
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe(IV) alkylidenes via protonation of Fe(II) vinyl chelates and a comparative Mössbauer spectroscopic study†
Brian M.
Lindley
a,
Ala'aeddeen
Swidan
a,
Emil B.
Lobkovsky
a,
Peter T.
Wolczanski
*a,
Mario
Adelhardt
b,
Jörg
Sutter
b and
Karsten
Meyer
b
aDepartment of Chemistry & Chemical Biology, Baker Laboratory, Cornell University, Ithaca, NY 14850, USA. E-mail: ptw2@cornell.edu; Fax: +1 607 255 4137; Tel: +1 607 255 7220
bDepartment of Chemistry & Pharmacy, Friedrich Alexander University Erlangen-Nürnberg (FAU), Egerlandstr. 1, D-91058 Erlangen, Germany
First published on 20th May 2015
Abstract
Treatment of cis-Me2Fe(PMe3)4 with di-1,2-(E-2-(pyridin-2-yl)vinyl)benzene ((bdvp)H2), a tetradentate ligand precursor, afforded (bdvp)Fe(PMe3)2 (1-PMe3) and 2 equiv. CH4, via C–H bond activation. Similar treatments with tridentate ligand precursors PhCH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) NCH2(E-CHCHPh) ((pipp)H2) and PhCHN(2-CCMe-Ph) ((pipa)H) under dinitrogen provided trans-(pipp)Fe(PMe3)2N2 (2) and trans-(pipvd)Fe(PMe3)2N2 (3), respectively; the latter via one C–H bond activation, and a subsequent insertion of the alkyne into the remaining Fe–Me bond. All three Fe(II) vinyl species were protonated with H[BArF4] to form the corresponding Fe(IV) alkylidene cations, [(bavp)Fe(PMe3)2][BArF4] (4-PMe3), [(piap)Fe(PMe3)3][BArF4] (5), and [(pipad)Fe(PMe3)3][BArF4] (6). Mössbauer spectroscopic measurements on the formally Fe(II) and Fe(IV) derivatives revealed isomer shifts within 0.1 mm s−1, reflecting the similarity in their bond distances.
NCH2(E-CHCHPh) ((pipp)H2) and PhCHN(2-CCMe-Ph) ((pipa)H) under dinitrogen provided trans-(pipp)Fe(PMe3)2N2 (2) and trans-(pipvd)Fe(PMe3)2N2 (3), respectively; the latter via one C–H bond activation, and a subsequent insertion of the alkyne into the remaining Fe–Me bond. All three Fe(II) vinyl species were protonated with H[BArF4] to form the corresponding Fe(IV) alkylidene cations, [(bavp)Fe(PMe3)2][BArF4] (4-PMe3), [(piap)Fe(PMe3)3][BArF4] (5), and [(pipad)Fe(PMe3)3][BArF4] (6). Mössbauer spectroscopic measurements on the formally Fe(II) and Fe(IV) derivatives revealed isomer shifts within 0.1 mm s−1, reflecting the similarity in their bond distances.
Introduction
Homogeneous alkylidene compounds that catalyze olefin metathesis1–5 typically contain 2nd row transition metals that have modest limitations regarding functionality tolerance (e.g., Mo),1,2 and relative abundance (e.g., Ru).3,4 Applications for commodity chemicals production have been hampered by these factors, and inexpensive first row transition metal alternatives hold great promise for solving some of the problems. Thus far, the synthesis of 1st row transition metal alkylidene complexes has presented a significant challenge to the organometallic community, especially in the case of iron.Electronic structure analysis by Hoffmann et al.6 suggests that metathesis catalysts need to be dn (n ≤ 4), hence Fe(IV) alkylidenes are the target of interest, especially in analogy to their 2nd row congeners. Several Fe(IV) alkylidenes have been synthesized, with two routes utilized in the cases of those structurally characterized (Fig. 1): (1) conversion of [CpLL′FeC(OR)R′]+via hydride or alkyl anion reagents,7–9 and (2) the addition of diazoalkanes, typically Ph2CN2, to coordinatively unsaturated complexes or labile precursors.10–13 The subsequent chemistry has been limited to carbene transfers, and some transformations that hint at radical reactions.
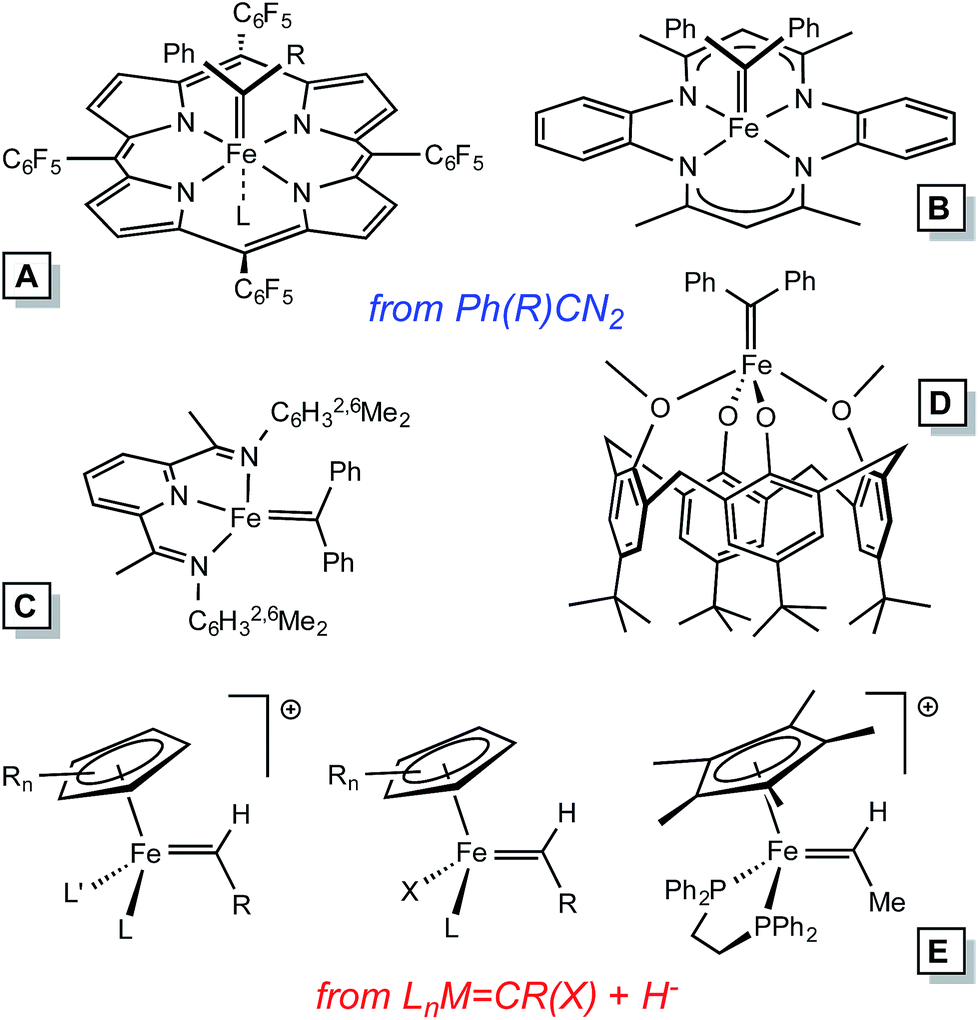 | ||
| Fig. 1 Some iron alkylidenes and methods of synthesis. | ||
In an effort to expand the scope of Fe(IV) alkylidenes, and to develop new synthetic paths, Fe(II) vinyl chelates have been explored as potential precursors to cationic Fe(IV) alkylidenes via protonation.14–18 Entry into ferrous vinyl derivatives was implemented via precedented C–H bond activations by Karsch's cis-Me2Fe(PMe3)4 (ref. 19) complex.20–23 While viable olefin metathesis catalysts containing Fe have not yet been realized, the generality of this approach suggests that incremental advances may yet prove successful.
