
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cobalt catalyzed sp3 C–H amination utilizing aryl azides†
Omar
Villanueva‡
,
Nina Mace
Weldy
,
Simon B.
Blakey
* and
Cora E.
MacBeth
*
Department of Chemistry, Emory University, USA. E-mail: sblakey@emory.edu; cmacbet@emory.edu
First published on 30th July 2015
Abstract
A dinuclear Co(II) complex supported by a modular, tunable redox-active ligand system is capable of selective C–H amination to form indolines from aryl azides in good yields at low (1 mol%) catalyst loading. The reaction is tolerant of medicinally relevant heterocycles, such as pyridine and indole, and can be used to form 5-, 6-, and 7-membered rings. The synthetic versatility obtained using low loadings of an earth abundant transition metal complex represents a significant advance in catalytic C–H amination technology.
Introduction
The direct amination of C–H bonds in a mild, functional group tolerant manner represents an important technology for the synthesis of biologically active molecules, including natural products and pharmaceutical agents.1 Significant progress has been made in the development of catalysts capable of promoting metallonitrene initiated C–H amination utilizing carbamate,2–6 sulfamate,2,7–12 sulfonamide,13,14 sulfamide,15–18 and phosphoramide19 reagents for both intra- and intermolecular C–H amination, and in some cases, useful levels of enantio-induction can also be achieved.20–24On occasion, these C–H amination reactions install the desired nitrogen functionality, but more frequently deprotection and subsequent elaboration are required. Robust C–H functionalization reactions to directly incorporate alkyl or aryl amines into a target molecule have proven to be more challenging and are the focus of this study.25
Alkyl and aryl azides have been recognized as important reagents for these reactions, offering the potential to deliver the C–H amination product in the presence of an appropriate catalyst, with nitrogen gas as the only byproduct. In early work, Driver and coworkers described [(cod)Ir(OMe)]2 as uniquely effective in catalyzing the intramolecular sp3 C–H insertion of a presumed metallonitrene intermediate obtained by aryl azide decomposition.26 Subsequent studies have shown that Rh2(esp)2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 27 and Fe(TPP)28 are also capable of inducing these reactions.
27 and Fe(TPP)28 are also capable of inducing these reactions.
The pioneering studies of Tim Warren and coworkers established β-diketimide copper complexes as effective catalysts for C–H amination reactions with alkyl azides, but these reactions are complicated by competitive α-hydride migration.29,30
Recent reports by Betley and coworkers have established the advantage of incorporating a dipyrromethene redox-active ligand system31–33 at Fe(II) centers to promote efficient catalytic C–H amination of aliphatic34 and olefinic35 substrates. Application of this ligand system in a Co(I) complex proved to stabilize a cobalt(III)-imido species derived from aryl and alkyl azides capable of carrying out C–H bond activation. However, this system was not reported to be catalytic.36
In this manuscript we report the development of a cobalt catalyst for C–H amination that is tolerant of medicinally relevant heterocycles, such as pyridine and indole, and is capable of delivering 5-, 6-, and 7-membered rings. This combination of robust functional group tolerance and versatility with respect to ring size obtained, with low loadings of an earth-abundant transition metal complex, represents a significant advance in C–H amination catalysis.
We have previously disclosed that Co(II) complexes supported by a coordinatively versatile redox-active ligand derived from the NH(o-PhNHC(O)iPr)2 scaffold are capable of promoting catalytic dioxygen activation to carry out O-atom transfer processes at impressive rates.37 The multi-electron processes required for this oxygen activation chemistry parallel those implicit in the azide activation/C–H amination sequence, suggesting that these novel dinuclear Co(II) complexes should be investigated as catalysts for metallonitrene chemistry (Fig. 1).
