
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn situ investigation of dissociation and migration phenomena at the Pt/electrolyte interface of an electrochemical cell†
Yeuk Ting
Law‡
a,
Spyridon
Zafeiratos
a,
Stylianos G.
Neophytides
b,
Alin
Orfanidi§
b,
Dominique
Costa
c,
Thierry
Dintzer
a,
Rosa
Arrigo
de,
Axel
Knop-Gericke
d,
Robert
Schlögl
de and
Elena R.
Savinova
*a
aInstitut de Chimie et Procédés pour l'Energie, l'Environnement et la Santé, UMR 7515-CNRS-UdS-ECPM, 25 rue Becquerel, 67087 Strasbourg, France. E-mail: elena.savinova@unistra.fr
bInstitute of Chemical Engineering Sciences, Stadiou Str., Platani GR-26504, Rion Achaias, Patras, Greece
cLaboratoire de Physico-Chimie des Surfaces, ENSCP, CHIMIE-PARISTECH, 11, rue Pierre et Marie CURIE, 75005 Paris, France
dFritz-Haber-Institut der MPG, Faradayweg 4-6, Dahlem, D-14195 Berlin, Germany
eMax-Planck-Institut for Chemical Energy Conversion, Stiftstr. 34-36, 45470 Mülheim, Germany
First published on 1st July 2015
Abstract
The development of efficient energy conversion systems requires precise engineering of electrochemical interfaces and thus asks for in situ techniques to probe the structure and the composition of the dynamic electrode/electrolyte interfacial region. This work demonstrates the potential of the near ambient pressure X-ray photoelectron spectroscopy (NAPXPS) for in situ studies of processes occurring at the interface between a metal electrode and a liquid electrolyte. By using a model membrane-electrode assembly of a high temperature phosphoric acid-imbibed proton exchange membrane fuel cell, and combining NAPXPS measurements with the density functional theory, it was possible to monitor such fundamental processes as dissociation and migration of the phosphoric acid within a nanostructured Pt electrode under polarization.
Introduction
Global pursuit of clean and sustainable energy is guiding the development of electrochemical energy conversion and storage technologies, including fuel cells, batteries, and electrolyzers, where the interface between an electronic and an ionic conductor (solid, liquid or polymer) plays a central role. The development of efficient energy conversion systems not only requires potent, durable and cost-effective materials, but also asks for precise engineering of electrochemical interfaces where molecular, ionic and electronic flows merge. Future progress in the field thus heavily relies on the availability of in situ techniques to probe structure and composition of the dynamic electrode/electrolyte interfacial region. While various spectroscopic and microscopic methods are nowadays available for the investigation of such interfaces, methods, which allow probing the chemical state of the interface under operation conditions are still limited.X-ray Photoelectron spectroscopy (XPS) is one of the most powerful techniques for studies of the chemical composition and the oxidation state of components located within the near-surface region. However, XPS is typically operating under ultra-high vacuum thus severely restricting the range of electrochemical systems which can be investigated. The capability of the XPS for probing the chemical state of surfaces and interfaces has inspired various specialized approaches for studies of electrode/electrolyte interfaces including those involving emersed electrodes,1,2 frozen electrolytes,3 ionic liquids,4etc. Recent advances in vacuum and analyzer technologies have resulted in the development of specialized instruments which allow performing the so-called near ambient pressure photoelectron spectroscopy (NAPXPS) measurements in the pressure range of millibars.5–7 NAPXPS has already proven its extraordinary capabilities to probe in situ reversible and irreversible transformations occurring at interfaces relevant to catalysis8–10 and electrocatalysis.11–15 By applying NAPXPS to a model membrane-electrode assembly (MEA) of a low temperature proton-exchange membrane fuel cell (PEMFC), Casalongue et al.14,16,17 were able to observe chemical changes on the electrode surface and detect reactive intermediates.
This work demonstrates that NAPXPS can also provide an insight into potential-driven processes occurring in a liquid electrolyte at the interface with a polarized metal electrode. By taking advantage of the low vapor pressure of phosphoric acid (PA) we investigate the interface between a nanostructured Pt electrode and a PA-imbibed membrane of the so-called high temperature (HT) PEMFC. HT PEMFC is a subkind of the proton exchange membrane fuel cells that uses PA-imbibed poly-benzimidazole or hydrocarbon-based polymer membranes and operates at temperatures up to 200 °C.18 Due to their high tolerance to carbon monoxide and a simplified water management, HT PEMFCs are highly promising for combined heat and power generation, and can be directly fed with a reformate-based gas, avoiding expensive fuel purification processes. Although currently HT PEMFCs utilize Pt at the anode and at the cathode, non-platinum group catalysts may become a viable alternative, especially considering faster reaction kinetics under the high temperature operation.19 PA being weakly bound to the membrane, its dynamics in the catalytic layer and in the membrane-electrode assembly plays a key role in the operation and the degradation of HT PEMFCs. While conventional diagnostic tools provide valuable information regarding the global performance of a fuel cell (such as current–voltage characteristics, cell resistance, etc.20), they can only deliver indirect information on the PA distribution within a membrane-electrode assembly (MEA) under the operation conditions. Post-mortem analyses (porosimetry, acid-base titrations, gravimetric analyses21) are usually used to analyze the PA distribution in a HT PEMFC. In this work, by using a model MEA of a HT PEMFC and combining NAPXPS with the density functional theory (DFT) calculations, we observe potential-induced dissociation of PA and its redistribution in the catalytic layer in real time.
