
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvader probes: harnessing the energy of intercalation to facilitate recognition of chromosomal DNA for diagnostic applications†
Dale C.
Guenther
a,
Grace H.
Anderson
ab,
Saswata
Karmakar
a,
Brooke A.
Anderson
a,
Bradley A.
Didion
c,
Wei
Guo
c,
John P.
Verstegen
c and
Patrick J.
Hrdlicka
*a
aDepartment of Chemistry, University of Idaho, 875 Perimeter Dr, Moscow, ID 83844-2343, USA. E-mail: Hrdlicka@uidaho.edu
bDepartment of Biological Sciences, Montana Tech of the University of Montana, 1300 W Park St, Butte, MT 59701-8997, USA
cMoFA, PO Box 930187, 419 Venture Ct., Verona, WI 53593, USA
First published on 12th June 2015
Abstract
Development of probes capable of recognizing specific regions of chromosomal DNA has been a long-standing goal for chemical biologists. Current strategies such as PNA, triplex-forming oligonucleotides, and polyamides are subject to target choice limitations and/or necessitate non-physiological conditions, leaving a need for alternative approaches. Toward this end, we have recently introduced double-stranded oligonucleotide probes that are energetically activated for DNA recognition through modification with +1 interstrand zippers of intercalator-functionalized nucleotide monomers. Herein, probes with different chemistries and architectures – varying in the position, number, and distance between the intercalator zippers – are studied with respect to hybridization energetics and DNA-targeting properties. Experiments with model DNA targets demonstrate that optimized probes enable efficient (C50 < 1 μM), fast (t50 < 3 h), kinetically stable (>24 h), and single nucleotide specific recognition of DNA targets at physiologically relevant ionic strengths. Optimized probes were used in non-denaturing fluorescence in situ hybridization experiments for detection of gender-specific mixed-sequence chromosomal DNA target regions. These probes present themselves as a promising strategy for recognition of chromosomal DNA, which will enable development of new tools for applications in molecular biology, genomic engineering and nanotechnology.
Introduction
There is an unmet need for chemical probes capable of recognizing biological DNA for identification, regulation, and manipulation of genes.1–7 Considerable progress has been made towards this end with triplex forming oligonucleotides (TFOs),8 polyamides9,10 peptide nucleic acids (PNA),11,12 and – more recently – engineered proteins,3,13,14 though significant limitations exist with all of these approaches. For example, TFOs only form Hoogsteen base pairs in the major groove of DNA duplexes containing long purine tracts, which reduces the number of suitable targets sites within a genome.8,12 Pyrrole-imidazole (Py/Im) polyamides, on the other hand, bind through base pair specific contacts in the minor groove of DNA duplexes, but typically only recognize short target regions (<8 bp), which may impede recognition of unique genomic sites.9,10,15 This is less of a concern with engineered nucleases but the construction requires the use of advanced molecular cloning techniques,14 and there are mounting concerns regarding the high frequency of off-target effects.16 PNAs, in which canonical nucleobases are attached to an N-(2-aminoethyl)glycine backbone, display strong affinity towards complementary single-stranded DNA (cDNA), allowing for strand invasion of double-stranded DNA (dsDNA) through simultaneous Watson–Crick and Hoogsteen base pairing. Thus, recognition of dsDNA using regular PNAs is typically subject to similar sequence limitations as TFOs,11,12 although alternative PNA-based strategies with more relaxed sequence requirements have been developed.17,18 The use of a conformationally restricted PNA backbone, γ-PNA, substantially increases the binding affinity towards cDNA, presumably due to strand preorganization and reduced entropic penalties. Single-stranded γ-PNAs have been shown to recognize mixed-sequence dsDNA target regions (150–300 bp) at low ionic strengths via duplex invasion, resulting in the formation of a D-loop, in which a segment of one of the DNA target strands is unhybridized.19,20 Nonetheless, invasion is inefficient at physiological ionic strengths.Double-duplex invasion mechanisms, in which a double-stranded probe binds to both strands of a DNA target, are desirable due to the potential stability gain from having both target strands engaged in Watson–Crick base-pairing. In order for a double-stranded probe to invade the buried Watson–Crick face of a dsDNA target, both probe strands must have substantially higher affinity toward their targets than they have towards themselves. Introduction of pseudocomplementary base pairs is one approach to achieve a favorable energetic gradient for recognition of dsDNA. This is accomplished through the use of modified base pairs such as 2-thiothymine and 2-aminoadenine, which are destabilized due to steric interactions between the sulfur atom and the additional exocyclic amino group, but maintain adequate affinities towards canonical nucleotide binding partners.21 This concept has been used with PNA backbones, and these pseudocomplementary PNA were shown to recognize internal regions of mixed-sequence dsDNA.22–24 Although the requirement of low salt conditions remains in order for a stable recognition complex to form, it may be partially overcome under conditions that mimic molecular crowding in the nucleus.25
We have recently introduced a fundamentally different strategy for recognition of dsDNA, which is based on double-stranded oligodeoxyribonucleotide (ON) probes that are energetically activated through modification with +1 interstrand zippers of intercalator-functionalized nucleotides (Fig. 1; for a description of the zipper nomenclature, see ESI†).26,27 This particular motif forces the intercalators into the same region of the probe duplex resulting in unwinding and destabilization, as the nearest neighbor exclusion principle28 is violated, which is why we have coined the term energetic hotspot to describe this motif. According to this principle, the two sites neighboring a bound intercalator will remain unoccupied due to limitations in local helix expandability (every intercalation event unwinds the duplex by ∼3.4 Å)29 and/or to avoid disruption of highly stable stacking interactions between nucleobases and the first bound intercalator.30 In contrast, each of the two strands comprising the energetically activated probes, display very high affinity toward cDNA, since duplex formation is accompanied by strongly stabilizing stacking interactions between intercalators and nucleobases (Fig. 1). The energy difference between the reactants (i.e., the double-stranded probe and DNA target) and products (i.e., the two probe-target duplexes formed as part of the recognition complex), provides the driving force for dsDNA-recognition (Fig. 1).
 | ||
| Fig. 1 (a) Illustration of the Invader approach for recognition of dsDNA. (b) Structure of Invader monomers discussed herein. | ||
We initially used 2′-N-(pyren-1-yl)methyl-2′-amino-α-L-LNA monomers as the key activating components of these Invader probes, but recently discovered that ONs modified with the simpler 2′-O-(pyren-1-yl)methyl-RNA and 2′-N-(pyren-1-yl)methyl-2′-N-methyl-2′-amino-DNA monomers display very similar hybridization properties (Fig. 1).27 We have utilized Invader probes based on 2′-O-(pyren-1-yl)methyl-RNA monomers for diagnostic proof-of-concept applications. For example, we have developed a colorimetric sandwich assay based on Invader capture/signaling probes for recognition of 28-mer mixed-sequence dsDNA fragments specific to important food pathogens. Targets are detected at concentrations down to 20 pM with excellent binding specificity.31 In another study, we demonstrated that Invader probes can detect chromosomal DNA target regions in fixed interphase or metaphase nuclei under non-denaturing conditions.32
In the present study, Invader probes with different architectures – varying in the position, number and distance between energetic hotspots – based on either 2′-O-(pyren-1-yl)methyluridine monomer X or 2′-N-(pyren-1-yl)methyl-2′-N-methyl-2′-aminouridine monomer Y, are characterized with respect to denaturation and thermodynamic properties, and dsDNA recognition efficiency, kinetics, and specificity. Informed by these insights, optimized Invader probes were used in non-denaturing fluorescence in situ hybridization (nd-FISH) experiments for detection of gender-specific chromosomal DNA target regions at near physiological conditions.