Results and discussion
Di-1,2-(E-2-(pyridin-2-yl)vinyl)benzene: tetradentate ligand precursor
As Scheme 1 illustrates, incorporation of two vinyl groups into a tetradentate chelate precursor was predicated on successful implementation of a Horner–Wadsworth–Emmons reaction to achieve the requisite E-stereochemistry. The modified 2-pyridinyl-methyl reagent was prepared according to a literature procedure24 from Na[OP(OEt)2] and 2-pyridyl-methylchloride in 70% yield. Its addition to 1,2-benzene-dialdehyde afforded di-1,2-(E-2-(pyridin-2-yl)vinyl)benzene ((bdvp)H2, E/Z > 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) in 37% yield upon crystallization from ether/hexane.
:1) in 37% yield upon crystallization from ether/hexane.
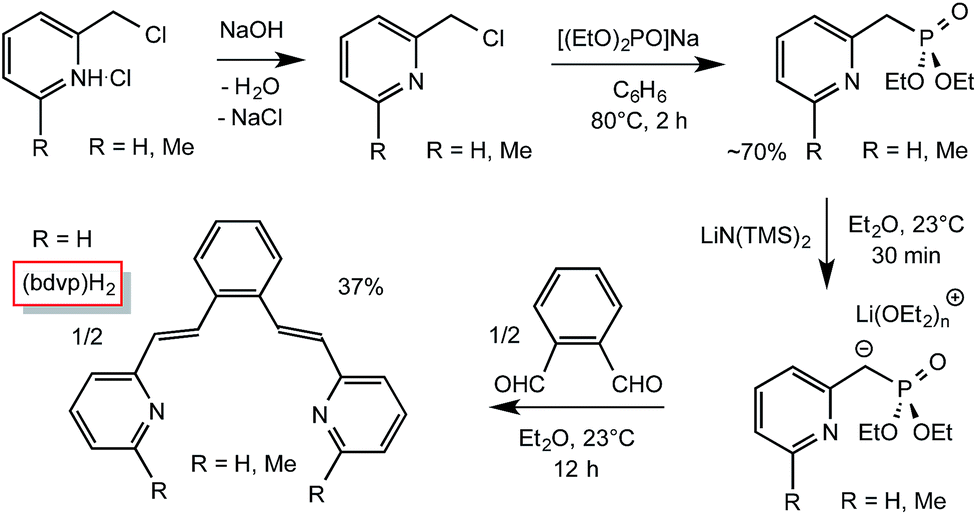 | ||
| Scheme 1 Preparation of the divinyl ligand precursor, 1,2-(E-2-(pyridin-2-yl)vinyl)benzene ((bdvp)H2). | ||
Tridentate ligand precursors: PhCHNCH2(E-CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h3_char_e001.gif) CHPh) and PhCHN(2-CCMe-Ph)
CHPh) and PhCHN(2-CCMe-Ph)
Condensation routes afforded the two additional tridentate ligands used in this study, as shown in Scheme 2. Cinnamyl amine25 and benzaldehyde were used to synthesize PhCHNCH2(E-CHCHPh) ((pipp)H2),26 while 2-propynyl-aniline, prepared from Pd-catalyzed cross-coupling27 of propyne and 2-iodo-aniline, and benzaldehyde were used to generate PhCHN(2-CCMe-Ph) ((pipa)H). A number of other potential imine and pyridine-containing tridentate ligand precursors were similarly made, but the subsequent C–H bond activations proved to be too slow or ineffective, allowing for competitive cis-Me2Fe(PMe3)4 degradation.
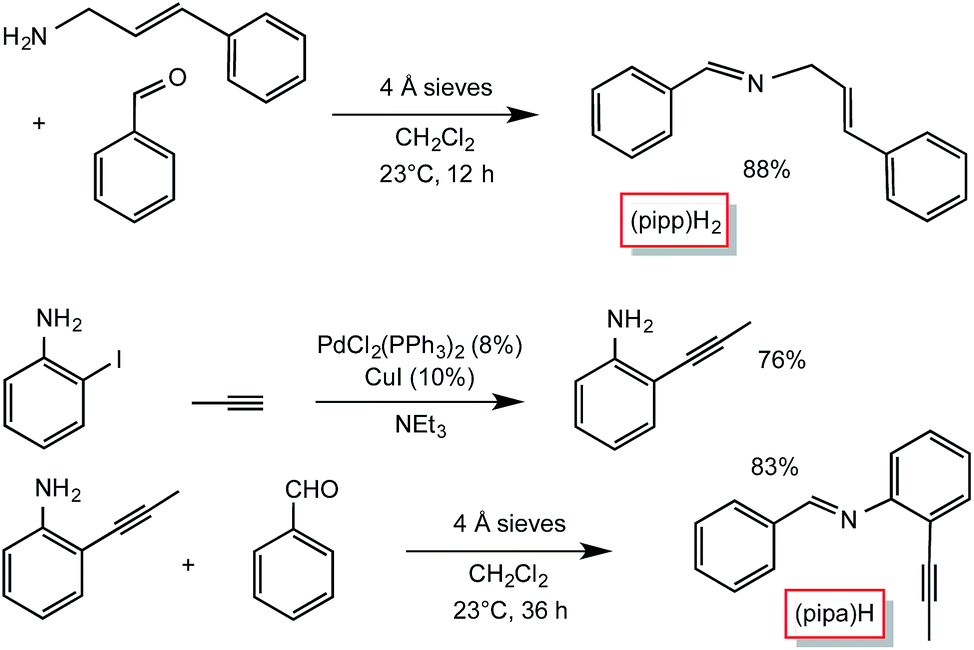 | ||
| Scheme 2 Preparation of the tridentate precursors, PhCHNCH2(E-CHCHPh) ((pipp)H2) and PhCHN(2-CCMe-Ph) ((pipa)H). | ||
Metalation via C–H bond activation and insertion
Treatment of cis-Me2Fe(PMe3)4 (ref. 19) with the tetradentate precursor 1,2-(E-2-(pyridin-2-yl)vinyl)benzene ((bdvp)H2) was undertaken at −20 °C in toluene. After 10 h, the solution was warmed to 23 °C and concentrated to afford (bdvp)Fe(PMe3)2 (1-PMe3) in 84% yield as purple microcrystals (Scheme 3). The reaction is quite sensitive to steric bulk, as use of a precursor possessing o-Me groups on the pyridines (Scheme 1) failed to metalate, and decomposition of cis-Me2Fe(PMe3)4 was instead observed.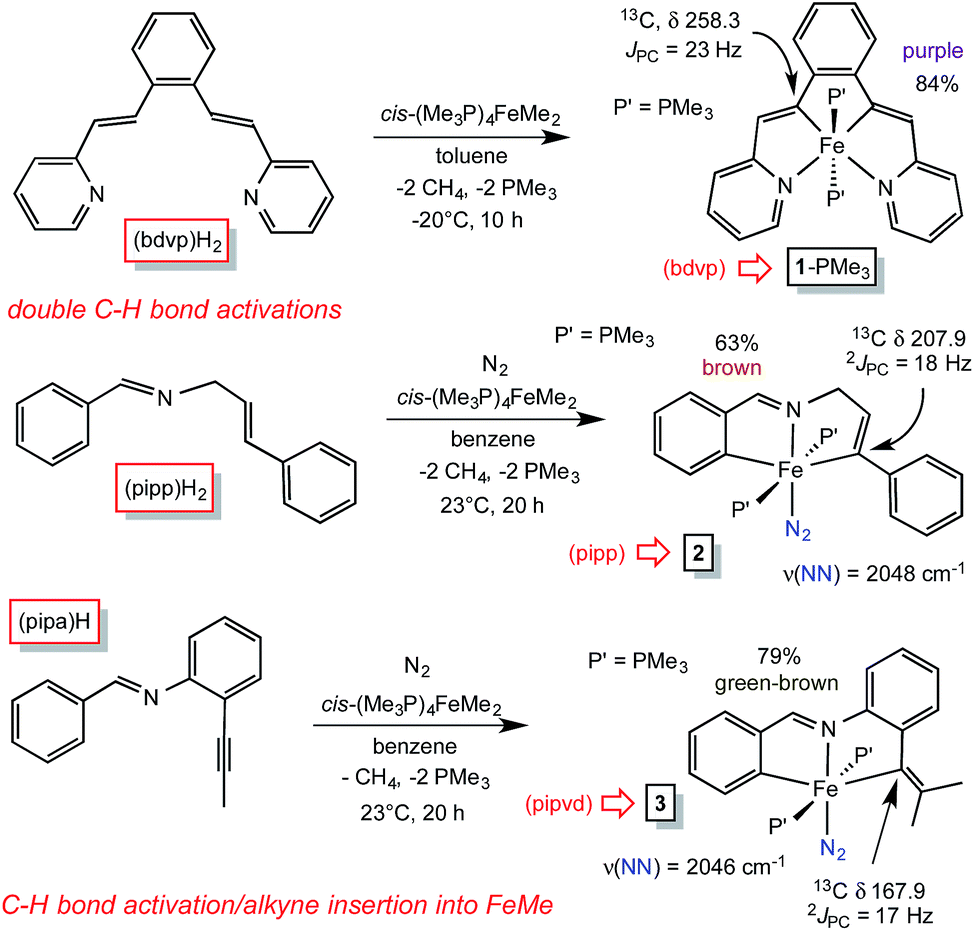 | ||
| Scheme 3 Methods employed in synthesizing vinyl precursors derived from CH-bond activation/metalation of cis-Me2Fe(PMe3)4 and acetylene insertion. | ||
A similar exposure of cis-Me2Fe(PMe3)4 (ref. 19) to PhCHNCH2(E-CHCHPh) ((pipp)H2) in benzene at 23 °C after 20 h afforded a purple solid upon subsequent concentration (Scheme 3). Dissolution in THF under a dinitrogen atmosphere provided brown trans-(pipp)Fe(PMe3)2N2 (2) in 63% yield after removal of solvent. It is likely that the tris-PMe3 derivative is formed initially, but N2 replaces PMe3 in a probable dissociative process. Previous examples have shown that steric factors – in this case the phenyl substituent – labilize the phosphine opposite the imine.23 The dinitrogen ligand is readily discerned via its IR spectrum, which reveals a ν(NN) at 2048 cm−1.28,29