Results and discussion
Fig. 1 shows the sketch of the model MEA, integrated in the measurement chamber of a NAPXPS spectrometer (A), and the SEM image of the working electrode (WE) side of the MEA (B). Pt nanoparticles, with the average size of ca. 3 nm (see TEM images in ref. 22) were assembled in ca. 30 nm agglomerates. The cyclic voltammogram of the MEA shown in Fig. 1C, exhibits features characteristic of a Pt electrode. Negative currents at VWE–CE < −0.7 V correspond to the hydrogen evolution (confirmed by the mass spectrometry (MS) data), while positive currents at VWE–CE > 0.5 V correspond to the Pt surface oxide formation. In what follows this will be corroborated using the NAPXPS. Note that an absence of hydrogen adsorption/desorption peaks is typical for a high temperature low humidity operation.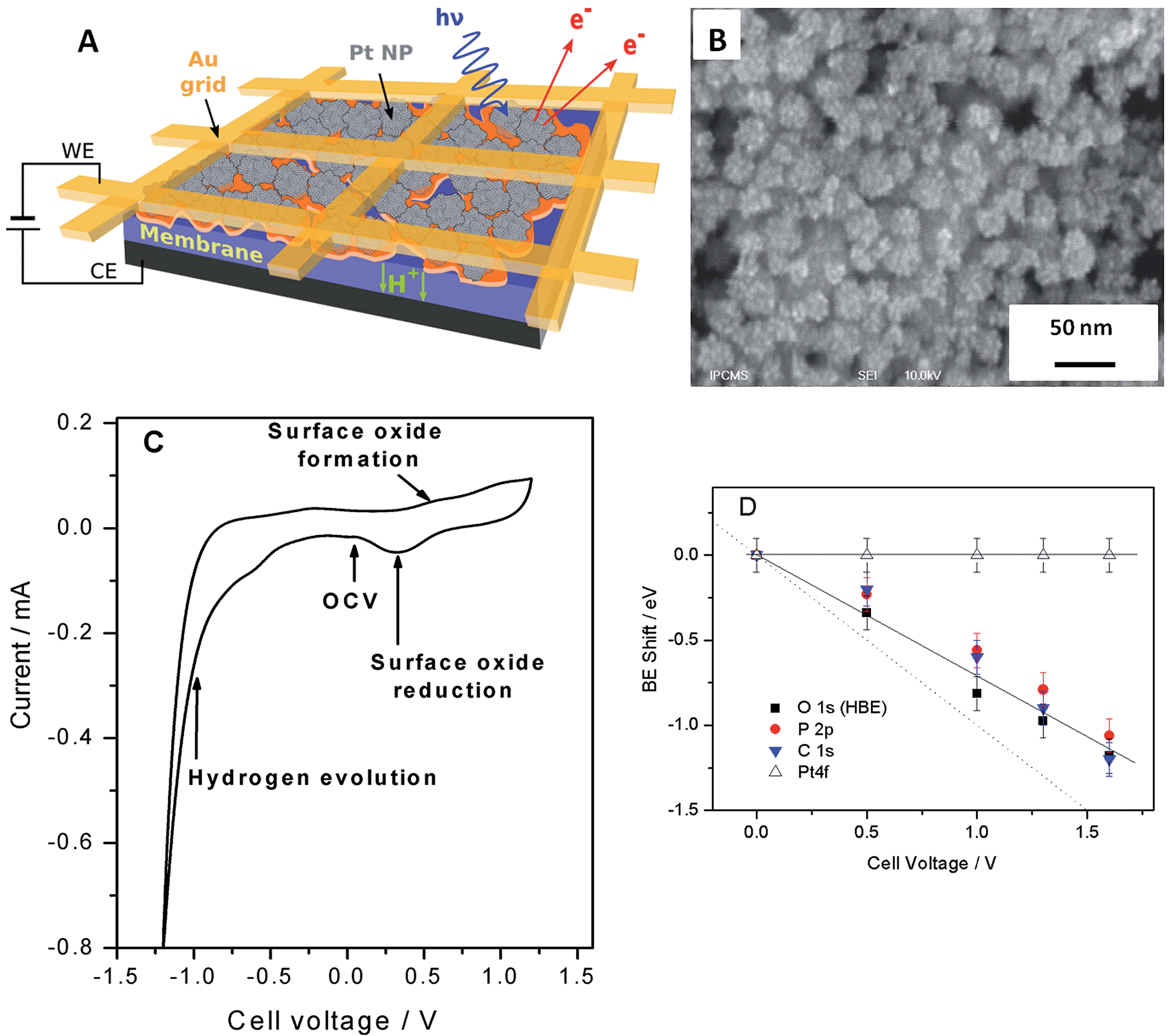 | ||
Fig. 1 (A) Schematic representation of a model membrane-electrode assembly. (B) Top view SEM image of the porous nanostructured Pt film applied as the working electrode. (C) Cyclic voltammogram recorded in the chamber of the NAPXP spectrometer at 180 °C and 0.3 mbar H2O at a sweep rate of 20 mV s−1. (D) BE shifts relative to the OCV for Pt 4f, C 1s, O 1s and P 2p extracted from the analysis of the experimentally measured NAPXP spectra at 180 °C and 0.3 mbar H2O. Solid lines are guides for the eye. Dotted line corresponds to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 correlation between the BE shift and the cell bias. :1 correlation between the BE shift and the cell bias. | ||
We now apply voltage between the WE and the counter electrode (CE), in a stepwise manner, and acquire XP spectra of the elements present on the surface of the working electrode. Fig. 2 demonstrates Pt 4f, O 1s, P 2p and C 1s spectra at different cell voltages. Note that in this work we focus on phenomena occurring under positive voltage bias, and thus relevant to the operation conditions of a cathode of a fuel cell or an anode of an electrolyzer. The first important question is whether the interface between Pt particles and the electrolyte is polarized under the conditions of a NAPXPS experiment. Ladas et al.,23 when studying a model solid oxide fuel cell with XPS, at that time under ultra-high vacuum conditions, observed binding energy shifts (ΔBE) following, in a 1:1 correlation, the potential of a WE. Since in electrochemical systems the rates of interfacial processes are related to the exponent of the potential difference between an electronic and an ionic conductor, the in situ information on the interfacial potentials is vital.