Results and discussion
Thermal denaturation properties of Invader probes
A library of twenty different 13-merX- or Y-modified Invader probes was synthesized, in which the position, number, and distance between energetic hotspots were systematically varied (Table 1). Thermal denaturation temperatures (Tm's) were determined for each component of the Invader-mediated dsDNA-recognition process, i.e., the double-stranded Invader probe, the corresponding dsDNA target region, and the two probe-target duplexes (i.e., 5′-Inv:cDNA and 3′-Inv:cDNA). The Tm's were used to calculate the thermal advantage [TA = Tm (5′-Inv:cDNA) + Tm (3′-Inv:cDNA) − Tm (Invader) − Tm (dsDNA target region)], which serves as a first approximation to describe the energy difference between the ‘products’ and ‘reactants’ of the recognition process, with more positive values signifying greater thermodynamic dsDNA recognition potential.| ONs | Sequence | M = Monomer X | M = Monomer Y | ||||
|---|---|---|---|---|---|---|---|
| T m [ΔTm] (°C) | T m [ΔTm] (°C) | ||||||
| Invader | 5′-Inv:cDNA | TA (°C) | Invader | 5′-Inv:cDNA | TA (°C) | ||
| 3′-Inv:cDNA | 3′-Inv:cDNA | ||||||
| a ΔTm = change in Tm relative to unmodified dsDNA (Tm = 37.5 °C); thermal denaturation curves were recorded in medium salt buffer ([Na+] = 110 mM, [Cl−] = 100 mM, pH 7.0 (NaH2PO4/Na2HPO4), [EDTA] = 0.2 mM) and [ON] = 1.0 μM; see main text for definition of TA. A = adenin-9-yl DNA monomer, C = cytosin-1-yl DNA monomer, G = guanin-9-yl DNA monomer, T = thymin-1-yl DNA monomer. | |||||||
| M1 | 5′-GG![[M with combining low line]](https://www.rsc.org/images/entities/b_char_004d_0332.gif) ATATATAGGC ATATATAGGC |
36.5 [−1.0] | 44.5 [+7.0] | +18.0 | 33.5 [−4.0] | 45.5 [+8.0] | +22.0 |
| M2 | 3′-CCAATATATCCG |
47.5 [+10.0] | 47.5 [+10.0] | ||||
| M3 | 5′-GGTAATATAGGC |
36.5 [−1.0] | 47.5 [+10.0] | +22.0 | 40.5 [+3.0] | 48.5 [+11.0] | +21.5 |
| M4 | 3′-CCATAATATCCG |
48.5 [+11.0] | 51.0 [+13.5] | ||||
| M5 | 5′-GGTATAATAGGC |
36.5 [−1.0] | 48.5 [+11.0] | +22.0 | 38.5 [+1.0] | 49.5 [+12.0] | +22.5 |
| M6 | 3′-CCATATAATCCG |
47.5 [+10.0] | 49.0 [+11.5] | ||||
| M7 | 5′-GGTATATAAGGC |
35.5 [−2.0] | 47.5 [+10.0] | +21.0 | 36.5 [−1.0] | 48.0 [+10.5] | +22.0 |
| M8 | 3′-CCATATATACCG |
46.5 [+9.0] | 48.0 [+10.5] | ||||
| M9 | 5′-GGAATATAGGC |
40.0 [+2.5] | 51.5 [+14.0] | +29.5 | 43.5 [+6.0] | 51.5 [+14.0] | +25.0 |
| M10 | 3′-CCAAATATCCG |
55.5 [+18.0] | 54.5 [+17.0] | ||||
| M11 | 5′-GGATAATAGGC |
49.0 [+11.5] | 53.5 [+16.0] | +23.5 | 48.0 [+10.5] | 56.0 [+18.5] | +29.0 |
| M12 | 3′-CCAATAATCCG |
56.5 [+19.0] | 58.5 [+21.0] | ||||
| M13 | 5′-GGATATAAGGC |
49.0 [+11.5] | 52.5 [+15.0] | +21.5 | 45.0 [+7.5] | 55.5 [+18.0] | +32.0 |
| M14 | 3′-CCAATATACCG |
55.5 [+18.0] | 59.0 [+21.5] | ||||
| M15 | 5′-GGTAAATAGGC |
45.0 [+7.5] | 55.5 [+18.0] | +28.5 | 38.5 [+1.0] | 55.5 [+18.0] | +35.0 |
| M16 | 3′-CCATAAATCCG |
55.5 [+18.0] | 55.5 [+18.0] | ||||
| M17 | 5′-GGTATAAAGGC |
47.5 [+10.0] | 54.5 [+17.0] | +24.0 | 46.5 [+9.0] | 56.0 [+18.5] | +28.0 |
| M18 | 3′-CCATATAACCG |
54.5 [+17.0] | 56.0 [+18.5] | ||||
| M19 | 5′-GGAAAAGGC |
50.0 [+12.5] | 65.5 [+28.0] | +45.5 | 39.5 [+2.0] | 66.5 [+29.0] | +56.5 |
| M20 | 3′-CCAAAACCG |
67.5 [+30.0] | 67.0 [+29.5] | ||||
Singly modified ONs display greatly increased affinity towards cDNA relative to unmodified ONs, with Y-modified ONs forming slightly more stable duplexes (ΔTm = 7.0–11.0 °C vs. 8.0–13.5 °C, for X1–X8 and Y1–Y8, respectively, Table 1). Incorporation of a second pyrene-functionalized nucleotide in the same strand results in further stabilization (ΔTm for M9–M18 = 14.0–21.5 °C). However, the Tm increases are less than additive, suggesting that intercalation of the first pyrene moiety negatively impacts the energetics of the second intercalation event (e.g., compare ΔTm of M1:cDNA and M3:cDNA relative to M9:cDNA).
Invader duplexes with one +1 interstrand zipper arrangement – or energetic hotspot – of X or Y monomers display low Tm's (ΔTm = −1.0 to +3.0 °C) and are, accordingly, activated for recognition of dsDNA targets (TA ≫ 0 °C for M1:M2–M7:M8, Table 1). In accordance with previous results,27 double-stranded probes with other interstrand zipper arrangements of X- or Y-monomers are not activated for dsDNA-recognition (compare TA values, Tables 1 and S3–S5†). This is because the intercalators only are forced to occupy the same region – leading to violation of the nearest neighbor principle – when the corresponding monomers are placed in +1 interstrand zipper arrangements.33 DNA duplexes with two energetic hotspots are moderately stabilized (ΔTm for M9:M10–M17:M18 = 1.0–11.5 °C), with higher Tm's being observed for X-modified probes and probes with two non-consecutive energetic hotspots. These trends mirror our results for Invader probes modified with 2′-N-(pyren-1-yl)methyl-2′-amino-α-L-LNA.26 All of the double hotspot Invader probes are strongly activated for dsDNA-recognition due to the very high cDNA affinity of the individual strands, with Y-modified probes generally being more strongly activated (TA for M9:M10–M17:M18 = 21.5–35.0 °C). The results with X19:X20 and Y19:Y20, having four consecutive intercalator zippers, underscore the above conclusions.