 | ||
| Fig. 1 Left: family of dinuclear Co(II) complexes investigated in this study (R = iPr, tBu, Ph, CF3); right: molecular structure of 4 determined by single crystal X-ray diffraction. | ||
Results and discussion
ortho-Homobenzyl substituted aryl azide 5 was selected as a test substrate to explore the possibility of the cobalt(II)-catalyzed intramolecular amination of aryl azides and establish effective reaction conditions (Table 1). No reaction was observed at room temperature with complex 1 and azide 5 over the course of 24 hours (entry 1). However, performing the reaction at elevated temperature (110 °C) produced indoline 6 as the only product in 43% yield (entry 2). To take advantage of the modularity of the ligand, dinuclear Co(II) complexes with varying acyl substituents (R = CF3, Ph, tBu) were evaluated for catalytic activity for intramolecular C–H amination using aryl azide 5. tButyl- and phenyl-substituted Co(II) complexes 2 and 3 furnished lower indoline yields of 20% and 10%, respectively (entries 3 and 4). Substitution of an electron-withdrawing CF3 group on the ligand in complex 4 resulted in trace amounts of 6 (entry 5). Carrying out the reaction with lower catalyst loading (1 mol%) of the best-performing catalyst 1 resulted in a yield of 11% 6 over 24 hours (entry 6). Extending the reaction time to 48 hours resulted in an optimal yield of 83% 6 (entry 7). Control experiments show that under these reaction conditions azide 5 remains intact in the absence of a Co(II) catalyst (entry 8). Furthermore, performing this reaction in the presence of a simple Co(II) salt (CoBr2) resulted in no product formation (entry 9), suggesting that the ligand framework is essential to promote this transformation using Co(II). It is important to note that this system is sensitive to water, and molecular sieves are required to achieve optimal indoline yields. In the absence of molecular sieves, a cyclized ligand byproduct (see ESI†) was observed, along with diminished indoline yields.
Under optimized conditions, azide 5 was converted to indoline in 80% isolated yield, notably without formation of the corresponding aniline or over-oxidation to the 2-phenylindole (Table 2, entry 1). The reaction was found to be robust with respect to the electronic nature of the homobenzylic aryl ring (83% and 81%, entries 2 and 3, respectively). Modulation of the aryl azide substitution resulted in a slight decrease in yield to 72% for CF3-substituted azide (entry 4). Notably, OMe substitution of the aryl azide resulted in 60% yield of 5-methoxy-2-phenylindole (entry 5). A 2:1 mixture of indoline to indole was observed by crude 1H NMR of the reaction mixture, but the indoline was oxidized upon exposure to silica gel chromatography during purification. The previously reported [(cod)Ir(OMe)]2 did not catalyze indoline formation with this particular substrate.26