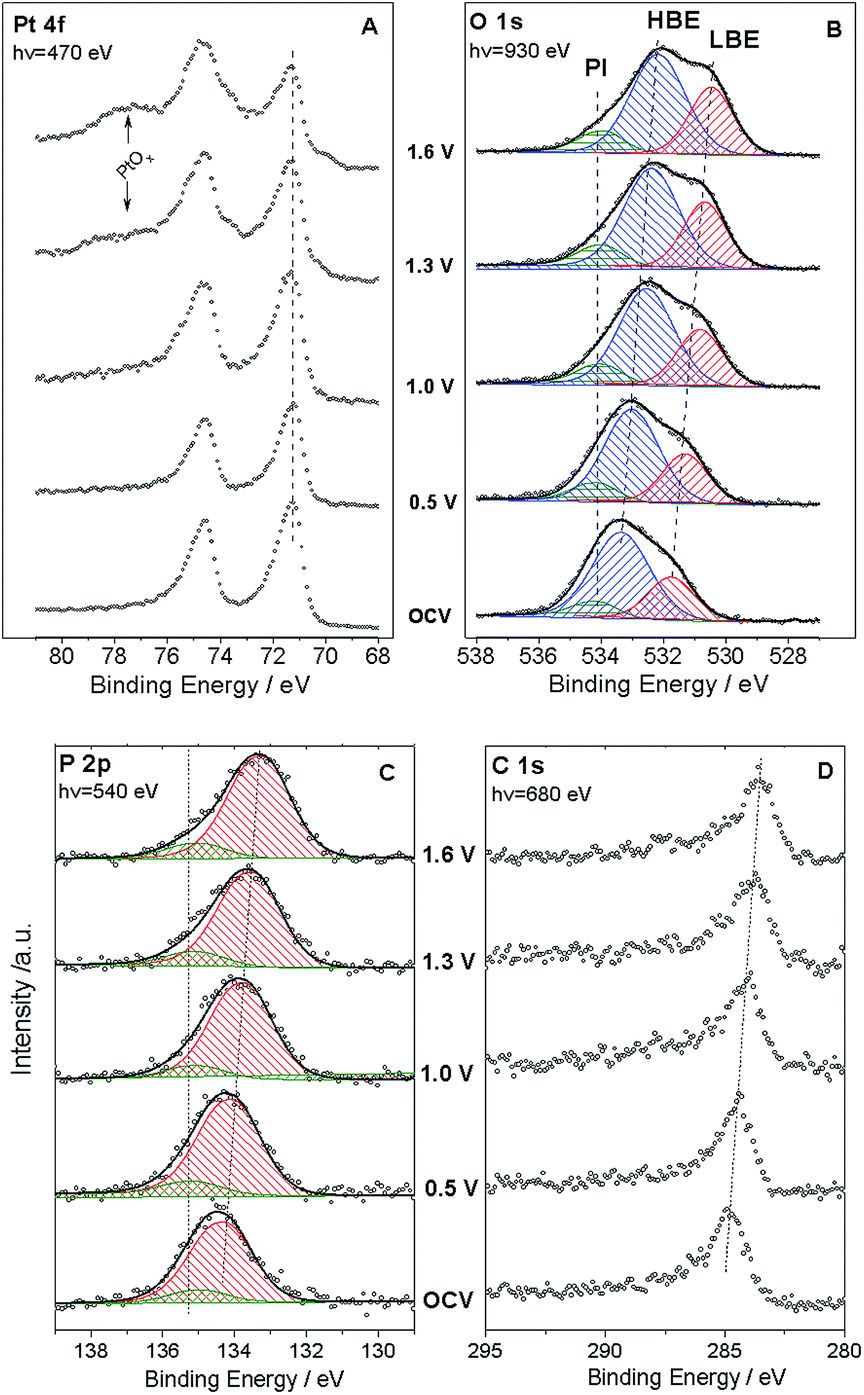 | ||
| Fig. 2 Pt 4f (A), O 1s (B), P 2p (C), and C 1s (D) spectra acquired in situ under 0.3 mbar H2O and 180 °C at various cell voltages. Panels B and C also show the results of the O 1s and P 2p spectra deconvolution. See text for details. | ||
The analysis of spectra presented in Fig. 2A reveals that when a positive bias is applied between the WE and the CE, the position of the main Pt 4f peaks (corresponding to metallic Pt) stays constant. This is not surprising, since the WE is grounded to the spectrometer through the Au grid, and thus Fermi level of Pt is pinned to the Fermi level of the spectrometer. This also means that XP spectra of the electrode species do not provide direct information on the electrode polarization.
Meanwhile, the BEs of the elements originating from the electrolyte (O and P) and the HT membrane (C), shift to lower BEs, following polarization of the electrode/electrolyte interface. This shift arises from the fact that the binding energy scale is referenced to the Fermi level of the analyzer, and thus an increase of the potential difference at the electrode/electrolyte interface induces higher photoelectron kinetic energies, which translates into a decrease of the apparent BE of electrolyte species. We further note that the correlation between the cell bias and the BE shifts of the elements originating from the electrolyte is close but not quite equal to the 1:1 correlation observed by Ladas et al.23 This is due to the fact that unlike in ref. 23 no reference electrode (RE) was used in this work. The observed deviation of the BE shifts from the dotted line in Fig. 1D can be explained by the contribution of the ohmic losses in the membrane and overpotential at the counter electrode (see ESI†). Changes in the chemical environment of P and O also contribute to the deviation of their BE shifts from a straight line (vide infra).
Thus, NAPXPS provides a unique tool for probing the interfacial potential difference between an electrode and an electrolyte. Note that if some Pt clusters are eventually disconnected from the current collector, a potential-dependent doublet can be detected in the Pt 4f spectra (see a shoulder at the low BE side of the Pt 4f spectra at high voltage bias). At high positive potentials VWE–CE ≥ 1 V, the surface of Pt is oxidized, as shown by the modification of the peak shape and the appearance of a shoulder at the high BE side of Pt 4f peak in Fig. 2A. Detailed analysis of Pt oxidation, and its dependence on the gas ambience will be subject of a forthcoming publication.
We now turn to the analysis of spectra corresponding to electrolyte components. Quantitative elemental analysis indicates that the O 1s peak intensity is dominated by the PA electrolyte, the contribution of oxygen from either Pt oxides or polymer membrane being negligible (below 6%, see ESI†). At the open circuit voltage (OCV) the O 1s spectrum (Fig. 2B) shows at least two components: a low BE (LBE), and a high BE (HBE). These peaks simultaneously shift to lower BEs as positive voltage is applied to the WE, suggesting that they both belong to the electrolyte species. Also, as the voltage across the MEA exceeds 1.0 V, a tailing at the high BE side becomes clearly visible. This tailing can be fit with a potential independent (PI) peak at 534.1 eV. The independence of this peak of the voltage bias suggests that it originates from species which are electrically equilibrated with the Pt electrode and disconnected from the electrolyte. Likewise, a potential independent (PI) component can be observed in the P 2p spectra (Fig. 2C). Since our experimental set-up has limited spatial resolution, NAPXPS data alone do not allow one to unveil the nature of the PI peaks. The insight comes from our recent scanning photoelectron microscopy study22 which suggests that part of the Pt/PA interface is not polarized. This may be attributed to the presence of PA droplets (or a PA film) on top of the WE, which are disconnected from the bulk of the electrolyte and result in the appearance of potential-independent components in the O 1s and P 2p spectra. Note that this is characteristic of the high temperature dry operation conditions, since at lower operation temperature of 150 °C (not shown) we did not observe the PI component.
Deconvolution of the O 1s spectra into three (LBE, HBE, and PI) components indicates that while the contribution of the PI peak stays fairly constant at ca. 7–9%, the intensity ratio of the LBE and the HBE peaks strongly increases with the positive bias (Fig. 3A) evidencing significant transformations of the PA electrolyte in the interfacial region. Meanwhile, the P:O atomic ratio is potential-independent and amounts to 0.25 ± 0.03 (Fig. S2†). This value is in perfect agreement with the composition of ortho-phosphoric (ortho-PA) H3PO4 (P:O = 0.25), or (pyro-PA) acid H4P2O7 (P:O = 0.29) containing one water per H4P2O7 molecule. In what follows the abbreviation PA will be applied to both ortho- and pyro-phosphoric acid.