Thermodynamic parameters for duplex formation
The available free energy for the prototypical dsDNA recognition process can also be parameterized as ΔG293rec = ΔG293 (5′-Inv:cDNA) + ΔG293 (3′-Inv:cDNA) − ΔG293 (Invader) − ΔG293 (dsDNA target region). Thermodynamic parameters for duplex formation were obtained from thermal denaturation curves via the van't Hoff method (Tables S6–S11†). Consistent with the Tm-based conclusions, Invader probes are strongly activated for dsDNA-recognition (i.e., ΔG293rec ≪ 0 kJ mol−1, Fig. 2a) due to the low stability of the probe duplexes (i.e., ΔΔG293 between −6 and +11 kJ mol−1, Fig. 2b) and the high stability of the probe-target duplexes (i.e., ΔΔG293 between −52 and −6 kJ mol−1, Fig. 2c and d). Recognition of dsDNA is very strongly enthalpically favored (ΔHrec ≪ 0 kJ mol−1, Tables S8, S9 and Fig. S2†), further underlining that forced intercalation is the main driving force (stabilizing in probe-target duplexes and destabilizing in Invader probes).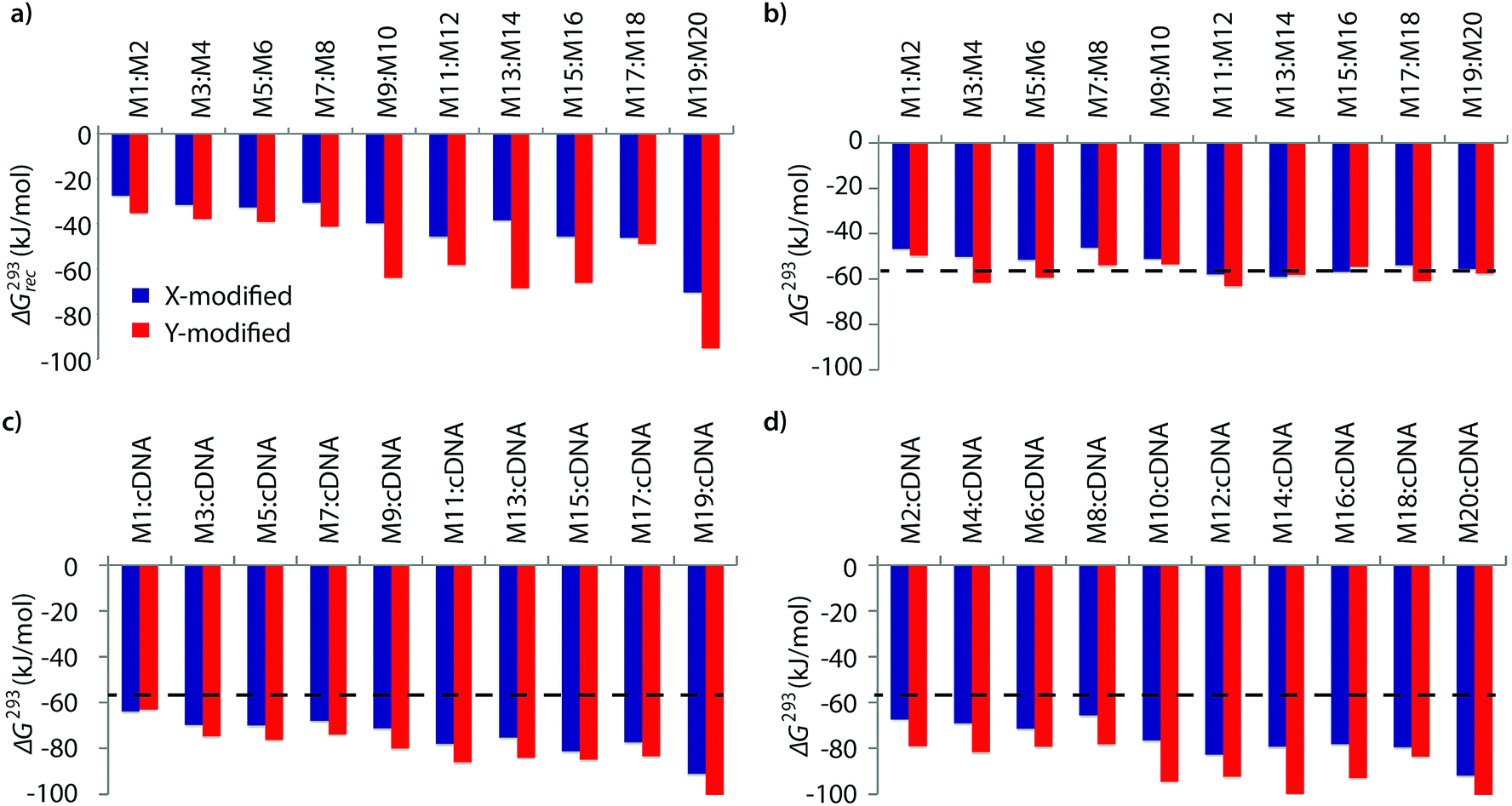 | ||
| Fig. 2 (a) Available Gibbs free energy at 293 K (ΔG293rec) for Invader-mediated recognition of isosequential dsDNA targets, and (b–d) change in Gibbs free energy upon formation of X- or Y-modified DNA duplexes. The ΔG293 for the dsDNA reference is shown as a dotted line at −57 kJ mol−1. See Table 1 for experimental conditions. See Tables S6 and S7† for tabulated data. | ||
Invader probes with multiple energetic hotspots display more favorable dsDNA-recognition thermodynamics than single hotspot probes (compare ΔG293rec for M1:M2–M7:M8vs.M9:M10–M19:M20, Fig. 2a), due to the exceptionally high cDNA affinity of the individual strands (note the highly negative ΔG293 values for duplexes between M9–M20 and cDNA, Fig. 2c and d). Y-modified Invader probes are more strongly activated for dsDNA-recognition than corresponding X-modified probes (ΔG293rec more favorable by 3–31 kJ mol−1, Fig. 2a), due to the higher stability of Y-modified probe-target duplexes (compare blue and red bars in Fig. 2c and d). Interestingly, X-/Y-modified Invader probes are more strongly activated for dsDNA recognition than isosequential probes based on the original 2′-N-(pyren-1-yl)methyl-2′-amino-α-L-LNA monomers (ΔG293rec more favorable by 1–29 kJ mol−1).26
Recognition of model dsDNA targets
The dsDNA recognition characteristics of the Invader probes were evaluated using an electrophoretic mobility shift assay (EMSA) that we developed in our preliminary studies (Fig. 3a).32 A 3′-digoxigenin (DIG) labeled DNA hairpin (DH), comprised of a 13-mer isosequential double-stranded stem that is connected on one side by a T10 linker, was used as a model dsDNA target. Successful recognition of the stem by an Invader probe is expected to result in the formation of a ternary complex with decreased mobility during non-denaturing polyacrylamide gel electrophoresis (nd-PAGE). All twenty Invader probes were screened at a concentration of 6.88 μM (i.e., 200-fold molar excess with respect to DH1) to identify probe architectures and monomer chemistries that result in efficient dsDNA recognition (Fig. 3b). All of the Invader probes result in recognition of the mixed-sequence stem when incubated at room temperature for 17 h (Fig. S4†). As expected, dsDNA recognition efficiency increases with more highly modified probes, with M19:M20 resulting in virtually complete recognition of DH1 (compare e.g.M1:M2 < M9:M10 < M19:M20, Fig. 3b). | ||
| Fig. 3 (a) Illustration of the electrophoretic mobility shift assay (EMSA) used to evaluate dsDNA recognition. (b) dsDNA recognition by Invader probes. DIG-labeled DH1 (34.4 nM) was incubated with 6.88 μM of a pre-annealed Invader probe in HEPES buffer (50 mM HEPES, 100 mM NaCl, 5 mM MgCl2, pH 7.2, 10% sucrose, 1.44 mM spermine tetrahydrochloride) for 17 h at room temperature. For representative electrophoretograms see Fig. S4.† | ||
Closer inspection of the results reveals that single hotspot Invader probes M1:M2 and M3:M4, in which the hotspot is located toward the ‘left’ terminus, recognize DH1 more efficiently than M5:M6 and M7:M8, in which the hotspot is located toward the ‘right’ terminus, despite having similar ΔG293rec values (Fig. 3b). To determine if these trends are due to fraying at the ‘left’ terminus of the target, X1:X2–X7:X8 were incubated with DH8 in which the ‘left’ side of the stem is connected via a T10 loop instead (Fig. S5†). Indeed, DH8 is recognized more efficiently by X5:X6 and X7:X8 but the trend is not fully reversed, suggesting that additional factors, such as the higher GC-content at the ‘right’ end, also impact recognition efficiency. Along similar lines, Invader probe X9:X10, featuring two consecutive energetic hotspots near the ‘left’ terminus, results in more efficient recognition of DH1 than X17:X18 where two consecutive energetic hotspots are located near the ‘right’ terminus (Fig. 3b). This trend is partially reversed when these Invaders are incubated with DH8 (Fig. S5†). On the other hand, Invader probe X15:X16, featuring two central consecutive hotspots, recognizes DH1 and DH8 with similar efficiency, while Invader probes M11:M12 and M13:M14, having two separated energetic hotspots, result in slightly less efficient dsDNA recognition (Fig. 3b and S5†). In summary, these results suggest that Invader probes with multiple hotspots – irrespective of the substitution pattern – enable recognition of dsDNA model targets, although targets with minimally fraying termini are more challenging.