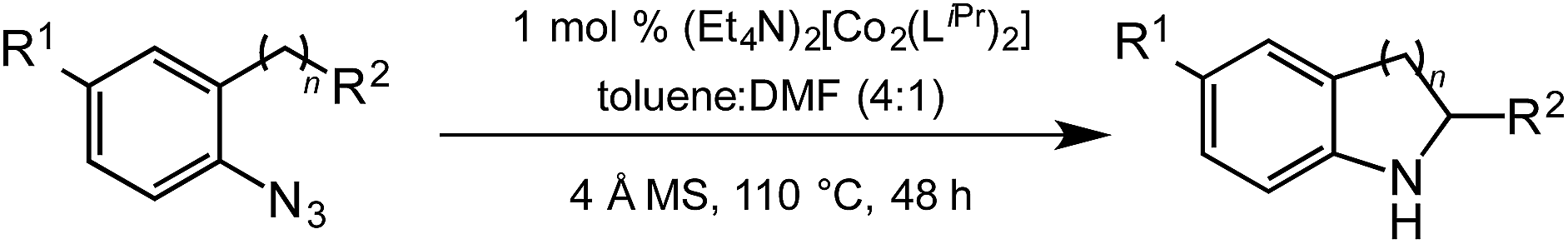



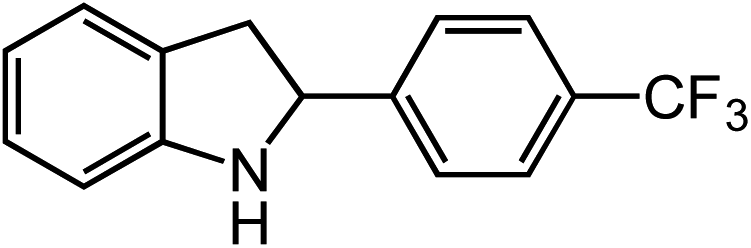





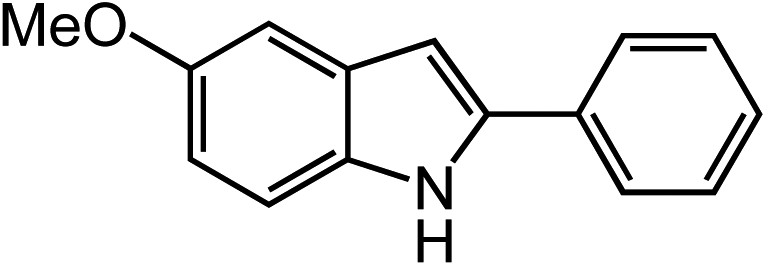











Extending the alkyl linker resulted in selective formation of the tetrahydroquinoline under these conditions, demonstrating six-membered ring formation is possible via this catalytic C–H amination reaction (Table 2, entry 6). Additionally, by extending the alkyl chain further, the seven-membered tetrahydrobenzoazepine was formed, albeit in diminished yield (30%, entry 7).
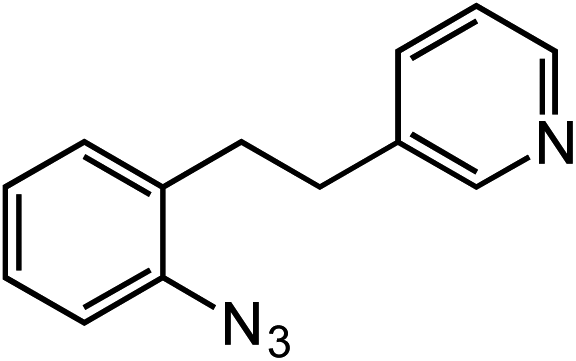
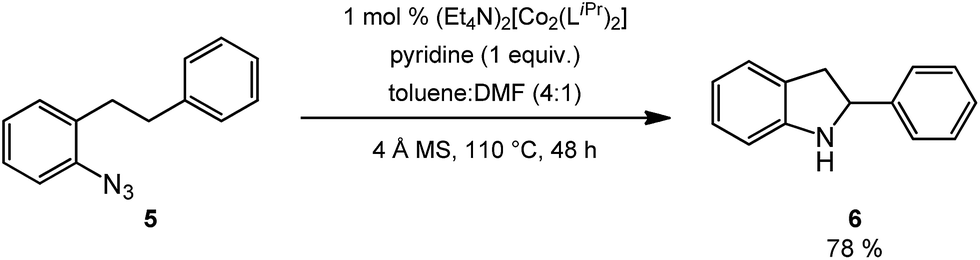
Novel heteroaromatic-containing substrates were also investigated with this catalyst system (Table 2, entries 8 and 9). Prior to investigating insertion at the benzylic position adjacent to pyridine, 1 equivalent of pyridine was added to the standard reaction of 1-azido-2-phenethylbenzene (5), and it was observed that the reaction proceeded normally (Scheme 1). This is notable due to the expectation that the high binding affinity of pyridine to transition metals might inhibit reactivity, as has been observed for other azide amination systems in the presence of exogenous coordinating ligands.34 Based on these results, the ortho-homopyridinyl-substituted aryl azide was synthesized and found to undergo amination in identical yield to the comparable benzylic insertion (entry 8). This tolerance of pyridine functionality, a privileged heterocycle for drug discovery, demonstrates the usefulness of this catalyst system for targets of pharmaceutical interest.38
 | ||
| Scheme 1 C–H amination in the presence of pyridine. | ||
In expanding to other biologically-relevant heteroaromatics, we investigated amination at the benzylic position adjacent to a protected indole and observed excellent conversion to product (85%, Table 2, entry 9). This also demonstrates the reaction is tolerant of an oxidation-sensitive substrate. However, in testing insertion into a benzylic 3° C–H center (Table 2, entry 10), we found amination was suppressed, resulting in 90% recovery of unreacted azide. Notably, when the alkyl chain was extended by one carbon, 3° C–H insertion was achieved to form the tetrahydroquinoline in 85% yield (Table 2, entry 11).
