between the B–O bonds in OBBO and the B–C bonds in B2(NHCMe)2 is a mistaken application of the principle of isolable relationship.
Acknowledgements
We appreciate critical and constructive comments of the referees which were very helpful to improve the paper. This work was supported by the Deutsche Forschungsgemeinschaft.References
- H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420 CrossRef CAS PubMed.
- P. Pyykkö, S. Riedel and M. Patzschke, Chem.–Eur. J., 2005, 11, 3511 CrossRef PubMed.
- N. Holzmann, A. Stasch, C. Jones and G. Frenking, Chem.–Eur. J., 2011, 17, 13517 CrossRef CAS PubMed.
- N. Holzmann, A. Stasch, C. Jones and G. Frenking, Chem.–Eur. J., 2013, 19, 6467 CrossRef CAS PubMed.
- R. Köppe and H. Schnöckel, Chem. Sci., 2015, 6, 1199 RSC.
- G. Frenking and N. Holzmann, Science, 2012, 336, 1394 CrossRef CAS PubMed.
- An enlightening example for the specific stabilization of NHC via donor–acceptor interactions and the difference to a normal covalent (electron-sharing) bond has been reported: K. C. Mondal, H. W. Roesky, M. C. Schwarzer, G. Frenking, S. Neudeck, I. Tkach, H. Wolf, D. Kratzert, R. Herbst-Irmer, B. Niepötter and D. Stalke, Angew. Chem., Int. Ed., 2013, 52, 1801 CrossRef CAS PubMed.
-
(a) A striking example is the phosphane complex N2(PPh3) which was synthesized already in 1964, without the bonding situation being recognized: R. Appel and R. Schöllhorn, Angew. Chem., Int. Ed. Engl., 1964, 3, 805 CrossRef PubMed;
(b) Recent theoretical studies showed that the compound N2(PPh3) which melts at 184–186 °C is thermodynamically unstable toward loss of N2 by ∼90 kcal mol−1: D. J. D. Wilson, S. A. Couchman and J. L. Dutton, Inorg. Chem., 2012, 51, 7657 CrossRef CAS PubMed;
(c) The high kinetic stability of N2(PPh3) could be explained in terms of strong donor–acceptor interactions between the phosphane donor ligands and N2 in the doubly excited (1)1Γg state which has a N–N single bond and two lone pairs at each nitrogen atom: N. Holzmann, D. Dange, C. Jones and G. Frenking, Angew. Chem., Int. Ed., 2013, 52, 3004 CrossRef CAS PubMed.
- One referee pointed out that previous theoretical studies showed already that the assignment of a B–B single bond for OBBO is not appropriate: (a) B. Ruscic, C. A. Mayhew and J. Berkowitz, J. Chem. Phys., 1988, 88, 5580 CrossRef CAS PubMed; (b) A. V. Neumukhin and F. Weinhold, J. Chem. Phys., 1993, 98, 1329 CrossRef PubMed; (c) M. T. Nguyen, M. H. Matus, V. T. Ngan, D. J. Grant and D. A. Dixon, J. Phys. Chem. A, 2009, 113, 4895 CrossRef CAS PubMed.
- (a) A. R. W. McKellar, P. R. Bunker, T. J. Sears, K. M. Evenson, R. J. Saykally and S. R. Langhoff, J. Chem. Phys., 1983, 79, 5251 CrossRef CAS PubMed; (b) D. G. Leopold, K. K. Murray and W. C. Lineberger, J. Chem. Phys., 1984, 81, 1048 CrossRef CAS PubMed.
- R. Hoffmann, Angew. Chem., Int. Ed. Engl., 1982, 21, 711 CrossRef PubMed.
- D. A. Dixon, K. D. Dobbs, A. J. Arduengo III and G. Bertrand, J. Am. Chem. Soc., 1991, 113, 8782 CrossRef CAS.
- W. A. Herrmann and C. Köcher, Angew. Chem., 1997, 109, 2256 ( Angew. Chem., Int. Ed. Engl. , 1997 , 36 , 2162 ) CrossRef PubMed.
- G. Frenking and R. Tonner, Pure Appl. Chem., 2009, 81, 597 CrossRef CAS.
- (a) R. Tonner and G. Frenking, Angew. Chem., Int. Ed., 2007, 46, 8695 CrossRef CAS PubMed; (b) R. Tonner, G. Heydenrych and G. Frenking, ChemPhysChem, 2008, 9, 1474 CrossRef CAS PubMed; (c) W. Petz, F. Öxler, B. Neumüller, R. Tonner and G. Frenking, Eur. J. Inorg. Chem., 2009, 4507 CrossRef CAS PubMed.