Six Invader probes – featuring one, two or four consecutive hotspots based on either monomer X or Y – were selected from this initial screen for more thorough characterization. Dose response experiments were performed to determine C50 values, i.e., the probe concentration that results in 50% recognition of DH1 (Fig. 4 and Table 2). Increasing the number of energetic hotspots progressively decreases the C50 values from single digit micromolar to submicromolar ranges. Probes based on 2′-N-(pyren-1-yl)methyl-2′-N-methyl-2′-aminouridine monomer Y display lower C50 values than probes based on 2′-O-(pyren-1-yl)methyluridine monomer X, which is in line with the observed ΔG293rec values.
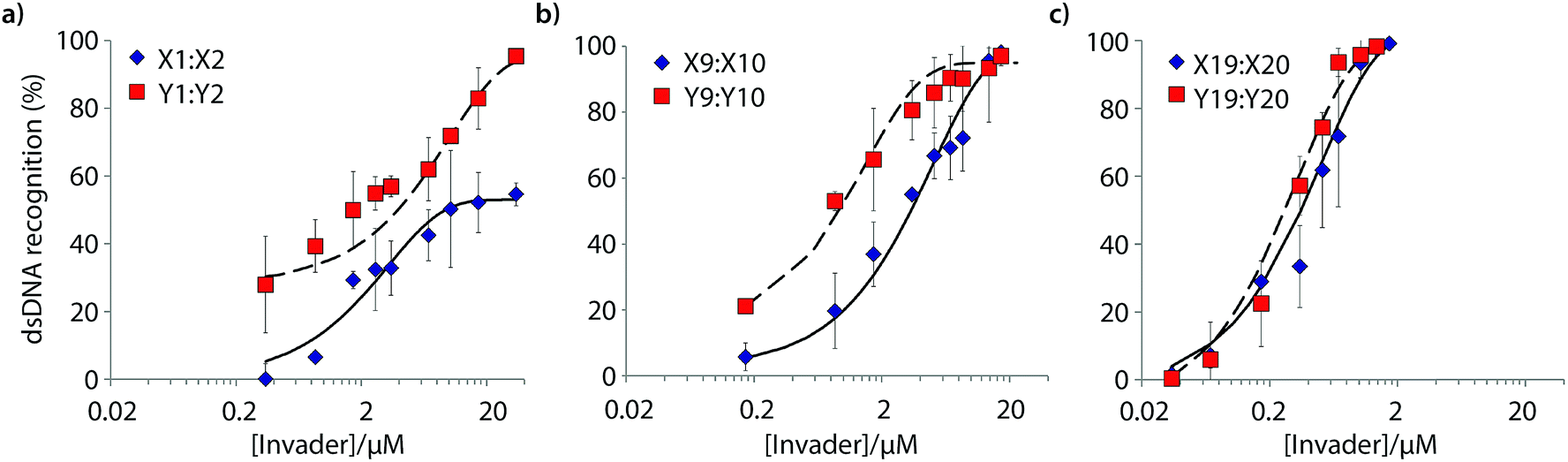 | ||
| Fig. 4 Dose-response curves for recognition of DH1 using (a) M1:M2, (b) M9:M10, or (c) M19:M20. For experimental conditions, see Fig. 3. | ||
| Invader | X-modified Invader | Y-modified Invader | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Rec200 (%) | C 50 (μM) | t 50 (min) | k obs (min−1) | k rel | Rec200 (%) | C 50 (μM) | t 50 (min) | k obs (min−1) | k rel | |
| a Rec200 denotes degree of recognition when using Invader at a 200-fold molar excess. C50, t50 and kobs values obtained from Fig. 4, 5, and S7, respectively. krel are calculated relative to the pseudo-first order rate constant for X1:X2. | ||||||||||
| M1:M2 | 46 | 9.4 | ND | 1.1 × 10−3 | 1 | 51 | 3.9 | 498 | 1.4 × 10−3 | 1.3 |
| M9:M10 | 74 | 3.4 | 185 | 7.5 × 10−3 | 6.8 | 92 | 0.9 | 211 | 4.3 × 10−3 | 3.9 |
| M19:M20 | >95 | 0.4 | 7 | 9.7 × 10−2 | 88 | >95 | 0.3 | 47 | 1.5 × 10−2 | 13.5 |
Recognition of DH1 using individual probe strands was also examined. Incubation of DH1 with 1000-fold molar excess of M1 or M2 or 500-fold molar excess of M9 or M10 results only in trace formation of recognition complexes, demonstrating that both strands of an Invader probe normally are necessary for efficient dsDNA-recognition (Fig. S6†). However, the use of 50-fold molar excess of X19, X20 or Y20 results in complete recognition of DH1. Clearly, the cDNA affinity of these strands is able to overcome the enthalpic penalty associated with opening the Watson–Crick base-pairs of the stem region and leaving one hairpin arm unhybridized.
Kinetics of dsDNA-recognition
Time-course experiments were performed, in which Invader probes (200-fold molar excess) were incubated with model target DH1 and quenched at specific time-points to elucidate dsDNA-recognition kinetics (Fig. 5 and Table 2). Recognition of DH1 proceeds incrementally faster using more highly modified Invader probes, with 50% recognition (t50) being attained within ∼3 h with double hotspot probe M9:M10 and within 10–50 min with quadruple hotspot probe M19:M20. It is noteworthy that X-modified probes have faster recognition kinetics than the corresponding Y-modified probes despite less favorable C50 values. Similar conclusions are reached based on pseudo-first order rate constants (Fig. S7† and Table 2).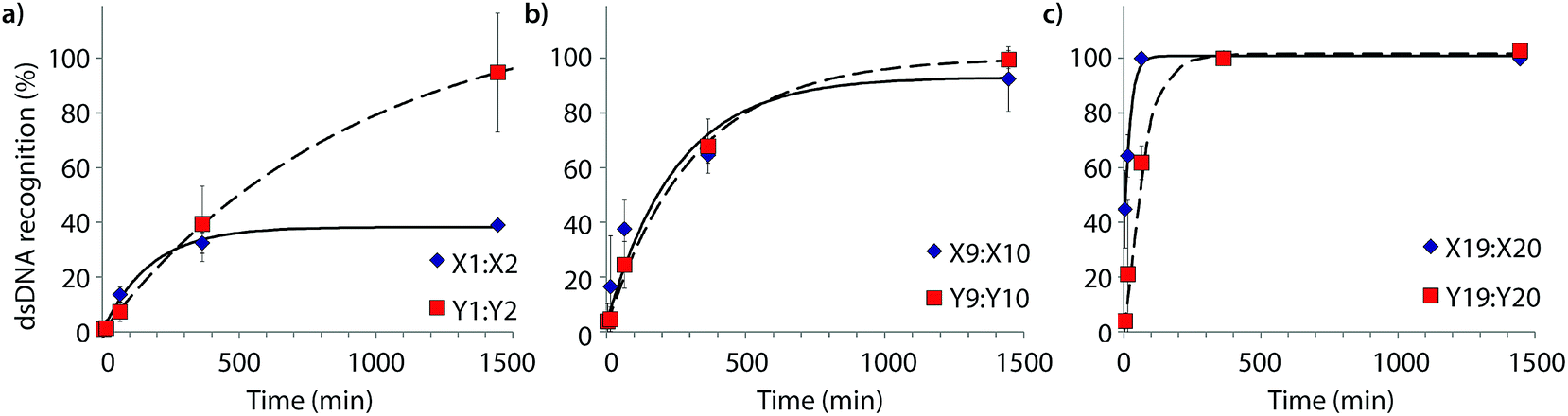 | ||
| Fig. 5 Kinetic profile of DH1 recognition using 200-fold molar excess of (a) M1:M2, (b) M9:M10, or (c) M19:M20. For incubation conditions, see Fig. 3. Aliquots were taken at specific time points, flash frozen in liquid N2, and stored at −76 °C until analysis. | ||
The reaction kinetics are highly dependent on the incubation temperature (Fig. 6). Thus, incubation of DNA hairpin DH1 with probes X9:X10 or Y9:Y10 at ∼8 °C fails to result in any dsDNA recognition (data not shown), while incubation at 35 °C or 45 °C results in major rate enhancements relative to room temperature incubation (e.g., ∼20%, 50% and 80% recognition after 10 min using X9:X10 at room temperature, 35 °C and 45 °C, respectively). The rate enhancements are, most likely, due to increased denaturation of the probes (Tm's of X9:X10 or Y9:Y10 = 40.0 and 43.5 °C, respectively, Table 1) rather than denaturation of DH1 (Tm = 58.5 °C, Table S12†). Incubation at 55 °C results in fast, but less pronounced, product formation, presumably because the recognition complex is partially denatured at this temperature (Tm of X9/X10/Y9/Y10vs. cDNA = 51.5–55.5 °C, Table 1). These results demonstrate that dsDNA recognition can be accelerated by increasing experimental temperatures to 5–10 °C below the Tm of the probe-target recognition duplexes, when practically possible.