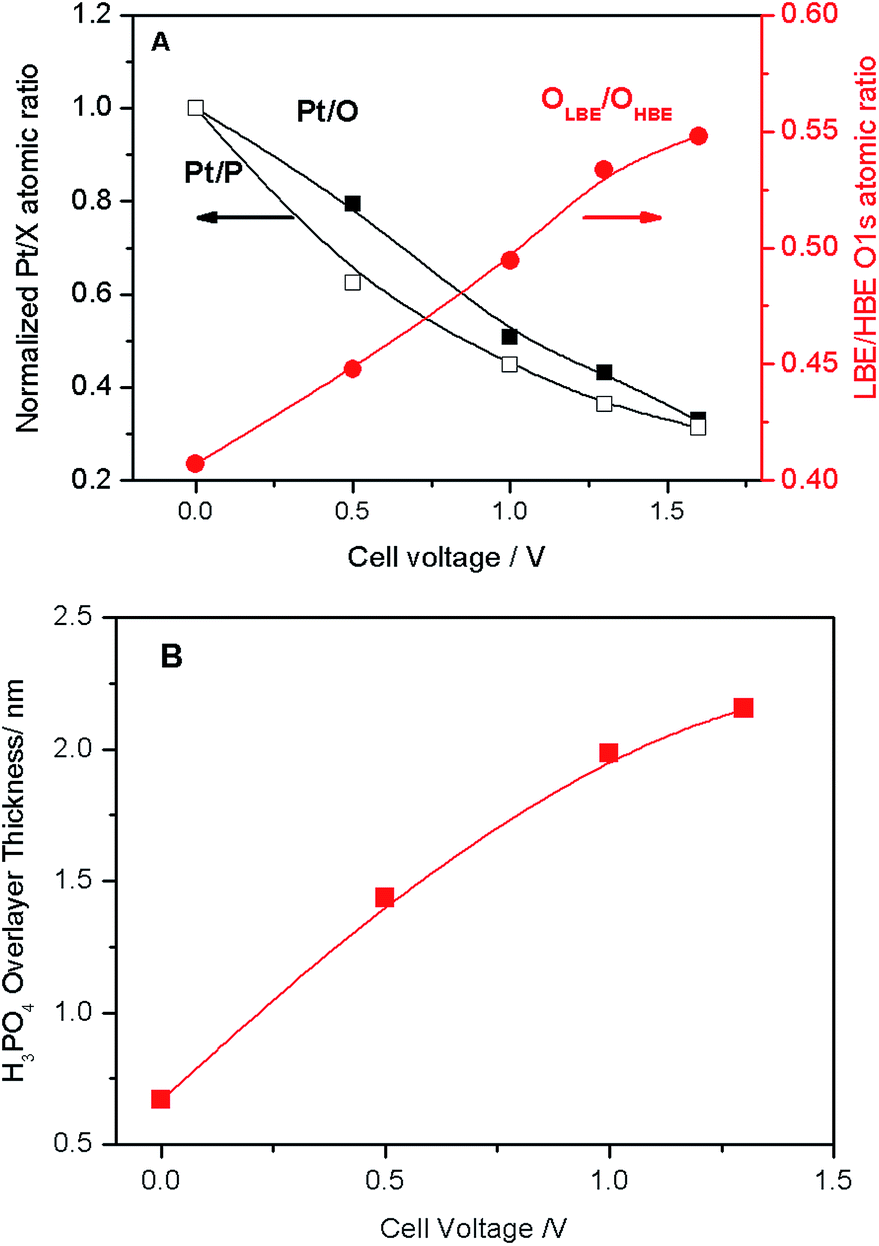 | ||
| Fig. 3 (A) Pt/O and Pt/P atomic ratios normalized to the corresponding values at the OCV (black left-hand-side axis), and LBE (OLBE) to HBE (OHBE) oxygen intensity (red right-hand-side axis) obtained via deconvolution of spectra presented in Fig. 2B as a function of the cell voltage. (B) Apparent thickness of the PA film calculated as a function of the applied cell voltage. | ||
In order to disentangle the complex O 1s spectra and to unveil their evolution with the voltage bias, O 1s and P 2p core levels were calculated by means of DFT for several model systems. Four types of phenomena were investigated: condensation of H3PO4 (eqn (1)), acid-base dissociation of H3PO4 (eqn (2)) and H4P2O7, hydrogen bonding between PA molecules and/or its anions, and adsorption of PA and its anionic fragments on the Pt surface.
| 2H3PO4 = H4P2O7 + H2O | (1) |
| 2H3PO4 = H2PO4− + H4PO4+ | (2) |
Table 1 summarizes BE shifts calculated using DFT for various molecular/ionic configurations, while Fig. 4 shows spectra simulated using the calculated BE shifts (for more details the reader could refer to Table S1 and Fig. S4 in the ESI†).
| Molecule/ion | Functional groupa | O 1s BE shiftb, eV |
|---|---|---|
a Respective O atom is underlined.
b The O 1s BE shifts are referred to P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O of a H3PO4 molecule in the gas phase. O of a H3PO4 molecule in the gas phase.
|
||
| H3PO4 | P![[O with combining low line]](https://www.rsc.org/images/entities/char_004f_0332.gif) |
0 |
| P––H |
2.0–2.1 | |
| H4P2O7 | PO |
0 |
| P––P |
2.3 | |
| P––H |
2.1 | |
| H2O | H2 |
1.2 |
| H3O+ | H3+ |
8 |
| H2PO4− or H3P2O7− | P–− |
−0.2–0 |
| P––H |
2.2 | |
| H4PO4+ | P––H+ |
8 |
| H-bonded H3PO4 and H4P2O7 | P⋯H |
0.5–1.2 |
| P––H⋯O |
1.6 | |
| H3PO4, H2PO4− or HPO42− chemisorbed on Pt | Pt––P |
1.45 ± 0.15 |
| H4P2O7 chemisorbed on Pt | Pt––P |
2.2 |
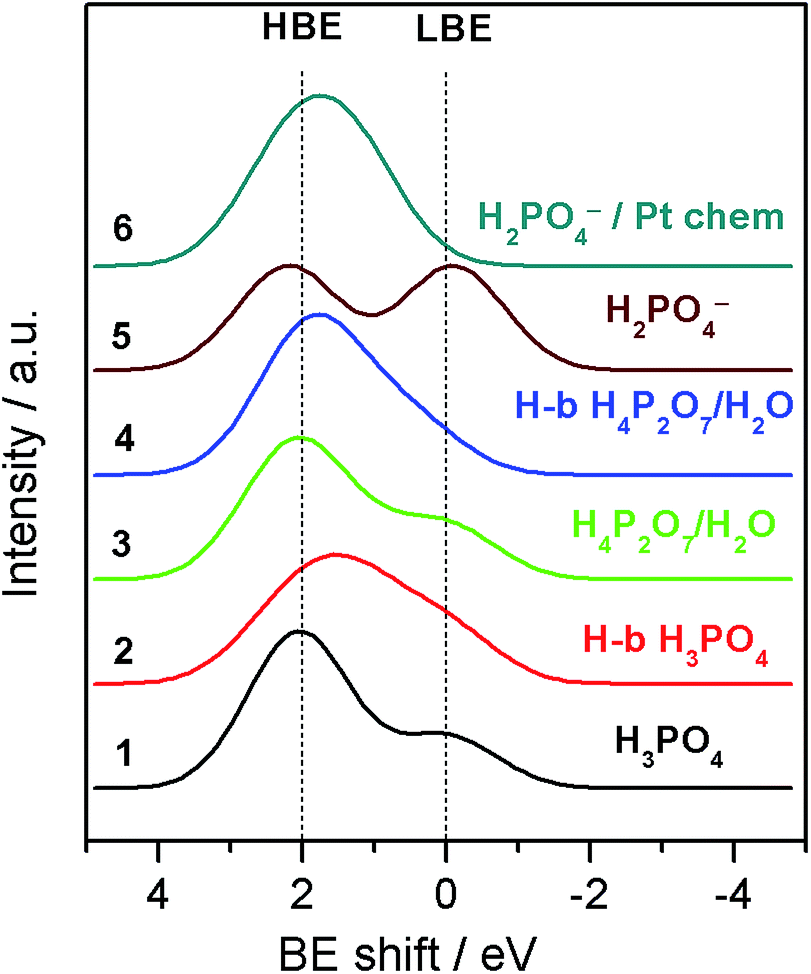 | ||
| Fig. 4 Simulated O 1s XP spectra for various molecular/ionic configurations using convolution of Gaussian shape peaks with relative BE shifts calculated by means of the DFT. For more details see the ESI.† | ||
For a free H3PO4 molecule the O 1s BE shift between P–OH and PO is around 2.0–2.1 eV in agreement with the experimental data.24,25 Thus, for a H3PO4 molecule in the gas phase the DFT predicts an O 1s spectrum consisting of two well defined HBE and LBE peaks with a ratio of 3:1 (Spectrum 1 in Fig. 4). The O 1s shift for P–O–P bond of the gaseous dimer H4P2O7 (pyro-PA) is 2.3 eV, only slightly (0.2 eV) higher than for the P–O–H bond. This calculation is in agreement with experimental measurements of O 1s spectra in ortho- and meta-phosphates, although no precise assignment of the P–O–H and P–O–P bonds was proposed in these studies.26,27 Close O 1s BE values for P–O–H and P–O–P bonds complicate spectroscopic separation of H4P2O7 from H3PO4. In agreement with the literature,28 hydrogen bonding also affects the BE shifts, leading to a decrease of the BE for an H bond donor, and its increase for a H-bond acceptor (see Tables 1 and S1†). H-bonding of H3PO4 and H4P2O7 molecules is expected to result in broadening of the HBE peak (see simulated spectra in Fig. 4 and S4†) due to the appearance of multiple O states with high (P–O–H, P–O–P) and intermediate (PO⋯H, P–O–H⋯O, H2O) values of the BE. Dissociation of the O–H bond in H3PO4 (eqn (2)) or H4P2O7 strongly affects the O 1s BE. For a negatively charged anion, the O 1s BE drops from ca. 2.1 (characteristic of P–O–H) down to ca. 0 eV (for P–O− fragment). Thus, for a H2PO4− anion DFT predicts an O 1s spectrum consisting of two fairly similar peaks separated by 2.2 eV (spectrum 5 in Fig. 4). Meanwhile, for a H4PO4+ cation, DFT predicts an O 1s BE shift to a very high value of ca. 8 eV. Chemisorption of phosphate anions on Pt results in a strong increase of the O 1s BE, the exact value depending on the type of anion, and on the adsorbate geometry. It is worth noting that according to our DFT calculations, the most stable configuration corresponds to HnPO4(3−n)− which maximizes the number of bonds with the Pt surface. The rationale behind the calculated BE shifts is an increase of the BE with the (i) coordination number of oxygen and (ii) covalency of the O–X bond.29