In order to probe the reaction pathway, the deuterated ortho-homobenzyl-aryl azide 7 was synthesized27 to measure the kinetic isotope effect (KIE) of this reaction. The deuterated substrate was subjected to the standard reaction conditions, and analysis of the crude reaction mixture by 1H NMR provided a KIE of 3.4 (Scheme 2). This kinetic isotope effect is lower than those observed for benzylic insertion by Driver and coworkers for the aryl azide system with [(cod)Ir(OMe)]2 (5.06)26 and Rh2(esp)2 (6.0)27 and by Betley and coworkers for their Fe-catalyzed alkyl azide amination (5.3).34 Zhang and coworkers observed a similarly greater value (6.2) for allylic insertion of a sulfonyl azide.17 While the value measured for the Co system is lower than these examples, it is higher than the KIE of 1.9 observed for dirhodium(II) catalyzed intramolecular benzylic amination with sulfamate esters.39 These data suggest a hydrogen-atom abstraction/radical recombination mechanism maybe operative in this system. Further studies to propose a mechanism for these reactions are currently underway in our laboratories.
 | ||
| Scheme 2 Kinetic isotope effect of intramolecular amination. | ||
Conclusions
In conclusion, we have shown a Co(II) complex with a modular, redox-active ligand is capable of selectively catalyzing benzylic C–H amination with aryl azides utilizing a variety of substrates in good yields. This robust catalyst system holds potential for application in pharmaceutical and total synthesis, as well as extension to other substrate classes.Acknowledgements
This research was supported by the National Science Foundation under the Center for Chemical Innovation in Selective C–H Functionalization (CHE-1205646).Notes and references
- J. L. Jeffrey and R. Sarpong, Chem. Sci., 2013, 4, 4092 RSC.
- C. G. Espino, P. M. Wehn, J. Chow and J. Du Bois, J. Am. Chem. Soc., 2001, 123, 6935 CrossRef CAS.
- H. Lu, V. Subbarayan, J. Tao and X. P. Zhang, Organometallics, 2009, 29, 389 CrossRef.
- R. D. Grigg, J. W. Rigoli, S. D. Pearce and J. M. Schomaker, Org. Lett., 2012, 14, 280 CrossRef CAS PubMed.
- H. Lebel, K. Huard and S. Lectard, J. Am. Chem. Soc., 2005, 127, 14198 CrossRef CAS PubMed.
- H. Lebel, C. Spitz, O. Leogane, C. Trudel and M. Parmentier, Org. Lett., 2011, 13, 5460 CrossRef CAS PubMed.
- C. G. Espino, K. W. Fiori, M. Kim and J. Du Bois, J. Am. Chem. Soc., 2004, 126, 15378 CrossRef CAS PubMed.
- K. W. Fiori and J. Du Bois, J. Am. Chem. Soc., 2007, 129, 562 CrossRef CAS PubMed.
- D. E. Olson and J. Du Bois, J. Am. Chem. Soc., 2008, 130, 11248 CrossRef CAS PubMed.
- J. L. Roizen, D. N. Zalatan and J. Du Bois, Angew. Chem., Int. Ed., 2013, 52, 11343 CrossRef CAS PubMed.
- P. M. Wehn, J. H. Lee and J. Du Bois, Org. Lett., 2003, 5, 4823 CrossRef CAS PubMed.
- D. E. Olson, D. A. Roberts and J. Du Bois, Org. Lett., 2012, 14, 6174 CrossRef CAS PubMed.
- J. V. Ruppel, R. M. Kamble and X. P. Zhang, Org. Lett., 2007, 9, 4889 CrossRef CAS PubMed.
- T. Kang, Y. Kim, D. Lee, Z. Wang and S. Chang, J. Am. Chem. Soc., 2014, 136, 4141 CrossRef CAS PubMed.