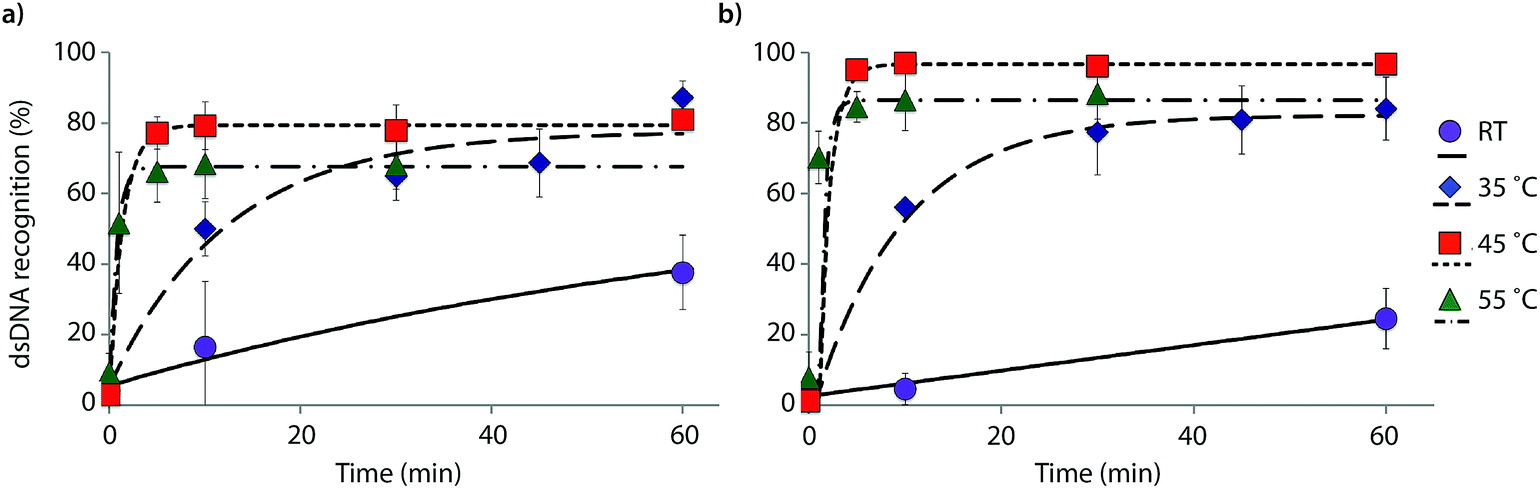 | ||
| Fig. 6 Temperature dependence of dsDNA recognition kinetics using (a) X9:X10 or (b) Y9:Y10 having two consecutive energetic hotspots. Experiments were performed as described in Fig. 3 with the exception of different incubation temperatures. | ||
Stability of recognition complexes
The kinetic stability of the recognition complexes with DH1 was studied next. The recognition complex was allowed to form and was then incubated with a large excess of linear competitor dsDNA target that rapidly sequesters any dissociating Invader strands,26 preventing their re-association with DH1 (Fig. 7a). Indeed gradual disappearance of the recognition complex is observed with time (Fig. 7b). The recognition complex between DH1 and X1:X2 dissociates within 8 h, while the complex between DH1 and Y1:Y2 requires ∼24 h for complete dissociation. In contrast, the recognition complexes between DH1 and M9:M10 are very stable, as evidenced by the small amounts of DH1 formed after 24 h (∼60% and ∼85% of the recognition complexes with X9:X10 and Y9:Y10, respectively, remaining). It is noteworthy that the collapse of the recognition complexes between DH1 and M9:M10, in all likelihood, proceeds via a binary complex in which only one of the two probe-target recognition duplexes has dehybridized (notice the band immediately below the recognition complex band, Fig. 7b). Thus, although single-stranded M9 or M10 cannot overcome the activation energy of the recognition process and invade the stem of DH1 (Fig. S6†), it appears that an Invader strand can remain bound to the hairpin after dissociation of the other strand.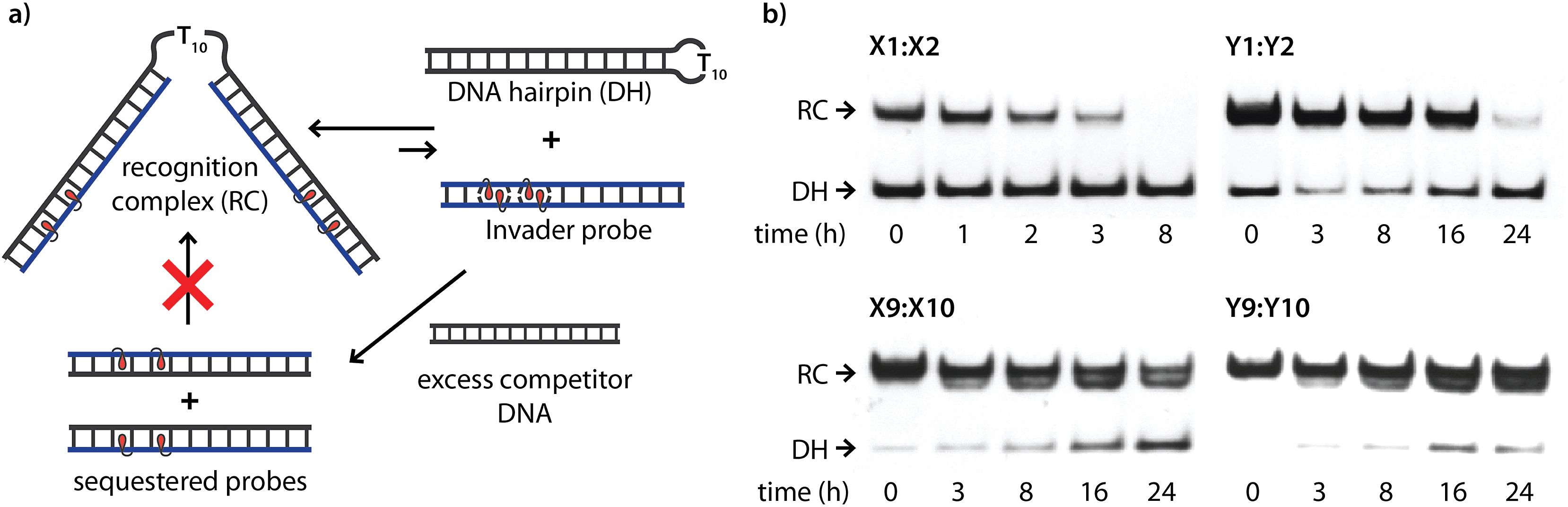 | ||
Fig. 7 Dissociation kinetics of recognition complexes between DNA hairpins and Invader probes. (a) Illustration of competition assay. (b) Representative gel electrophoretograms for dissociation reactions. Recognition complexes were formed (incubation of 34.4 nM DH1 with 200-fold molar excess of M1:M2 or M9:M10 for 17 h at room temperature), followed by addition of 2000-fold molar excess of 5′-GGTATATATAGGC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3′-CCATATATATCCG. Incubation conditions are as described in Fig. 3. Reactions were quenched at specific times points as described in Fig. 5. :3′-CCATATATATCCG. Incubation conditions are as described in Fig. 3. Reactions were quenched at specific times points as described in Fig. 5. | ||
Binding specificity of dsDNA recognition
The above results show that it is possible to design energetically activated duplexes that enable efficient (C50 < 1 μM), fast (t50 < 3 h), and kinetically stable (>24 h) recognition of mixed-sequence dsDNA targets under physiologically relevant buffer conditions. To assess the specificity of the recognition process, the six selected Invader probes were incubated with DNA hairpins DH2–DH7 (Fig. 8), which have stem regions that differ in the nucleotide sequence relative to the probes at either the 6- or 8-position of the stem. Probes with one or two hotspots generally display excellent discrimination of the mismatched targets (<10% recognition of DH5 with M9:M10) at concentrations resulting in very efficient recognition of DH1 using sequence-matched probes (1000- and 500-fold molar excess of M1:M2 and M9:M10, respectively). In contrast, the very high dsDNA affinity of the constructs containing four energetic hotspots compromises the specificity of the recognition process. Only X19:X20 exhibits partial discrimination when the non-complementary base pairs are centrally positioned in the target region (55–80% recognition of DH2–DH4), which indicates that the initial nucleation site is distal to the loop region. | ||
| Fig. 8 Discrimination of non-complementary DNA hairpins (DH2–DH7) using 1000-, 500- and 50-fold excess of M1:M2, M9:M10 and M19:M20, respectively. For experimental conditions, see Fig. 3. Tm's of DH1–DH7 are between 58.5–63.5 °C (Table S12†). | ||
Detection of chromosomal DNA