Examination of the calculated P 2p signal with the DFT reveals that it is virtually independent either of the condensation of molecules (pyro- vs. ortho-phosphate), the PA dissociation, or the adsorption of PA or its anions onto the Pt surface. The BE difference between the P2p and the O 1s (gas molecule reference) was equal to 383.5 eV ± 0.2 eV regardless the type of species considered, suggesting that the P 2p peak is rather insensitive to the chemical state of oxygen due to the electron redistribution in the P–O bond.
Armed with the theoretical values of the BE shifts for various states of PA and its anions we analyze the O 1s spectra and their evolution with the applied voltage bias. Even if close values of the BE shifts for certain PA configurations prevent us from an unambiguous assignment of each of the O-containing species, the results of the DFT calculations allow us to explain the potential-induced evolution of the LBE to the HBE O 1s peak ratio. First, we conclude that even if H3PO4 condensation to H4P2O7 leads to a certain increase of the LBE/HBE O 1s peak ratio, the latter is too small to fully account for the observed potential evolution of the shape of the O 1s spectra (cf. spectra 1 and 3 in Fig. 4). This goes in line with the potential independence of the P:O atomic ratio (Fig. S2†).
Second, broader HBE vs. LBE O 1s peak in the experimental spectra (Fig. 2B) is likely to originate from the H-bonding of PA molecules. However, considering the shape of the simulated spectra for H-bonded PA molecules, one can safely conclude that the potential-induced increase in the LBE/HBE ratio cannot be explained by the variation in the extent of H-bonding in the PA electrolyte. Finally, we arrive at the conclusion that significant increase of the LBE to the HBE peak intensity ratio can only be rationalized through dissociation of the O–H bond (eqn (2)) to produce negatively charged phosphate anions (spectrum 5 in Fig. 4). This, of course, does not rule out condensation and/or H-bonding of the PA molecules, but rather means that, contrary to the PA dissociation, these processes are virtually independent of the interfacial polarization. Formation of PA anions can be justified by the electroneutrality considerations. Indeed, under positive applied bias Pt particles are charged positively, and thus negatively charged species are required to compensate the positive electrode charge by forming the so-called electric double layer at the electrode/electrolyte interface. However, if negatively charged phosphate anions were chemisorbed, forming P–O–Pt bonds, the LBE peak would disappear, since bonding of oxygen to Pt results in an increase of the O 1s BE (cf. spectra 5 and 6 in Fig. 4).
Thus, we conclude that an increase of the relative intensity of the LBE O 1s peak at positive working electrode potentials can only be explained by the formation of PA anions which do not form chemical bonds with the Pt surface. This can occur if the anions are screened from the surface by their solvation shells. Electrochemists usually name this type of adsorption (physisorption) as non-specific, contrary to specific adsorption (chemisorption) accompanied by the chemical bond formation.