A third, different metalation was conducted with PhCHN(2-CCMe-Ph) ((pipa)H) and cis-Me2Fe(PMe3)4. The precedented imine-directed Ar-H activation occurred, followed by insertion of the pendant acetylene into the Fe–Me bond. The resulting complex, trans-(pipvd)Fe(PMe3)2N2 (3), contains a dimethyl-vinyl group as the precursor to a potential alkylidene. Green-brown 3 was prepared in 79% yield after metalation for 20 h at 23 °C, and as in the previous case, it is likely an initially formed tris-PMe3 complex lost a phosphine in the presence of N2 to afford the dinitrogen complex,23 whose ν(NN) is at 2046 cm−1.
All three precursors feature downfield 13C NMR chemical shifts for the vinyl carbons bound to iron. A triplet (JPC = 23 Hz) corresponding to the Fe–C(Ar) unit in (bdvp)Fe(PMe3)2 (1-PMe3) was located at δ 258.3, an unusual shift that may be intrinsic to the metrics of its tetradentate chelation, i.e., reflecting a very short d(Fe–C). The Fe–vinyl carbon of the tridentate chelate in trans-(pipp)Fe(PMe3)2N2 (2) also manifests a significant downfield shift at δ 207.9 (t, JPC = 18 Hz), while the Fe–C(Ar) carbon of trans-(pipvd)Fe(PMe3)2N2 (3), the most sterically hindered vinyl, resonates at δ 167.9 (t, JPC = 17 Hz).
Structure of (bdvp)Fe(PMe3)2 (1-PMe3)
Shown in Fig. 2 is the molecular structure of (bdvp)Fe(PMe3)2 (1-PMe3), as determined by single crystal X-ray crystallography. The tetradentate ligand essentially resides in a plane of the pseudo-octahedral structure (∠C/N–Fe–P = 90.0(11)° (ave); ∠P1–Fe–P2 = 179.24(3)°), accompanied by trans-PMe3 groups at d(Fe–P) = 2.2283(8) Å (ave). The d(Fe–N) of 2.060(4) (ave) are normal, but there is a splay in the N1–Fe–N2 angle (112.43(11)°) indicative of a strain in the chelate. The bite angles of the vinyl-pyridine are 81.01(13)° (ave), and the phenyl-divinyl bite angle is 85.61(14)°, hence the chelate angles sum to 360.1°.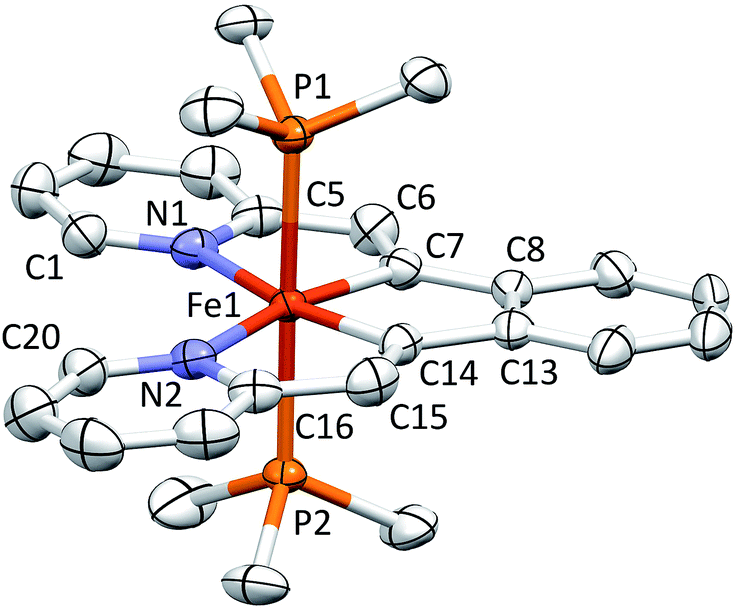 | ||
| Fig. 2 Molecular view of (bdvp)Fe(PMe3)2 (1-PMe3). Interatomic distances (Å) and angles (°): Fe–N1, 2.057(3); Fe–N2, 2.062(3); Fe–C7, 1.877(3); Fe–C14, 1.888(3); Fe–P1, 2.2285(8); Fe–P2, 2.2280(8); N1–Fe–C7, 81.10(13); N1–Fe–C14, 166.62(13); N1–Fe–N2, 112.43(11); N1–Fe–P1, 89.45(8); N1–Fe–P2, 91.17(8); C7–Fe–C14, 85.61(14); C7–Fe–N2, 166.35(13); C7–Fe–P1, 90.06(10); C7–Fe–P2, 89.61(10); C14–Fe–N2, 80.91(13); C14–Fe–P1, 89.03(9); C14–Fe–P2, 90.26(9); N2–Fe–P1, 91.84(8); N2–Fe–P2, 88.32(8); P1–Fe–P2, 179.24(3). | ||
Considerable distortion in the chelate is evident, as the iron–carbon bonds are quite short at 1.883(8) Å (ave), while the C6–C7–C8 and C13–C14–C15 angles of 127.4(4)° (ave) deviate significantly from 120°. Fig. 3 illustrates the chelate distances and angles in comparison to those of the related alkylidene complex (vide infra). All the angles about the Fe–C bonds are distorted in response to the proximity of the vinyl carbons to the iron. Note that the pyridines are not perfectly aligned as donors, as the Fe–N–C angles open to an average of 130.6(5)°.