- T. Kurokawa, M. Kim and J. Du Bois, Angew. Chem., Int. Ed., 2009, 48, 2777 CrossRef CAS PubMed.
- H. Lu, H. Jiang, L. Wojtas and X. P. Zhang, Angew. Chem., Int. Ed., 2010, 49, 10192 CrossRef CAS PubMed.
- H. Lu, H. Jiang, Y. Hu, L. Wojtas and X. P. Zhang, Chem. Sci., 2011, 2, 2361 RSC.
- H. Lu, Y. Hu, H. Jiang, L. Wojtas and X. P. Zhang, Org. Lett., 2012, 14, 5158 CrossRef CAS PubMed.
- H. Lu, J. Tao, J. E. Jones, L. Wojtas and X. P. Zhang, Org. Lett., 2010, 12, 1248 CrossRef CAS PubMed.
- R. P. Reddy and H. M. L. Davies, Org. Lett., 2006, 8, 5013 CrossRef CAS PubMed.
- E. Milczek, N. Boudet and S. Blakey, Angew. Chem., Int. Ed., 2008, 47, 6825 CrossRef CAS PubMed.
- D. N. Zalatan and J. Du Bois, J. Am. Chem. Soc., 2008, 130, 9220 CrossRef CAS PubMed.
- V. Subbarayan, J. V. Ruppel, S. Zhu, J. A. Perman and X. P. Zhang, Chem. Commun., 2009, 4266 RSC.
- Y. Nishioka, T. Uchida and T. Katsuki, Angew. Chem., Int. Ed., 2013, 52, 1739 CrossRef CAS PubMed.
- A. McNally, B. Haffemayer, B. S. L. Collins and M. J. Gaunt, Nature, 2014, 510, 129 CrossRef CAS PubMed.
- K. Sun, R. Sachwani, K. J. Richert and T. G. Driver, Org. Lett., 2009, 11, 3598 CrossRef CAS PubMed.
- Q. Nguyen, K. Sun and T. G. Driver, J. Am. Chem. Soc., 2012, 134, 7262 CrossRef CAS PubMed.
- Y. Liu, J. Wei and C.-M. Che, Chem. Commun., 2010, 46, 6926 RSC.
- Y. M. Badiei, A. Dinescu, X. Dai, R. M. Palomino, F. W. Heinemann, T. R. Cundari and T. H. Warren, Angew. Chem., Int. Ed., 2008, 47, 9961 CrossRef CAS PubMed.
- M. J. B. Aguila, Y. M. Badiei and T. H. Warren, J. Am. Chem. Soc., 2013, 135, 9399 CrossRef CAS PubMed.
- A. Vlcek, Coord. Chem. Rev., 2010, 254, 1357 CrossRef CAS.
- O. R. Luca and R. H. Crabtree, Chem. Soc. Rev., 2013, 42, 1440 RSC.
- V. K. K. Praneeth, M. R. Ringenberg and T. R. Ward, Angew. Chem., Int. Ed., 2012, 51, 10228 CrossRef CAS PubMed.
- E. T. Hennessy and T. A. Betley, Science, 2013, 340, 591 CrossRef CAS PubMed.
- E. T. Hennessy, R. Y. Liu, D. A. Iovan, R. A. Duncan and T. A. Betley, Chem. Sci., 2014, 5, 1526 RSC.
- E. R. King, G. T. Sazama and T. A. Betley, J. Am. Chem. Soc., 2012, 134, 17858 CrossRef CAS PubMed.
- S. K. Sharma, P. S. May, M. B. Jones, S. Lense, K. I. Hardcastle and C. E. MacBeth, Chem. Commun., 2011, 47, 1827 RSC.
- J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 Search PubMed.
- K. W. Fiori, C. G. Espino, B. H. Brodsky and J. Du Bois, Tetrahedron, 2009, 65, 3042 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1057198–1057200. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01162k |
| ‡ Present Address: Department of Natural Sciences, Dalton State College, USA. |
| This journal is © The Royal Society of Chemistry 2015 |