It is interesting to note that even at the OCV, where Pt surface is metallic (see cyclic voltammogram in Fig. 1C) and where, according to the literature data,30–32 the coverage of chemisorbed phosphate anions is expected to be at its maximum, the shape of the experimental O 1s spectrum does not match the one calculated for H2PO4− anion chemisorbed on Pt (cf. the OCV spectrum in Fig. 2B and spectrum 6 in Fig. 4). Thus, we conclude that even at the OCV the surface coverage of the Pt electrode with chemisorbed PA anions is relatively low. We attribute this to the high operation temperature of 180 °C (the phosphate coverage decreasing strongly with the temperature30), and the small size of Pt particles (3 nm). Indeed, it was found that the adsorption of phosphate anions on metallic Pt is structure-sensitive, for low index planes decreasing in the order of Pt(111) > Pt(100) > Pt(110).33 Hence, chemisorption of phosphate anions on the surface of metallic Pt is expected to decrease with the decrease of the particle size due to the decrease of the size of (111) facets. As the voltage bias is increased above the OCV, Pt surface oxide is formed. Literature data suggest that the coverage of specifically adsorbed (chemisorbed) phosphate anions decreases upon oxidation of the electrode surface.30–32 Meanwhile, the coverage of physisorbed solvated PA anions in the electric double layer will increase to compensate the positive electrode charge. The experimentally measured LBE/HBE O 1s ratio is indicative of a potential-dependent extent of PA dissociation in the electric double layer, roughly estimated as ca. 20% at the applied bias of 1.6 V. Even if, given the complexity of O 1s spectra illustrated by Fig. S4,† this estimation is very rough, and neglects the attenuation of photoelectrons in the electrolyte film, it is clear that this value much exceeds the degree of self-ionization of ortho-PA of ca. 7.4%.34
We note in passing that we observed no experimental trace of H4PO4+ cations at 8 eV vs. the PO reference. At positive electrode bias this may be explained by migration of cations towards the negatively charged counter electrode. Puzzled by the fact that no O 1s peak was found at 8 eV even at negative electrode bias (not shown), we used DFT to estimate the BE of H4PO4+ chemisorbed on Pt. It appeared that P–O–Pt bond formation results in the charge re-distribution and in the concomitant BE shift from 8 eV for a free H4PO4+ down to 3 or even 2 eV for a chemisorbed cation (depending on its orientation relative to the Pt surface). The latter value is close to that characteristic of either P–O–H or P–O–P fragment. It is thus likely that at negative electrode bias H4PO4+ cations, which compensate the negative charge of Pt, are chemisorbed on the electrode surface, which hinders their detection.
The voltage bias not only affects the ratio between the LBE and the HBE O 1s peaks, but also the Pt/P and Pt/O atomic ratios. Fig. 3A shows that Pt/P and Pt/O ratios strongly decrease with the positive polarization suggesting that Pt is covered by electrolyte species comprising PA and its anions. Reversal of the cell voltage induces an increase of the Pt/P atomic ratio, even though the reverse scan usually shows a hysteresis suggesting slow kinetics of the underlying processes (Fig. S5†). Tuning the photon energy allows one to vary the electron penetration depth and thus to estimate the apparent thickness of the PA/phosphate overlayer covering Pt nanoparticles. This strongly depends on the applied cell voltage increasing from ca. 0.8 nm at the OCV to ca. 2.2 nm at VWE–CE = 1.6 V (Fig. 3B). An effective monolayer thickness being estimated as 0.44 nm, the thickness of the PA overlayer at VWE–CE = 1.6 V corresponds to ca. 5 monolayers.
Thus, we infer that application of the positive bias to the Pt working electrode not only leads to the migration of phosphate anions to the surface of Pt particles, to form an electric double layer, but, since they move together with their solvation shells, also to the transport of PA molecules towards the working electrode. Note that the Pt/P(O) atomic ratio is also influenced by the potential-dependent surface tension changes.22
The experimental data supported with the DFT calculations allow us to propose the following model summarized in Fig. 5. As the voltage across the MEA is increased above the OCV, the positive charge density on Pt nanoparticles increases. Since the degree of self-ionization of ortho-PA is small (7.4%),34 the applied voltage induces dissociation of PA (eqn (2)) to provide negatively charged ions (H2PO4− and possibly H3P2O7−) required for the compensation of the positive charge of the WE. Negatively charged phosphate anions migrate to the working electrode, while positively charged species (H4PO4+) migrate to the counter electrode. Since phosphate anions are solvated by PA molecules,¶ their migration towards the positively charged Pt electrode leads to the transport of PA to the working electrode via electroosmotic effect and results in the formation of a film composed from PA molecules and its anions.
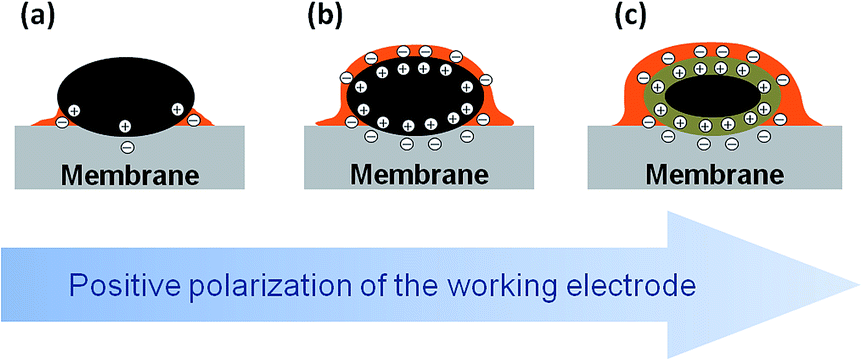 | ||
| Fig. 5 Schematic representation of Pt/PA interface under the potential control. The figure schematically shows a metallic Pt particle (black), phosphoric acid electrolyte (orange), and membrane (gray) at the OCV (a), and at an increasing positive bias (b) and (c). One may see accumulation of the positive charge at the surface of Pt particles (shown as encircled plus signs), and negative charge at the electrolyte side (encircled minus signs) from (a) to (b), and then (c), “flooding” of the surface of Pt particles with PA electrolyte in (b) and (c), and formation of the oxide layer (taupe) on the surface of Pt particles in (c). | ||
It is widely accepted that the penetration of PA into the working electrode has major influence on the HT PEMFC performance. Either excessive penetration of the PA into the WE (the so-called “flooding”), or the electrode drying have detrimental effects on the cell performance.20 This work shows that the thickness of the PA layer on the Pt surface strongly depends on the electrode potential. Therefore, controlling the cell voltage (along with the temperature and humidity) in such a way that an optimum film thickness is maintained could be used as a means to increase the HT PEMFC performance.
This work opens up an opportunity to visualize the interface between the catalyst and a liquid or polymer electrolyte under operation conditions and thus to help in better understanding of the chemical state of materials of electrochemical power sources and electrolyzers. Further studies will be performed in the presence of reactive H2, O2, and other gases.