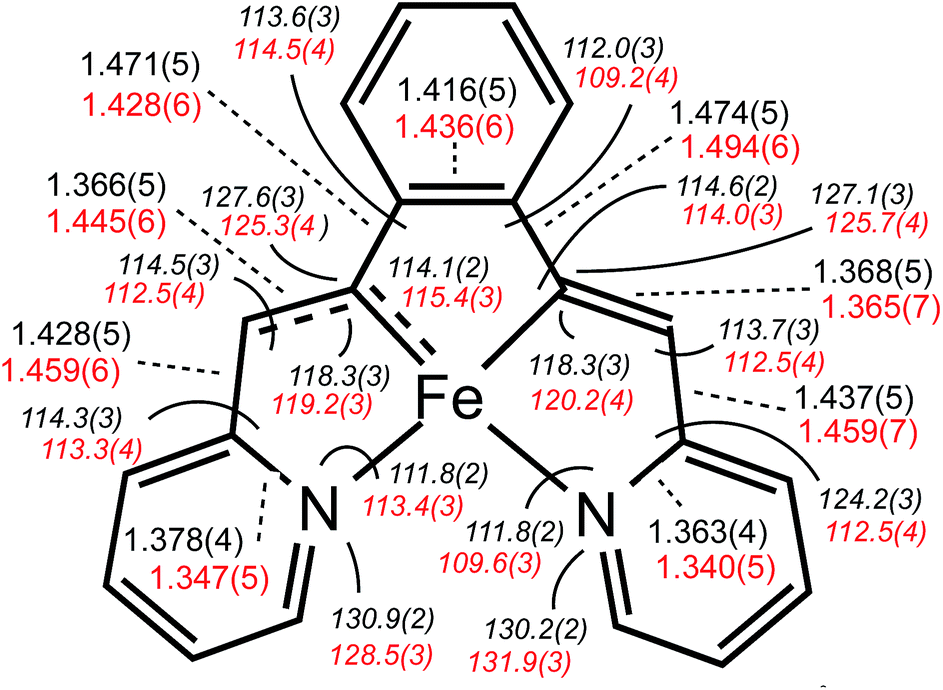 | ||
| Fig. 3 Comparative ligand metric parameters (distances (Å), dashed black lines; angles (°, italics), curved black lines) for Fe(II) (bdvp)Fe(PMe3)2 (1-PMe3, black) vs. Fe(IV) [(bavp)Fe(PMe3)2][BArF4] (4-PMe3, red). | ||
Vinyl protonations lead to Fe(IV) alkylidenes
The vinyl precursors synthesized via the C–H bond activation and insertion processes were protonated14–18 to yield cationic Fe(IV) alkylidenes, as illustrated in Scheme 4. The tetradentate chelate complex (bdvp)Fe(PMe3)2 (1-PMe3) was treated with H[BArF4]30 in THF to afford orange [(bavp)Fe(PMe3)2][BArF4] (4-PMe3) in 80% yield. The lability of 1-PMe3 was tested with excess PMe2Ph (10 equiv.), and repeated thermolyses in refluxing toluene, including periodic removal of PMe3, were required to generate (bdvp)Fe(PMe2Ph)2 (1-PMe2Ph). The dimethylphenylphosphine derivative 1-PMe2Ph was not isolated, and characterization using NMR spectroscopy proved elusive due to broadened and overlapping resonances. As a consequence, it was generated in situ and treated with H[BArF4] in THF to yield analytically pure [(bavp)Fe(PMe2Ph)2][BArF4] (4-PMe2Ph, 94%).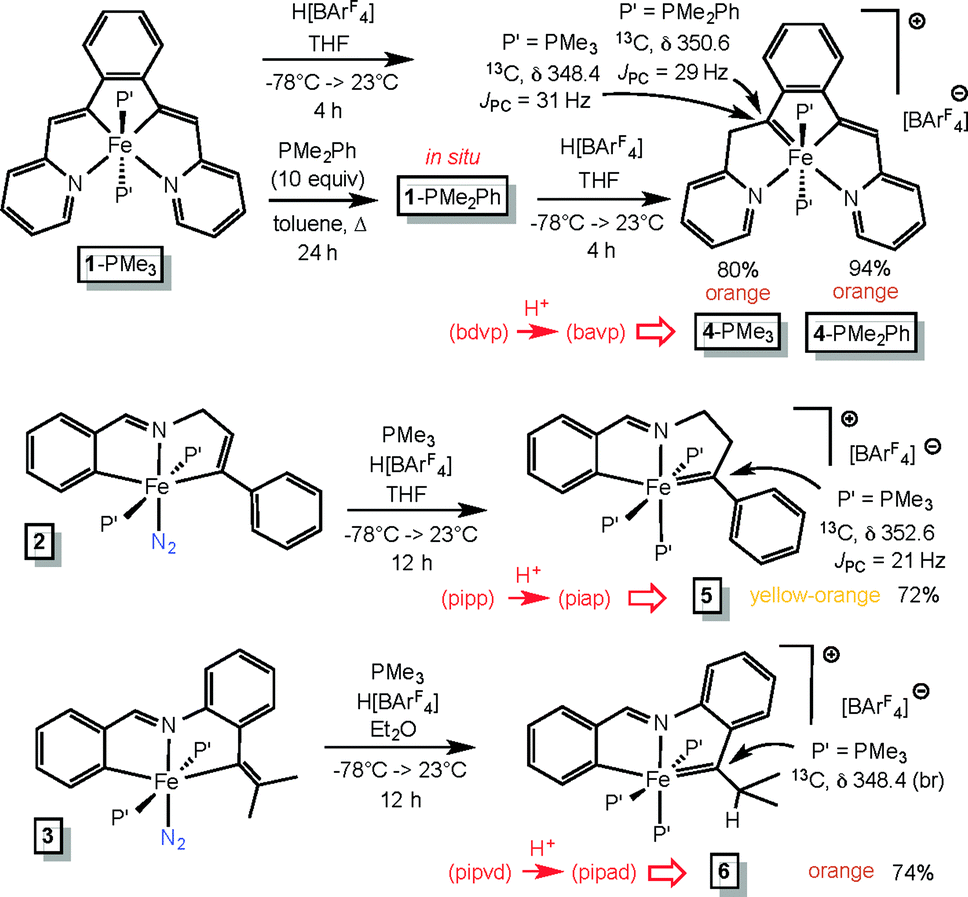 | ||
| Scheme 4 Protonation with H[BArF4] afforded cationic Fe(IV) alkylidenes. | ||
A related protonation of the tridentate chelate species trans-(pipp)Fe(PMe3)2N2 (2) in THF initially gave a complex mixture that exhibited 31P{1H} NMR spectral resonances consistent with starting material, a tri-phosphine complex, and degradation products. The addition of PMe3 to the reaction resulted in one major product, [(piap)Fe(PMe3)3][BArF4] (5) that was isolated as yellow-orange microcrystals in 72% yield. It is likely that an initial dinitrogen-containing Fe(IV) product, [(piap)Fe(PMe3)2N2][BArF4], readily loses N2, and through redistribution generates 5 along with decomposition products.
Protonation of (pipvd)Fe(PMe3)2N2 (3) was conducted with H[BArF4] in diethyl ether, and a mixture whose NMR spectra is related to that of the initial protonation of 2 was discerned. Again, the addition of PMe3 to the reaction mixture permitted the isolation of [(pipvd)Fe(PMe3)3][BArF4] (6) in 74% yield as orange microcrystals.
Definitive spectral characterization of the isolated Fe(IV) alkylidene complexes was predicated on observation of diagnostic downfield 13C NMR resonances31,32 attributed to the MCRR′ functionality (Scheme 4). The spectral signatures were difficult to detect, requiring indirect methods, but the alkylidene chemical shifts for 4-PMe3, 4-PMe2Ph, 5, and 6 were eventually observed at δ 348.4 (JPC = 31 Hz), δ 350.6 (JPC = 31 Hz), δ 352.6 (JPC = 21 Hz), and δ 348.4 (br), respectively.