Encouraged by these results, we set out to examine Invader probes based on 2′-O-(pyren-1-yl)methyl-RNA monomers as FISH probes for recognition of chromosomal DNA using non-denaturing conditions. Unlike conventional FISH assays, which require denaturation of chromosomal DNA by heat and/or formamide treatment,34 nd-FISH approaches enable mapping of chromosomal loci at mild conditions, thus offering the prospect of in vivo imaging. The majority of previously reported nd-FISH approaches are based either on classic dsDNA-targeting agents (i.e., TFOs, PNAs or polyamides)35–40 or the presence of uniquely accessible DNA regions,41,42 which has limited the widespread use of these approaches.Four Cy3-labeled Invader probes, varying in length as well as number and position of energetic hotspots (INV1–INV4, probe lengths: 11–14 nt, 2–3 hotspots, Table 3), were designed against a target region within the DYZ-1 satellite region (∼6 × 104 tandem repeats) of the bovine (Bos taurus) Y chromosome (NCBI code: M26067; target site: 562–575).43 We have previously used PNA FISH probes targeting this site to determine the gender of bovine somatic cells, spermatozoa, and embryos.44–46 However, single-stranded fluorophore-labeled PNAs fail to produce signals under non-denaturing conditions (Fig. S9†).
| Invader | Sequence | T m [ΔTm] (°C) | ΔG310rec (kJ mol−1) | |||
|---|---|---|---|---|---|---|
| 5′-Inv:cDNA | 3′-Inv:cDNA | Invader | dsDNA target | |||
a ΔTm = change in Tm values relative to corresponding unmodified and unlabeled reference duplex. For experimental details see Table 1. ![[A with combining low line]](https://www.rsc.org/images/entities/b_char_0041_0332.gif) , , ![[C with combining low line]](https://www.rsc.org/images/entities/b_char_0043_0332.gif) and and ![[U with combining low line]](https://www.rsc.org/images/entities/b_char_0055_0332.gif) denote 2′-O-(pyren-1-yl)methyladenosine,47 2′-O-(pyren-1-yl)methylcytidine47 and 2′-O-(pyren-1-yl)methyluridine (monomer X), respectively. denote 2′-O-(pyren-1-yl)methyladenosine,47 2′-O-(pyren-1-yl)methylcytidine47 and 2′-O-(pyren-1-yl)methyluridine (monomer X), respectively.
|
||||||
| INV1 | 5′-Cy3-AGCCCGGCCCTG |
69.5 [+9.0] | 74.5 [+14.0] | 58.0 [−2.5] | 60.5 | −46 |
| 3′-TCGGGAAGGGAC-Cy3 |
||||||
| INV2 | 5′-Cy3-CCGGCCCTG |
59.5 [+9.0] | 65.5 [+15.0] | 48.0 [−2.5] | 50.5 | −30 |
| 3′-GGAAGGGAC-Cy3 |
||||||
| INV3 | 5′-Cy3-CCGTGCCTG |
59.0 [+8.5] | 64.0 [+13.5] | 47.0 [−3.5] | 50.5 | −24 |
| 3′-GGAACGGGC-Cy3 |
||||||
| INV4 | 5′-Cy3-GCCCGTGCCTG |
69.5 [+9.0] | 75.5 [+15.0] | 61.5 [+1.0] | 60.5 | −29 |
| 3′-TGGGAACGGGC-Cy3 |
||||||
As expected, the designed Invader probes display low thermal stability relative to isosequential dsDNA target regions, while the individual strands form very stable duplexes with cDNA, resulting in prominent dsDNA-targeting potential for the probes (see Tm and ΔG310rec values, Table 3 – for additional thermodynamic parameters, see Tables S13–S15†).
Gratifyingly, incubation of these Cy3-labeled Invader probes with fixed interphase nuclei from a male bovine kidney cell line (MDBK (NBL-1) (ATCC® CCL-22™)) at non-denaturing conditions (3 h, 38.5 °C, 10 mM Tris–Cl, pH 8.0 and 1 mM EDTA), produces a single fluorescent signal that localizes to the heterochromatic region, consistent with the expected localization of the DYZ-1 satellite target (Fig. 9 upper panel and Fig. S10†). The high labeling coverage (∼90%), i.e., the proportion of nuclei with localized signals, is noteworthy. Localized signals are also observed when Cy3-labeled Invader probes are incubated with nuclei captured in metaphase (Fig. 9 middle panel and Fig. S10†). All of the probes result in robust Cy3-signals, with little variation between the different probes (Fig. S10†). Nuclei that were pre-treated with RNase A or proteinase K prior to incubation with Invader INV4 display similar signals as nuclei without pretreatment, while nuclei pre-treated with DNase I are devoid of localized signals (Fig. S11†), which verifies that DNA is the molecular target of the Invader probes. As expected, there is also an absence of localized signals when nuclei from a female bovine fibroblast cell line are incubated with a Y-chromosome specific Invader probe (Fig. 9 lower panel). This, along with observations from our initial studies showing lack of signal formation when triply mismatched Invaders are incubated with nuclei from the male bovine kidney cell line,32 strongly suggests that the Invader probes specifically bind to their intended targets.