Experimental
The MEA consisted of a working electrode comprised of a thin layer of Pt nanoparticles, a PA imbibed polymer membrane based on aromatic polyethers bearing pyridine groups (Advent Technologies S.A.), and a high loading carbon-supported Pt/C CE. Utilization of unsupported Pt at the WE is essential for minimizing formation of C–O bonds at positive electrode bias, which might overlap with the electrolyte-relevant O features in the O 1s XP spectra. Except for using an unsupported Pt on the WE side, the MEA was very similar to those used in a real HT PEMFC. For further details concerned with the preparation of the nanostructured Pt film and the MEA the reader is referred to ref. 22. A kapton film with a window in the middle was inserted between the membrane and the counter electrode to avoid electronic contact between the working and the counter electrode.Scanning electron microscopy (SEM)
SEM measurements were performed using Jeol JSM-6700F (Japan) electron microscope with the lattice resolution of 1 nm, at accelerating voltage of 10 kV.Near ambient pressure X-ray photoelectron spectroscopy (NAPXPS)
NAPXPS measurements were performed at the ISISS beamline of the BESSY synchrotron facility at the Helmholtz Zentrum Berlin, which allows measurements at pressures in the millibar range, in a setup described elsewhere.5,6 The samples were placed on a sample holder, where the electrical contact of the CE was made through the stainless steel back plate, while the contact of the WE was made through a 80 mesh gold grid, which exposed part of the platinum working electrode surface to the X-ray beam. The sample could be heated from the rear by an IR laser (cw, 808 nm). The temperature was calibrated in a separate experiment using a K-type thermocouple fixed onto the sample surface. H2O flow into the analysis chamber was controlled via calibrated mass flow controllers. A differentially pumped quadrupole mass spectrometer was connected through a leak valve to the experimental cell and the gas phase composition was monitored by online mass spectrometry simultaneously to the spectroscopic characterization of the surface. The temperature and the gaseous environment were maintained the same throughout the experiment, i.e. at 180 °C and 0.3 mbar H2O. We observed that the HT MEA was able to stably operate under these conditions for at least 24 hours, while in the absence of water the membrane dehydration resulted in a continuous increase of the cell resistance leading to a full collapse of the operation.If not otherwise stated, Pt 4f, P 2p, O 1s and C 1s spectra were recorded at selected photon energies, such that the photoelectrons were emitted at kinetic energy of 400 eV. XP spectra were acquired under applied constant voltage between the working and the counter electrode. Before performing potential-dependent measurements, dedicated experiments were made in order to rule out X-ray beam damage and the ensuing artifacts. The photoemission spectra were normalized by the storage ring current and the energy dependent incident photon flux, which was measured prior and after the measurements using a gold foil with known quantum efficiency. The photon flux obtained has been corrected for higher diffraction orders that contribute only to the background but not to the peak intensity in XPS. The BE scale was calibrated with respect to the Fermi level of the electron analyzer. Curve fitting of the O 1s peaks was performed based on a mixed Gaussian/Lorentzian function. Background subtraction was carried out using the Shirley method. Quantitative calculations were performed using normalized Pt 4f, P 2p, O 1s, C 1s intensities, taking into account the photon-energy dependence of the atomic subshell photoionization cross sections.35
Electrochemical measurements
A μ-AutoLab potentiostat from Metrohm was used to apply voltage between the WE and the CE (VWE–CE) and measure the current. Note that in this work the CE also served as a RE. In order to keep the potential of the RE/CE constant, the MEAs were formulated in such a way that the amount of Pt on the back side greatly exceeded that on the WE side.DFT calculations
DFT calculations of the binding energy (BE) shift were conducted using the VASP code36,37 for a number of test configurations, such as PA molecule and its oligomers, H-bonded molecules, phosphate ions, and phosphate ions adsorbed on Pt. The PBE GGA functional38,39 was used. The valence electrons are treated explicitly and their interactions with the ionic cores are described by the Projector Augmented-Wave method (PAW).40 The energy cutoff was set to 400 eV. Due to the large cell used, typically 11 Å × 11 Å × 40 Å, the calculations were performed at the Gamma point. Charged molecular systems were neutralized by a background charge. To compensate the negative charge of the adsorption of anionic phosphate on the Pt surface, protons were put on the bottom surface and relaxed. A dipolar correction was applied to eliminate spurious effects of cell–cell interactions along the z axis. BE shifts were evaluated at the initial state level (applying the Koopman's theorem) for various molecular configurations, as explained in previous ref. 41. To get an inner reference binding energy for O 1s, a PA molecule was introduced in the middle of the vacuum (without interactions with the studied molecules/systems) and the calculated O 1s BEs were systematically compared to the BE of oxygen in a PO functional group of the free PA molecule. For the H-bonded systems, the potential energy surface was not exhaustively explored. Instead, different local minima configurations were modeled in order to get some benchmarks on the binding energies. For each species, the number of configurations studied was sufficiently large to ensure that the calculated binding energies were representative.
Conclusions
This work demonstrates the potential of NAPXPS for in situ studies of processes occurring in a liquid electrolyte at its interface with a metal electrode. By performing NAPXPS measurements in a model membrane-electrode assembly of a HT PEMFC and combining them with the DFT calculations, it became possible to detect such fundamental processes as dissociation of PA, and migration of PA and its anions within a nanostructured Pt electrode. This work shows that the thickness of the PA film on the surface of Pt nanoparticles can be controlled by the cell bias (in addition to other parameters, like temperature and gas humidity). Since PA-imbibed HT PEMFC usually operate with unhumidified gases and at high temperatures, the current study is highly relevant to the “real” operation of a fuel cell/electrolyzer.Acknowledgements
We acknowledge the Helmholtz-Zentrum Berlin (electron storage ring BESSY II) for synchrotron radiation at ISISS beamline. Financial support from the International Center for Frontier Research in Chemistry (University of Strasbourg) and from the European Commission (FP7) under the projects DEMMEA, IRAFC and INSIDE is gratefully acknowledged.References
- G. J. Hansen and W. N. Hansen, J. Electroanal. Chem. Interfacial Electrochem., 1983, 150, 193 CrossRef CAS.
- E. M. Stuve, A. Krasnopoler and D. E. Sauer, Surf. Sci., 1995, 335, 177 CrossRef CAS.
- A. Shchukarev, Adv. Colloid Interface Sci., 2006, 122, 149 CrossRef CAS PubMed.
- R. Wibowo, L. Aldous, R. M. J. Jacobs, N. S. A. Manan and R. G. Compton, Chem. Phys. Lett., 2011, 517, 103 CrossRef CAS PubMed.
- M. Salmeron and R. Schlögl, Surf. Sci. Rep., 2008, 63, 169 CrossRef CAS PubMed.
- A. Knop-Gericke, E. Kleimenov, M. Hävecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schlögl, V. I. Bukhtiyarov, V. V. Kaichev, I. P. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, in Advances in Catalysis, ed. B. C. Gates and H. Knözinger, Elsevier, 2009, vol. 52, pp. 213–272 Search PubMed.
- F. Tao and M. Salmeron, Science, 2011, 331, 171 CrossRef CAS PubMed.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Révay, M. Hävecker, A. Knop-Gericke, S. D. Jackson and R. Schlögl, Science, 2008, 320, 86 CrossRef CAS PubMed.