Structure of [(bavp)Fe(PMe3)2][BArF4] (4-PMe3)
A molecular view of the cation pertaining to [(bavp)Fe(PMe3)2][BArF4] (4-PMe3) is illustrated in Fig. 4, showing its distorted octahedral structure, with the tetradentate chelate occupying a single plane. The P–Fe–C/N angles average 90.0(15)°, and there is a splay in the bavp ligand indicated by the N1–Fe–N2 angle of 110.98(14)°, and the trans-N–Fe–C angles of 168.31(19) and 167.48(17)°.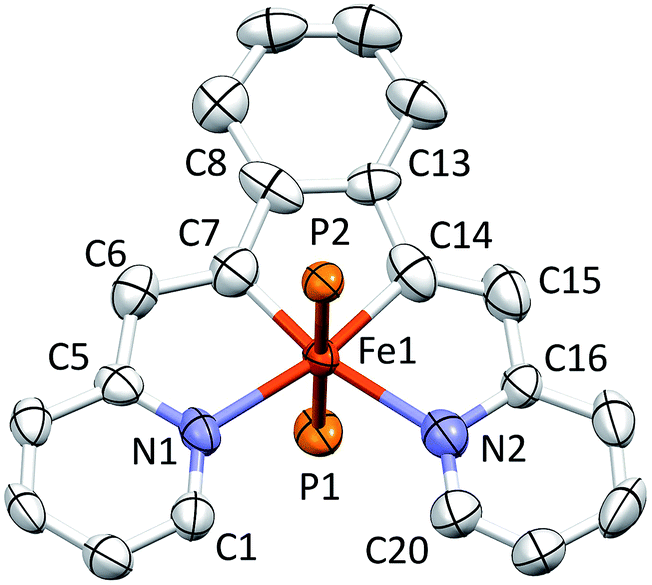 | ||
| Fig. 4 Molecular view of the cation pertaining to [(bavp)Fe(PMe3)2][BArF4] (4-PMe3); the PMe3 methyl groups have been removed for clarity. Interatomic distances (Å) and angles (°): Fe–N1, 2.083(3); Fe–N2, 2.129(4); Fe–C7, 1.809(4); Fe–C14, 1.858(4); Fe–P1, 2.2671(11); Fe–P2, 2.2725(11); N1–Fe–C7, 81.47(17); N1–Fe–C14, 168.31(19); N1–Fe–N2, 110.98(14); N1–Fe–P1, 88.53(9); N1–Fe–P2, 91.32(9); C7–Fe–C14, 86.9(2); N2–Fe–C7, 167.48(17); C7–Fe–P1, 91.87(13); C7–Fe–P2, 87.81(13); N2–Fe–C14, 80.71(18); C14–Fe–P1, 91.49(13); C14–Fe–P2, 88.60(13); N2–Fe–P1, 89.89(10); N2–Fe–P2, 90.44(10); P1–Fe–P2, 179.66(5). | ||
The critical d(FeC7) is 1.809(4) Å, which is ∼0.05 Å shorter than the adjacent iron–vinyl carbon distance of 1.858(4) Å. Both are shorter than the iron–carbon bond lengths in 1-PMe3, in contrast to the d(Fe–N), which are longer at 2.083(3) and 2.129(4) Å. As these changes and the comparison between 1-PMe3 and 4-PMe3 in Fig. 3 reveal, the chelate has pinched in to a slightly greater extent in the cation. The angles C6–C7–C8 and C13–C14–C15 are 2.3 and 1.4° less, respectively, than the corresponding angles in 1-PMe3, and the remaining chelate distances and angles change in concert.
Structure of [(piap)Fe(PMe3)3][BArF4] (5)
Fig. 5 displays a molecular view of the cation corresponding to [(piap)Fe(PMe3)3][BArF4] (5), and reveals its pseudo-octahedral geometry with the piap ligand occupying a mer-configuration about iron. The critical alkylidene distance, d(Fe–C10), is 1.867(7) Å, which is significantly shorter than d(Fe–C1) = 2.106(6) Å, but on par with the iron–vinyl carbon distances in 1-PMe3 (1.883(8) Å (ave)) and 4-PMe3 (1.858(4) Å). The tridentate chelate is strained, as the C1–Fe–C10 angle is 161.6(3)°, and the Fe–C10–C9 and Fe–C10–C11 angles of 115.7(5)° and 136.4(5)°, respectively, indicate that the alkylidene is not perfectly oriented. Note that the C10–Fe–P2 angles are 100.20(4)°; as a consequence, the d-orbital that comprises the iron portion of the FeC π-bond has some Fe–P σ* character that aids in producing better overlap with the carbon p-orbital.
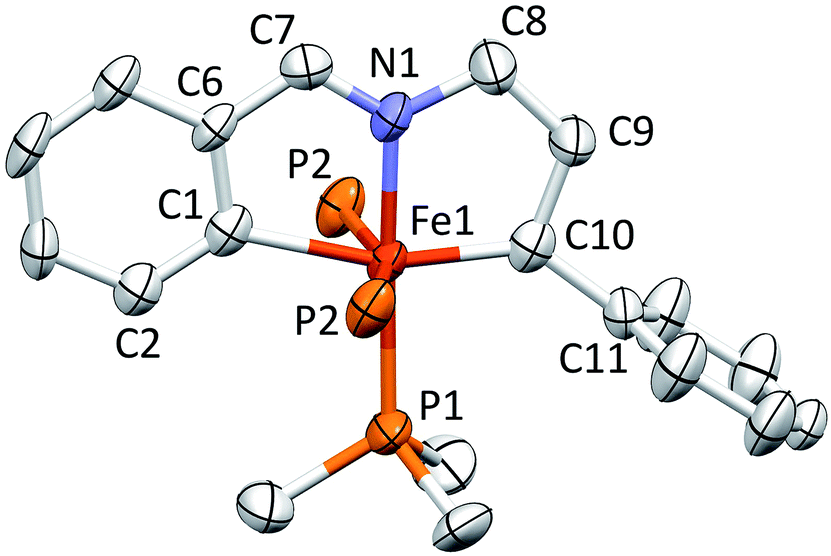 | ||
| Fig. 5 Molecular view of the cation pertaining to [(piap)Fe(PMe3)3][BArF4] (5); the methyl groups of the trans-PMe3 ligands have been removed for clarity. Interatomic distances (Å) and angles (°): Fe–C1, 2.106(6); Fe–N1, 1.949(6); Fe–C10, 1.867(7); Fe–P1, 2.281(2); Fe–P2, 2.2733(16); C7–N1, 1.307(9); C1–Fe–N1, 78.4(3); C1–Fe–C10, 161.6(3); C1–Fe–P1, 103.7(2); N1–Fe–C10, 83.2(3); N1–Fe–P1, 177.94(17); C10–Fe–P1, 94.8(2); C1–Fe–P2, 79.47(4); N1–Fe–P2, 88.54(5); C10–Fe–P2, 100.20(4); P1–Fe–P2, 91.83(5); P2–Fe–P2, 158.89(8); Fe–C1–C2, 133.8(5); Fe–C1–C6, 110.4(5); Fe–C10–C9, 115.7(5); Fe–C10–C11, 136.4(5). | ||
Structure of [(pipad)Fe(PMe3)3][BArF4] (6)
A molecular view of the cation of [(pipad)Fe(PMe3)3][BArF4] (6) is provided in Fig. 6, which indicates the mer-octahedral structure of the iron alkylidene. The tridentate pipad chelate is essentially planar, and the isopropyl-aryl alkylidene possesses a d(FeC) of 1.899(3) Å, which is longer than the iron–vinyl carbon distances in 1-PMe3 and 4-PMe3. Again, the chelate exhibits strain about the core, as the C1–Fe–C17 angle is 163.36(15)°, and its isopropyl group exerts a steric influence on the unique PMe3, as the C1–Fe–P1 angle is 104.46(12)°. The Fe–C1–C5 and Fe–C1–C2 angles are 112.8(2)° and 131.0(3)°, respectively, showing that the alkylidene is at an imperfect orientation with respect to the iron.