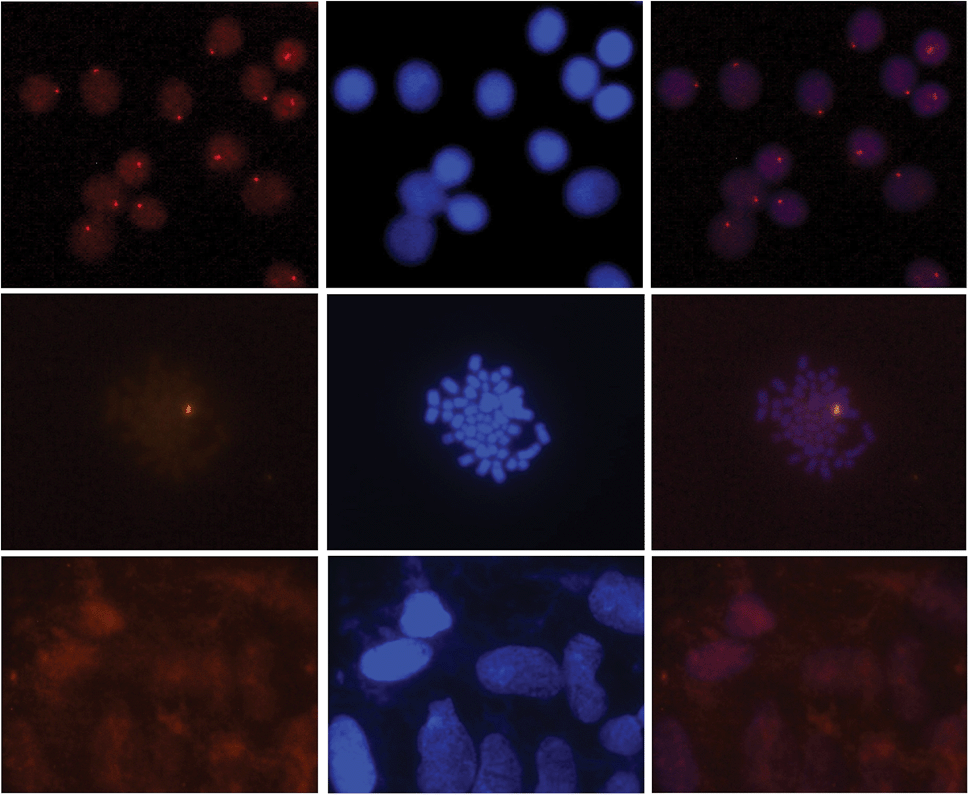 | ||
| Fig. 9 Images from fluorescence in situ hybridization using Y-chromosome specific Invader probes under non-denaturing conditions. Invader probe INV4 was added to nuclei from male bovine kidney cells in interphase (upper panel) or metaphase (middle panel), or to nuclei from female bovine fibroblast cells (lower panel). Images viewed using Cy3 (left column) or DAPI (middle column) filter settings; overlays are shown in the right column. Incubation: 3 h at 38.5 °C in 10 mM Tris–Cl, pH 8.0 and 1 mM EDTA. Cells were visualized at 400× magnification using a Zeiss AxioSkop 40 fluorescent microscope and images captured using a Zeiss AxioCam MRc5 camera. For additional images using probes INV1–INV4, see Fig. S10.† | ||
Binding mechanism
While our understanding of the recognition mechanism remains incomplete, we speculate that the distorted Invader probes trap regions of chromosomal DNA via a double duplex invasion mechanism analogous to that of pcPNAs (Fig. 10).24 Studies with γ-PNA19 have suggested that DNA is sufficiently dynamic to enable strand invasion at 37 °C, provided that the ligand has sufficient binding free energy. Presumably, nucleation of the Invader probes is initiated due to the exceptionally high cDNA affinity of the intercalator-functionalized nucleotides, and base-pairing proceeds until a stable double duplex invasion recognition complex is formed. Accordingly, many previously inaccessible mixed-sequence dsDNA target regions may become accessible to exogenous probes.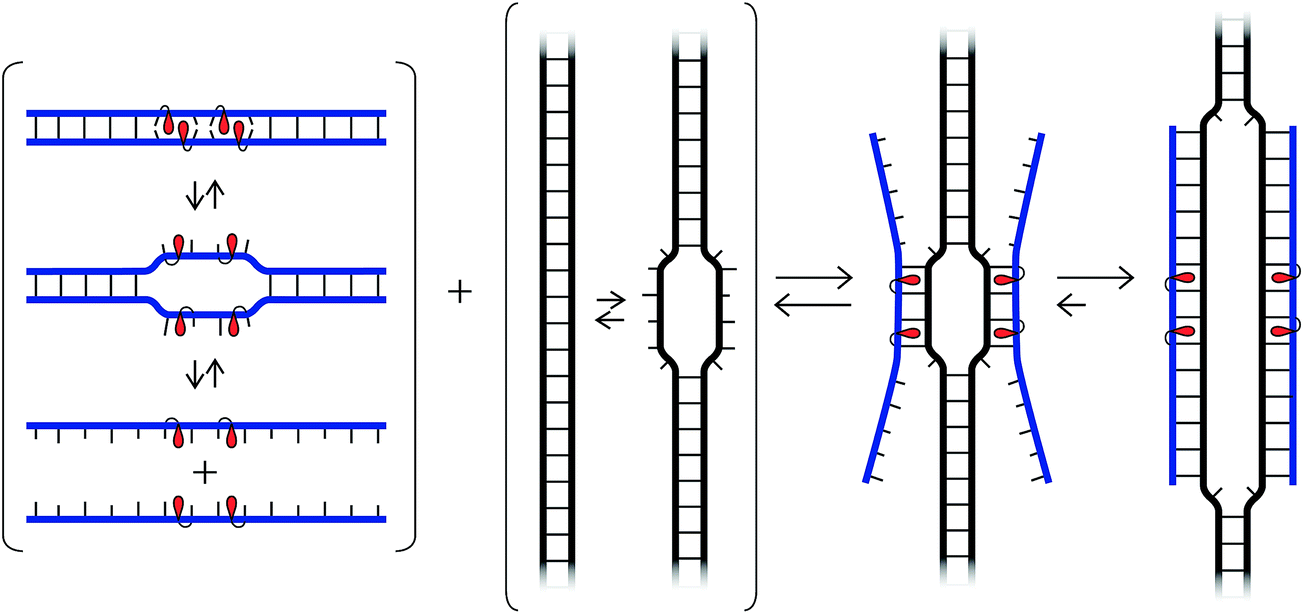 | ||
| Fig. 10 Illustration of hypothetical recognition mechanism. | ||
Conclusion
This study demonstrates that Invader probes can be designed to display efficient (C50 < 1 μM), fast (t50 < 3 h), kinetically stable (>24 h) and single nucleotide specific recognition of mixed-sequence DNA targets. Probe duplexes with energetic hotspots comprised of +1 interstrand zippers of 2′-O-(pyren-1-yl)methyl-RNA or 2′-N-(pyren-1-yl)methyl-2′-N-methyl-2′-amino-DNA monomers are thermodynamically activated for DNA recognition, whereas probes with other zipper motifs are not, which underscores the unique properties of +1 intercalator zipper motifs. Recognition of DNA targets proceeds progressively faster and with greater efficiency with every additional energetic hotspot that is incorporated into the Invader probes. These guidelines enabled the design of Invaders for successful detection of gender-specific mixed-sequence chromosomal DNA target regions in bovine kidney cells.The insights gained from this study will enable the design of efficient Invader probes for DNA-targeting applications including gene regulation via transcriptional interference, in vivo imaging of chromosomal DNA targets, cell-sorting of genotype-specific cells and development of artificial restriction ‘enzymes’.
Acknowledgements
This study has enjoyed support from NIH grant no. GM088697 (National Institute of General Medical Sciences); Higher Education Research Council, Idaho State Board of Education (Awards IF13-001 and IF14-012); The Office of Naval Research (N00014-10-1-0282); and INBRE Program, NIH grant no. P20 RR016454 (National Center for Research Resources) and P20 GM103408 (National Institute of General Medical Sciences). We thank Dr Lee Deobald (Murdock Mass Spectrometry Center, Univ. Idaho) for assistance with mass spectrometric analysis and Dr Carolyn J. Hovde (Food Science, Univ. Idaho) for access to gel documentation stations.Notes and references
- R. Besch, C. Giovannangeli and K. Degitz, Curr. Drug Targets, 2004, 5, 691–703 CrossRef CAS.
- F. A. Rogers, J. A. Lloyd and P. M. Glazer, Curr. Med. Chem.: Anti-Cancer Agents, 2005, 5, 319–326 CrossRef CAS.