- F. Tao, M. E. Grass, Y. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932 CrossRef CAS PubMed.
- V. Papaefthimiou, M. Shishkin, D. K. Niakolas, M. Athanasiou, Y. T. Law, R. Arrigo, D. Teschner, M. Hävecker, A. Knop-Gericke, S. Scholl, R. Schlögl, T. Ziegler, S. G. Neophytides and S. Zafeiratos, Adv. Energy Mater., 2013, 3, 690 CrossRef PubMed.
- C. Zhang, M. E. Grass, A. H. McDaniel, S. C. DeCaluwe, F. E. Gabaly, Z. Liu, K. F. McCarty, R. L. Farrow, M. A. Linne, Z. Hussain, G. S. Jackson, H. Bluhm and B. W. Eichhorn, Nat. Mater., 2010, 9, 944 CrossRef CAS PubMed.
- I. Valov, B. Luerssen, E. Mutoro, L. Gregoratti, R. A. De Souza, T. Bredow, S. Gunther, A. Barinov, P. Dudin, M. Martin and J. Janek, Phys. Chem. Chem. Phys., 2011, 13, 3394 RSC.
- R. Arrigo, M. Hävecker, M. E. Schuster, C. Ranjan, E. Stotz, A. Knop-Gericke and R. Schlögl, Angew. Chem., Int. Ed., 2013, 52, 11660 CrossRef CAS PubMed.
- H. S. Casalongue, S. Kaya, V. Viswanathan, D. J. Miller, D. Friebel, H. A. Hansen, J. K. Nørskov, A. Nilsson and H. Ogasawara, Nat. Commun., 2013, 4, 2817 Search PubMed.
- S. Axnanda, E. J. Crumlin, B. H. Mao, S. Rani, R. Chang, P. G. Karlsson, M. O. M. Edwards, M. Lundqvist, R. Moberg, P. Ross, Z. Hussain and Z. Liu, Sci. Rep., 2015, 5, 9788 CrossRef PubMed.
- H. G. S. Casalongue, J. D. Benck, C. Tsai, R. K. B. Karlsson, S. Kaya, M. L. Ng, L. G. M. Pettersson, F. Abild-Pedersen, J. K. Nørskov, H. Ogasawara, T. F. Jaramillo and A. Nilsson, J. Phys. Chem. C, 2014, 118, 29252 CAS.
- H. G. Sanchez-Casalongue, M. L. Ng, S. Kaya, D. Friebel, H. Ogasawara and A. Nilsson, Angew. Chem., Int. Ed., 2014, 53, 7169 CrossRef CAS PubMed.
- J. Zhang, Z. Xie, J. Zhang, Y. Tang, C. Song, T. Navessin, Z. Shi, D. Song, H. Wang, D. P. Wilkinson, Z. S. Liu and S. Holdcroft, J. Power Sources, 2006, 160, 872 CrossRef CAS PubMed.
- G. Papakonstantinou, M. Daletou, A. Kotsifa, F. Paloukis, K. Katerinopoulou, T. Ioannides and S. G. Neophytides, ECS Trans., 2009, 25, 181 CAS.
- A. Kamat, A. Huth, O. Klein and S. Scholl, Fuel Cells, 2010, 10, 983 CrossRef CAS PubMed.
- Y. Oono, A. Sounai and M. Hori, J. Power Sources, 2009, 189, 943 CrossRef CAS PubMed.
- W. H. Doh, L. Gregoratti, M. Amati, S. Zafeiratos, Y. T. Law, S. G. Neophytides, A. Orfanidi, M. Kiskinova and E. R. Savinova, ChemElectroChem, 2014, 1, 180 CrossRef CAS PubMed.
- S. Ladas, S. Kennou, S. Bebelis and C. G. Vayenas, J. Phys. Chem., 1993, 97, 8845 CrossRef CAS.
- A. M. Puziy, O. I. Poddubnaya, R. P. Socha, J. Gurgul and M. Wisniewski, Carbon, 2008, 46, 2113 CrossRef CAS PubMed.
- M. J. Ariza, E. Rodríguez-Castellón, R. Rico, J. Benavente, M. Muñoz and M. Oleinikova, Surf. Interface Anal., 2000, 30, 430 CrossRef CAS.
- J. A. Rotole and P. M. A. Sherwood, Chem. Mater., 2001, 13, 3933 CrossRef CAS.
- J. A. Rotole and P. M. A. Sherwood, Surf. Sci. Spectra, 1998, 5, 60 CrossRef CAS.
- B. Diawara, L. Joubert, D. Costa, P. Marcus and C. Adamo, Surf. Sci., 2009, 603, 3025 CrossRef CAS PubMed.
- S. J. Kerber, J. J. Bruckner, K. Wozniak, S. Seal, S. Hardcastle and T. L. Barr, J. Vac. Sci. Technol., A, 1996, 14, 1314 CAS.
- P. Zelenay, B. R. Scharifker, J. O. Bockris and D. Gervasio, J. Electrochem. Soc., 1986, 133, 2262 CrossRef CAS PubMed.
- M. Weber, F. C. Nart, I. R. de Moraes and T. Iwasita, J. Phys. Chem., 1996, 100, 19933 CrossRef CAS.
- A. Kamat, M. Herrmann, D. Ternes, O. Klein, U. Krewer and S. Scholl, Fuel Cells, 2011, 11, 511 CrossRef CAS PubMed.
- Q. He, X. Yang, W. Chen, S. Mukerjee, B. Koel and S. Chen, Phys. Chem. Chem. Phys., 2010, 12, 12544 RSC.
- R. A. Munson, J. Phys. Chem., 1964, 68, 3374 CrossRef CAS.
- J. J. Yeh and I. Lindau, At. Data Nucl. Data Tables, 1985, 32, 1 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Hafner, J. Phys.: Condens. Matter, 1994, 6, 8245 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- D. Costa and P. Marcus, Surf. Sci., 2010, 604, 932 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc01421b |
| ‡ Present Addresses: Yeuk Ting Law, University Ulm, Institute of Surface Chemistry and Catalysis, Albert-Einstein-Allee 47, D-89069 Ulm, Germany, e-mail: E-mail: yeuk-ting.law@uni-ulm.de. |
| § Present Addresses: Alin Orfanidi, Department of Chemistry, Technische Universität München, Lichtenbergstrasse 4, 85 748 Garching, Germany, e-mail: E-mail: alin.orfanidi@tum.de. |
| ¶ Note that the experimental O 1s and P 2p XP spectra do not allow to unambiguously distinguish H3PO4 from H4P2O7. However, the experimental P/O ratio suggests that if PA in the MEA is present in the form of a dimer (H4P2O7), it should also contain water, roughly in 1 H2O to 1 H4P2O7 ratio. |
| This journal is © The Royal Society of Chemistry 2015 |