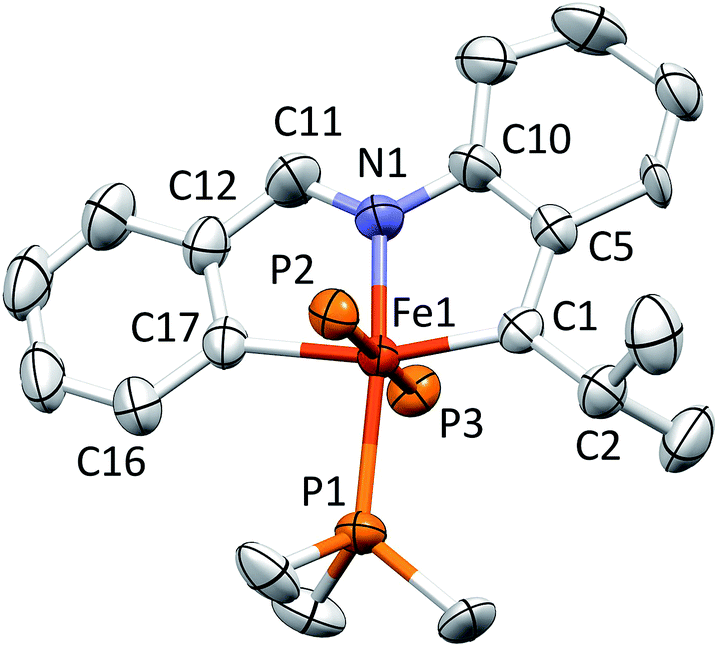 | ||
| Fig. 6 Molecular view of the cation pertaining to [(pipad)Fe(PMe3)3][BArF4] (6); the methyl groups of the trans-PMe3 ligands have been removed for clarity. Interatomic distances (Å) and angles (°): Fe–C1, 1.899(3); Fe–N1, 1.933(3); Fe–C17, 2.059(3); Fe–P1, 2.317(2); Fe–P2, 2.226(3); Fe–P3, 2.367(3); N1–C11, 1.307(5); C1–C2, 1.525(5); N1–Fe–C17, 80.04(14); C1–Fe–C17, 163.36(15); C17–Fe–P1, 92.03(12); C17–Fe–P2, 88.28(13); C17–Fe–P3, 87.16(12); N1–Fe–C1, 83.34(14); N1–Fe–P1, 167.04(14); N1–Fe–P2, 96.52(16); N1–Fe–P3, 79.34(14); C1–Fe–P1, 104.46(12); C1–Fe–P2, 92.87(12); C1–Fe–P3, 90.53(13); P1–Fe–P2, 93.45(14); P1–Fe–P3, 90.11(13); P2–Fe–P3, 174.32(13); Fe–C17–C16, 136.0(3); Fe–C17–C12, 110.8(3); Fe–C1–C5, 112.8(2); Fe–C1–C2, 131.0(3). | ||
Structural comparisons
In Table 1, a comparison of known Fe(IV) alkylidenes is given with reference to d(FeC) and 13C NMR chemical shift (δ).29,30 Paramagnetic derivatives are on the long side of the bond distance values, and the electronic structure analysis by Chirik et al.13 of the PDI derivatives suggests that these species are best considered carbene radicals.33 The π-interaction is construed as a carbene radical antiferromagnetically (AF) coupled to a metal dπ-electron of appropriate symmetry. Modern calculations have not been employed on Floriani's calix[4]arane diphenycarbene complexes,11 but they are high spin, and therefore are likely to conform to an AF coupling model.
C) and 13C NMR shift (δ)
| Compounda |
d(FeC) (Å) |
δ (13CFe) |
|---|---|---|
| a See Fig. 1 for ligand structural types corresponding to A–D. b Ref. 10. c Ref. 12. d Ref. 9. e Ref. 13. f Ref. 11. | ||
| (tmtaa)FeCPh2 (B)b |
1.794(3) | 313.2 |
| (TPFPP)FeCPh2 (A)c |
1.767(3) | 359.0 |
| [Cp*(dppe)FeCH(Me)]PF6 (E)d |
1.787(8) | 336.6 |
| [(bavp)Fe(PMe3)2][BArF4] (4-PMe3) | 1.809(4) | 350.6 |
| (TPFPP)Fe(CPh2)(MeIm) (A)c |
1.827(5) | 385.4 |
| [(piap)Fe(PMe3)3][BArF4] (5) | 1.867(7) | 352.6 |
| [(pipad)Fe(PMe3)3][BArF4] (6) | 1.899(3) | 348.4 |
| (EtPDI)FeCPh2 (C)e |
1.9205(19) | Para |
| (MeEtPDI)FeCPh2 (C)e |
1.9234(18) | Para |
| 1.9357(18) | Para | |
| [p-tBu-calix[4](O)2(OMe)2]FeCPh2 (D)f |
1.943(8) | Para |
| [p-tBu-calix[4](O)2(OSiMe3)2]FeCPh2 (D)f |
1.958(5) | Para |
| 1.973(5) | Para | |
Of the remaining diamagnetic complexes, some relative distances appear to be a clear consequence of the trans-influence. When no ligand is opposite the diphenylcarbene, the distance is short, as in the cases of (tmtaa)FeCPh2 (B)10 and (TPFPP)FeCPh2 (A).12 As the methylimidazole adduct of the latter (i.e., (TPFPP)Fe(CPh2)(MeIm) (A))12 indicates, the distance is increased by 0.55 Å. It is not surprising that the complexes herein have d(FeC) that range from 1.809–1.899 Å, given the presence of a strong trans-influence ligand (an aryl). There is no straightforward correlation of d(FeC) to its respective 13C NMR spectroscopic shift.