- I. Ghosh, C. I. Stains, A. T. Ooi and D. J. Segal, Mol. BioSyst., 2006, 2, 551–560 RSC.
- P. E. Nielsen, Chem. Biodiversity, 2010, 7, 786–804 CAS.
- A. Mukherjee and K. M. Vasquez, Biochimie, 2011, 93, 1197–1208 CrossRef CAS PubMed.
- Y. Aiba, J. Sumaoka and M. Komiyama, Chem. Soc. Rev., 2011, 40, 5657–5668 RSC.
- T. Vaijayanthi, T. Bando, G. N. Pandian and H. Sugiyama, ChemBioChem, 2012, 13, 2170–2185 CrossRef CAS PubMed.
- M. Duca, P. Vekhoff, K. Oussedik, L. Halby and P. B. Arimondo, Nucleic Acids Res., 2008, 36, 5123–5138 CrossRef CAS PubMed.
- P. B. Dervan and B. S. Edelson, Curr. Opin. Struct. Biol., 2003, 13, 284–299 CrossRef CAS.
- M. S. Blackledge and C. Melander, Bioorg. Med. Chem., 2013, 21, 6101–6114 CrossRef CAS PubMed.
- P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CAS.
- K. Kaihatsu, B. A. Janowski and D. R. Corey, Chem. Biol., 2004, 11, 749–758 CrossRef CAS PubMed.
- T. Gaj, C. A. Gersbach and C. F. Barbas III, Trends Biotechnol., 2013, 31, 397–405 CrossRef CAS PubMed.
- J. D. Sander and J. K. Joung, Nat. Biotechnol., 2014, 32, 347–355 CrossRef CAS PubMed.
- J. W. Trauger, E. E. Baird and P. B. Dervan, J. Am. Chem. Soc., 1998, 120, 3534–3535 CrossRef CAS.
- Y. Fu, J. A. Foden, C. Khayter, M. L. Maeder, D. Reyon, J. K. Joung and J. D. Sander, Nat. Biotechnol., 2013, 31, 822–826 CrossRef CAS PubMed.
- T. Bentin, H. J. Larsen and P. E. Nielsen, Biochemistry, 2003, 42, 13987–13995 CrossRef CAS PubMed.
- K. Kaihatsu, R. H. Shah, X. Zhao and D. R. Corey, Biochemistry, 2003, 42, 13996–14003 CrossRef CAS PubMed.
- S. Rapireddy, R. Bahal and D. H. Ly, Biochemistry, 2011, 50, 3913–3918 CrossRef CAS PubMed.
- R. Bahal, B. Sahu, S. Rapireddy, C.-M. Lee and D. H. Ly, ChemBioChem, 2012, 13, 56–60 CrossRef CAS PubMed.
- I. V. Kutyavin, R. L. Rhinehart, E. A. Lukhtanov, V. V. Gorn, R. B. Meyer Jr and H. B. Gamper Jr, Biochemistry, 1996, 35, 11170–11176 CrossRef CAS PubMed.
- J. Lohse, O. Dahl and P. E. Nielsen, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11804–11808 CrossRef CAS.
- T. Ishizuka, J. Yoshida, Y. Yamamoto, J. Sumaoka, T. Tedeschi, R. Corradini, S. Sforza and M. Komiyama, Nucleic Acids Res., 2008, 36, 1464–1471 CrossRef CAS PubMed.
- V. V. Demidov, E. Protozanova, K. I. Izvolsky, C. Price, P. E. Nielsen and M. D. Frank-Kamenetskii, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5953–5958 CrossRef CAS PubMed.
- J. Sumaoka and M. Komiyama, Chem. Lett., 2014, 43, 1581–1583 CrossRef CAS.
- S. P. Sau, T. S. Kumar and P. J. Hrdlicka, Org. Biomol. Chem., 2010, 8, 2028–2036 CAS.
- S. P. Sau, A. S. Madsen, P. Podbevsek, N. K. Andersen, T. S. Kumar, S. Andersen, R. L. Rathje, B. A. Anderson, D. C. Guenther, S. Karmakar, P. Kumar, J. Plavec, J. Wendel and P. J. Hrdlicka, J. Org. Chem., 2013, 78, 9560–9570 CrossRef CAS PubMed.
- D. M. Crothers, Biopolymers, 1968, 6, 575–584 CrossRef CAS PubMed.
- C. Tsai, S. C. Jain and H. M. Sobell, J. Mol. Biol., 1977, 114, 301–315 CrossRef CAS.
- L. D. Williams, M. Egli, Q. Gao and A. Rich, in Structure and Function, Volume 1: Nucleic Acids, ed. R. H. Sarma and M. H. Sarma, Adenine press, Albany, NY, 1992, pp. 107–125 Search PubMed.
- B. Denn, S. Karmakar, D. C. Guenther and P. J. Hrdlicka, Chem. Commun., 2013, 49, 9851–9853 RSC.
- B. A. Didion, S. Karmakar, D. C. Guenther, S. P. Sau, J. P. Verstegen and P. J. Hrdlicka, ChemBioChem, 2013, 14, 1534–1538 CrossRef CAS PubMed.
- S. Karmakar, A. S. Madsen, D. C. Guenther, B. C. Gibbons and P. J. Hrdlicka, Org. Biomol. Chem., 2014, 12, 7758–7773 CAS.
- J. M. Levsky and R. H. Singer, J. Cell Sci., 2003, 116, 2833–2838 CrossRef CAS PubMed.
- M. D. Johnson III and J. R. Fresco, Chromosoma, 1999, 108, 181–189 CrossRef.
- E. Schmitt, J. Schwarz-Finsterle, S. Stein, P. Mueller, A. Mokhir, R. Kraemer, C. Cremer and M. Hausmann, Methods Mol. Biol., 2010, 659, 185–202 CAS.
- C. Molenaar, K. Wiesmeijer, N. P. Verwoerd, S. Khazen, R. Eils, H. J. Tanke and R. W. Dirks, EMBO J., 2003, 22, 6631–6641 CrossRef CAS PubMed.
- S. Janssen, T. Durussel and U. K. Laemmli, Mol. Cell, 2000, 6, 999–1011 CrossRef CAS.
- M. P. Gygi, M. D. Ferguson, H. C. Mefford, K. P. Lund, C. O'Day, P. Zhou, C. Friedman, G. van der Engh, M. L. Stolowitz and B. J. Trask, Nucleic Acids Res., 2002, 30, 2790–2799 CrossRef CAS PubMed.
- A. N. Silahtaroglua, N. Tommerup and H. Vissing, Mol. Cell. Probes, 2003, 17, 165–169 CrossRef.
- K. Sugimura, S. Takebayashi, S. Ogata, H. Taguchi and K. Okumura, Biosci., Biotechnol., Biochem., 2007, 71, 627–632 CrossRef CAS PubMed.
- N. Cuadrado and N. Jouve, Chromosoma, 2010, 19, 495–503 CrossRef PubMed.
- J. Perret, Y. Shia, R. Fries, G. Vassart and M. Georges, Genomics, 1990, 6, 482–490 CrossRef CAS.
- R. Bleher, W. Erwin, A. M. Paprocki, C. M. Syverson, R. Koppang and B. A. Didion, Reprod., Fertil. Dev., 2009, 21, 227 CrossRef.
- B. A. Didion and R. Bleher, Reprod., Fertil. Dev., 2009, 21, 229 CrossRef.
- B. A. Didion, W. Erwin and R. Bleher, WO2009/079456A2, 2009.
- S. Karmakar, D. C. Guenther and P. J. Hrdlicka, J. Org. Chem., 2013, 78, 12040–12048 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: All experimental protocols; definition of zipper nomenclature, MS data for new modified ONs; representative Tm curves; additional thermal denaturation, thermodynamic parameter, dsDNA recognition and imaging data. See DOI: 10.1039/c5sc01238d |
| This journal is © The Royal Society of Chemistry 2015 |