- G. E. Hardy, J. I. Zink, W. C. Kaska and J. C. Baldwin, J. Am. Chem. Soc., 1978, 100, 8001 CrossRef CAS.
- (a) C. A. Dyker, V. Lavallo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2008, 47, 3206 CrossRef CAS PubMed; (b) A. Fürstner, M. Alcarazo, R. Goddard and C. W. Lehmann, Angew. Chem., Int. Ed., 2008, 47, 3210 CrossRef PubMed; (c) M. Alcarazo, W. Lehmann, A. Anoop, W. Thiel and A. Fürstner, Nat. Chem., 2009, 1, 295 CrossRef CAS PubMed.
- A. Szabó, A. Kovács and G. Frenking, Z. Anorg. Allg. Chem., 2005, 631, 1803 CrossRef PubMed.
- For a detailed analysis of the nature of the bonding in nonpolar single, double and triple bonds, see: A. Kovács, C. Esterhuysen and G. Frenking, Chem.–Eur. J., 2005, 11, 1813 CrossRef PubMed.
- (a) M. P. Mitoraj, A. Michalak and T. Ziegler, J. Phys. Chem. A, 2008, 112, 1933 CrossRef PubMed; (b) M. P. Mitoraj, A. Michalak and T. Ziegler, J. Chem. Theory Comput., 2009, 5, 962 CrossRef CAS.
- (a) T. Ziegler and A. Rauk, Inorg. Chem., 1979, 18, 1755 CrossRef CAS; (b) K. Morokuma, J. Chem. Phys., 1971, 55, 1236 CrossRef CAS PubMed.
- (a) M. P. Mitoraj and A. Michalak, Organometallics, 2007, 26, 6576 CrossRef CAS; (b) M. P. Mitoraj and A. Michalak, J. Mol. Model., 2008, 14, 681 CrossRef CAS PubMed.
- G. Frenking and F. M. Bickelhaupt, The Chemical Bond. 1. Fundamental Aspects of Chemical Bonding, ed. G. Frenking and S. Shaik, Wiley-VCH, Weinheim, 2014, pp. 121–158 Search PubMed.
- See the following paper and references cited therein: P. Jerabek, H. W. Roesky, G. Bertrand and G. Frenking, J. Am. Chem. Soc., 2014, 136, 17123 CrossRef CAS PubMed.
- (a) N. Holzmann and G. Frenking, Croat. Chem. Acta, 2014, 87, 413 CrossRef; (b) R. Kinjo, B. Donnadieu, M. A. Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610 CrossRef CAS PubMed; (c) M. A. Celik, R. Sure, S. Klein, R. Kinjo, G. Bertrand and G. Frenking, Chem.–Eur. J., 2012, 18, 5676 CrossRef CAS PubMed.
- See the following paper and references cited therein: G. Frenking and R. Tonner, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2011, 1, 869 CrossRef CAS PubMed.
- M. A. Celik, R. Sure, S. Klein, R. Kinjo, G. Bertrand and G. Frenking, Chem.–Eur. J., 2012, 18, 5676 CrossRef CAS PubMed.
- S. A. Couchman, N. Holzmann, G. Frenking, D. J. D. Wilson and J. L. Dutton, Dalton Trans., 2013, 42, 11375 RSC.
- G. Frenking, Angew. Chem., Int. Ed., 2014, 53, 6040 CrossRef CAS PubMed.
- (a) J. Böhnke, H. Braunschweig, P. Constandinidis, T. Dellermann, W. C. Ewing, I. Fischer, K. Hammond, F. Hupp, J. Mies, H.-C. Schmitt and A. Vargas, J. Am. Chem. Soc., 2015, 137, 1766 CrossRef PubMed; (b) F. A. Perras, W. C. Ewing, T. Dellermann, J. Böhnke, S. Ullrich, T. Schäfer, H. Braunschweig and D. L. Bryce, Chem. Sci., 2015, 6, 3378 RSC.
Footnotes |
| † This paper is dedicated to Professor Peter Paetzold on the occasion of his 80th birthday. |
| ‡ Electronic supplementary information (ESI) available: Details of the theoretical calculations and coordinates and energies of the calculated molecules. See DOI: 10.1039/c5sc01504a |
| This journal is © The Royal Society of Chemistry 2015 |