Mössbauer analysis of Fe(II) and Fe(IV) chelates
Shown in Fig. 7 are Mössbauer spectra of the related Fe(II) and Fe(IV) complexes trans-(pipvd)Fe(PMe3)2N2 (3) and [(pipad)Fe(PMe3)3][BArF4] (6), respectively. In Table 2, all Mössbauer parameters for the corresponding Fe(II) and Fe(IV) compounds are listed. The data in Table 2 reveal isomer shifts for the diamagnetic species all within Δδ of 0.1 mm s−1, and provide a textbook example of why they should not be simply correlated with formal oxidation state, but are strong indicators of covalency.34,35 “Iron–ligand bond lengths play a decisive role for the isomer shift of a compound”,35 and the data in Table 2 and Fig. 2–4 bear this out. Minimal bond distance changes occur in the protonation of (bdvp)Fe(PMe3)2 (1-PMe3) to afford [(bavp)Fe(PMe3)2][BArF4] (4-PMe3), and similarly small d(Fe-L/X) changes are likely in the related protonations, leading to small isomer shift differences.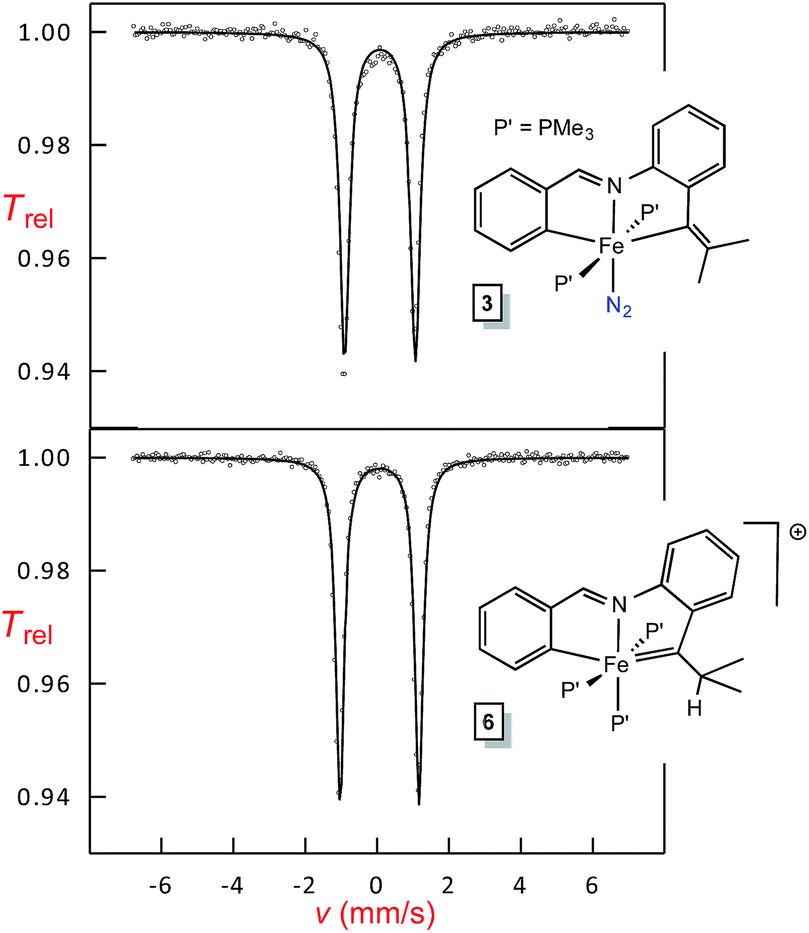 | ||
| Fig. 7 Mössbauer spectra of Fe(II) trans-(pipvd)Fe(PMe3)2N2 (3, δ = 0.07(1) mm s−1; ΔEQ = 1.97(1) mm s−1), and the corresponding Fe(IV) cation [(pipad)Fe(PMe3)3][BArF4] (6, δ = 0.07(1) mm s−1; ΔEQ = 2.20(1) mm s−1). | ||
| Compound | δ (mm s−1) | ΔEQ (mm s−1) | Γ FWHM (mm s−1) |
|---|---|---|---|
| a Sample contained 20% of a high spin Fe(II) species: δ = 1.23(1) mm s−1, ΔEQ = 2.40(1) mm s−1, ΓFWHM = 0.73(1) mm s−1. b Sample contained 18% of a high spin Fe(II) species: δ = 1.28(1) mm s−1, ΔEQ = 2.70(1) mm s−1, ΓFWHM = 0.54(1) mm s−1. c Sample contained 35% of a high spin Fe(II) species: δ = 1.25(1) mm s−1, ΔEQ = 2.42(1) mm s−1, ΓFWHM = 0.51(1) mm s−1. | |||
| (bdvp)Fe(PMe3)2 (1-PMe3) | 0.09(1) | 1.96(1) | 0.45(1) |
| trans-(pipp)Fe(PMe3)2N2 (2)a | 0.08(1) | 2.14(1) | 0.31(1) |
| trans-(pipvd)Fe(PMe3)2N2 (3) | 0.07(1) | 1.97(1) | 0.33(1) |
| [(bavp)Fe(PMe3)2][BArF4] (4-PMe3)b | 0.01(1) | 2.67(1) | 0.28(1) |
| [(piap)Fe(PMe3)3][BArF4] (5)c | 0.06(1) | 2.02(1) | 0.29(1) |
| [(pipad)Fe(PMe3)3][BArF4] (6) | 0.07(1) | 2.20(1) | 0.28(1) |
One counter argument regarding interpretation of isomer shifts pertains to the somewhat arbitrary formalism of treating a Schrock alkylidene as a (2−) ligand, whereas a Fischer carbene, in which conjugated lone pairs can donate to the carbon (i.e., MCX(R) ↔ M(−)–CX(+) (R)), is neutral. While one can argue there is some conjugation for 3 and 6, the other cases are less readily interpreted in this fashion, especially given the orientation of the phenyl group of 5 as roughly orthogonal to the FeC interaction. There can be little question that two pairs of electrons – one sigma and one pi – exist between iron and carbon in these compounds, and that the parameters of the Mössbauer spectra correlate with a strong degree of covalency. Previously characterized alkylidene species are limited to those reported by Chirik et al.,13 whose S = 1 systems are sufficiently different to be essentially incomparable.
Interpretation of the quadrupole splitting (ΔEQ), a measure of the electric field gradient at the nucleus,35 is less transparent. The changes in ligand coordination, principally PMe3 for N2 in the conversion of 2 and 3 to 5 and 6, respectively, are apparently significant enough to offset changes to the Ar-Fe(–Vy/C) axes. For 1-PMe3 and 4-PMe3, there is a consequential change from ΔEQ = 1.96(1) mm s−1 to 2.67(1) mm s−1, as the rhombicity of the complex is notably altered due to the change from a symmetric divinyl coordination to that of the alkylidene and vinyl arrangement.
Conclusions
Protonation of Fe(II) chelate complexes in which iron–vinyl bonds are present led to the formation of four cationic Fe(IV) alkylidene complexes, three of which are structurally characterized. Prior to this study, [Cp*(dppe)FeCH(Me)]PF6 was the only non-aryl Fe(IV) alkylidene that had undergone X-ray diffraction structural analysis, although numerous related [CpLL′FeCHR]+ complexes have been synthesized.7–9,14–16
This study confers confidence in iron–vinyl protonation as a viable, general route to Fe(IV) alkylidenes in non-Cp systems. The compounds herein (i.e., 4-PMe3, 5 and 6) were not active towards metathesis (e.g. cis-2-pentene and RCCR; R = Me, Ph) or cycloprotonation, primarily because PMe3 is not sufficiently labile, as expected. In order to implement this route toward viable olefin metathesis catalysts, future syntheses must address three factors: (1) complexes must be coordinatively unsaturated, with 14e− species the obvious targets based on ruthenium analogues; (2) complexes must be neutral or anionic, where the d-orbitals are less contracted; and (3) FeCHR moieties must be targeted.
Experimental section
Experimental details, full spectral characterizations, and a description of the Mössbauer spectroscopic analysis are given in the ESI.† For general descriptions, consult the schemes. Some crystallographic information is given below.Crystal data for 1-PMe3
C26H32N2P2Fe, M = 490.33, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.2138(8), b = 10.6014(8), c = 12.4208(10) Å, α = 88.674(4)° β = 67.062(3)°, γ = 89.687(4)°, V = 1238.24(17) Å3, T = 203(2) K, λ = 0.71073 Å, Z = 2, Rint = 0.0311, 22420 reflections, 6098 independent, R1(all data) = 0.0663, wR2 = 0.1766, GOF = 1.077.†
, a = 10.2138(8), b = 10.6014(8), c = 12.4208(10) Å, α = 88.674(4)° β = 67.062(3)°, γ = 89.687(4)°, V = 1238.24(17) Å3, T = 203(2) K, λ = 0.71073 Å, Z = 2, Rint = 0.0311, 22420 reflections, 6098 independent, R1(all data) = 0.0663, wR2 = 0.1766, GOF = 1.077.†
Crystal data for 4-PMe3
C58H45N2F24BP2Fe, M = 1354.56, monoclinic, P21/c, a = 19.6517(7), b = 12.5655(4), c = 25.3645(7) Å, β = 109.7450(10)°, V = 5895.1(3) Å3, T = 203(2) K, λ = 0.71073 Å, Z = 4, Rint = 0.0365, 49817 reflections, 12059 independent, R1(all data) = 0.0958, wR2 = 0.1782, GOF = 1.012.†
Crystal data for 5(THF)
C61H60NOF24BP3Fe, M = 1438.67, monoclinic, C2/m, a = 19.963(5), b = 17.492(6), c = 19.586(6) Å, β = 93.869(14)°, V = 6824(4) Å3, T = 203(2) K, λ = 0.71073 Å, Z = 4, Rint = 0.0579, 21278 reflections, 5076 independent, R1(all data) = 0.0899, wR2 = 0.1923, GOF = 1.155.†
Crystal data for 6
C58H55NF24BP3Fe, M = 1381.60, monoclinic, P21/c, a = 18.4232(6), b = 13.0618(4), c = 25.9802(8) Å, β = 99.5300(10)°, V = 6165.6(3) Å3, T = 233(2) K, λ = 0.71073 Å, Z = 4, Rint = 0.0393, 35706 reflections, 9178 independent, R1(all data) = 0.0780, wR2 = 0.1273, GOF = 1.050.†
Acknowledgements
Support from the National Science Foundation (CHE-1402149), Cornell University, and Friedrich Alexander University is gratefully acknowledged.Notes and references
- H. Jeong, J. M. John, R. R. Schrock and A. H. Hoveyda, J. Am. Chem. Soc., 2015, 137, 2239–2242 CrossRef CAS PubMed.
- (a) R. R. Schrock, Angew. Chem., Int. Ed., 2006, 45, 3748–3759 CrossRef CAS PubMed; (b) R. R. Schrock and A. H. Hoveyda, Angew. Chem., Int. Ed., 2003, 42, 4592–4633 CrossRef CAS PubMed.
- V. M. Marx, A. H. Sullivan, M. Melaimi, S. C. Virgil, B. K. Keitz, D. S. Weinberger, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 1919–1923 CrossRef CAS PubMed.
- R. H. Grubbs, Angew. Chem., Int. Ed., 2006, 45, 3760–3765 CrossRef CAS PubMed.
- Y. Chauvin, Angew. Chem., Int. Ed., 2006, 45, 3740–3747 CrossRef PubMed.
- O. Eisenstein, R. Hoffmann and A. R. Rossi, J. Am. Chem. Soc., 1981, 103, 5582–5584 CrossRef CAS.
- (a) M. Brookhart, J. R. Tucker and G. R. Husk, J. Am. Chem. Soc., 1983, 105, 258–264 CrossRef CAS; (b) M. Brookhart and J. R. Tucker, J. Am. Chem. Soc., 1981, 103, 979–981 CrossRef CAS; (c) M. Brookhart, D. Timmers, J. R. Tucker, G. D. Williams, G. R. Husk, H. Brunner and B. Hammer, J. Am. Chem. Soc., 1983, 105, 6721–6723 CrossRef CAS.
- G. Poignant, S. Nlate, V. Guerchais, A. J. Edwards and P. R. Raithby, Organometallics, 1997, 16, 124–132 CrossRef CAS.
- V. Mahias, S. Cron, L. Toupet and C. Lapinte, Organometallics, 1996, 15, 5399–5408 CrossRef CAS.
- A. Klose, E. Solari, C. Floriani, N. Re, A. Chiesi-Villa and C. Rizzoli, Chem. Commun., 1997, 2297–2298 RSC.
- V. Esposito, E. Solari, C. Floriani, N. Re, C. Rizzoli and A. Chiesi-Villa, Inorg. Chem., 2000, 39, 2604–2613 CrossRef CAS.
- Y. Li, J.-S. Huang, Z.-Y. Zhou, C.-M. Che and X.-Z. You, J. Am. Chem. Soc., 2002, 124, 13185–13193 CrossRef CAS PubMed.
- S. K. Russell, J. M. Hoyt, S. C. Bart, C. Milsmann, S. C. E. Stieber, S. P. Semproni, S. DeBeer and P. J. Chirik, Chem. Sci., 2014, 5, 1168–1174 RSC.
- (a) K. A. M. Kremer, G.-H. Kuo, E. J. O'Connor, P. Helquist and R. C. Kerber, J. Am. Chem. Soc., 1982, 104, 6119–6121 CrossRef CAS; (b) G.-H. Kuo, P. Helquist and R. C. Kerber, Organometallics, 1984, 3, 806–808 CrossRef CAS.
- T. Bodnar and A. R. Cutler, J. Organomet. Chem., 1981, 213, C31–C36 CrossRef CAS.
- (a) A. Davison and J. P. Selegue, J. Am. Chem. Soc., 1978, 100, 7763–7765 CrossRef CAS; (b) R. D. Adams, A. Davison and J. P. Selegue, J. Am. Chem. Soc., 1979, 101, 7232–7238 CrossRef CAS.
- M. I. Bruce and A. G. Swincer, Aust. J. Chem., 1980, 33, 1471–1483 CrossRef CAS.
- C. P. Casey, W. H. Miles, H. Tukada and J. M. O'Connor, J. Am. Chem. Soc., 1982, 104, 3761–3762 CrossRef CAS.
- H. H. Karsch, Chem. Ber., 1977, 110, 2699–2711 CrossRef CAS PubMed.
- (a) G. Xu, H. Sun and X. Li, Organometallics, 2009, 28, 6090–6095 CrossRef CAS; (b) X. Xu, J. Jia, H. Sun, Y. Liu, W. Xu, Y. Shi, D. Zhang and X. Li, Dalton Trans., 2013, 3417–3428 RSC.
- (a) R. Beck, T. Zheng, H. Sun, X. Li, U. Flörke and H.-F. Klein, J. Organomet. Chem., 2008, 693, 3471–3478 CrossRef CAS PubMed; (b) S. Camadanli, R. Beck, U. Flörke and H.-F. Klein, Organometallics, 2009, 28, 2300–2310 CrossRef CAS; (c) H.-F. Klein, S. Camadanli, R. Beck, D. Leukel and U. Flörke, Angew. Chem., Int. Ed., 2005, 44, 975–977 CrossRef CAS PubMed; (d) H.-F. Klein, S. Camadanli, R. Beck and U. Flörke, Chem. Commun., 2005, 381–382 RSC.
- E. C. Volpe, P. T. Wolczanski and E. B. Lobkovsky, Organometallics, 2010, 29, 364–377 CrossRef CAS.
- E. R. Bartholomew, E. C. Volpe, P. T. Wolczanski, E. B. Lobkovsky and T. R. Cundari, J. Am. Chem. Soc., 2013, 135, 3511–3527 CrossRef CAS PubMed.
- T. Yamamori, K. Nagata, N. Ishizuka and K. Hayashi, Utilities of Olefin Derivatives, US Patent Appl. No. 10/489, 365, 2004.
- N. Rodrigues, K. Bennis, D. Vivier, V. Pereira, C. F. Chatelain, E. Chapuy, H. Deokar, J. Busserolles, F. Lesage, A. Eschalier and S. Ducki, Eur. J. Med. Chem., 2014, 75, 391–402 CrossRef CAS PubMed.
- G. Wolf and E.-U. Würthwein, Chem. Ber., 1991, 124, 655–663 CrossRef CAS PubMed.
- (a) C. Shi, Q. Zhang and K. K. Wang, J. Org. Chem., 1999, 64, 925–932 CrossRef CAS PubMed; (b) H. Kusama, J. Takaya and N. Iwasawa, J. Am. Chem. Soc., 2002, 124, 11592–11593 CrossRef CAS PubMed.
- J. L. Crossland and D. R. Tyler, Coord. Chem. Rev., 2010, 254, 1883–1894 CrossRef CAS PubMed.
- N. Hazari, Chem. Soc. Rev., 2010, 39, 4044–4056 RSC.
- M. Brookhart, B. Grant and A. F. Volpe Jr, Organometallics, 1992, 11, 3920–3922 CrossRef CAS.
- J. Louie and R. H. Grubbs, Organometallics, 2002, 21, 2153–2164 CrossRef CAS.
- J. Feldman and R. R. Schrock, in Prog. Inorg. Chem., ed. S. J. Lippard, Wiley and Sons, New York, 1991, vol. 39, pp. 1–74 Search PubMed.
- W. I. Dzik, X. P. Zhang and B. de Bruin, Inorg. Chem., 2011, 50, 9896–9903 CrossRef CAS PubMed.
- F. Neese, Inorg. Chim. Acta, 2002, 337, 181–192 CrossRef.
- P. Gütlich, E. Bill and A. X. Trautwein, Mössbauer Spectroscopy and Transition Metal Chemistry: Fundamentals and Applications, Springer-Verlag, Berlin, 2011 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1057828–1057831. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01268f |
| This journal is © The Royal Society of Chemistry 2015 |