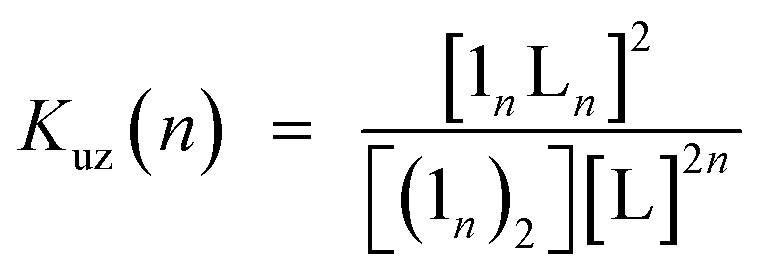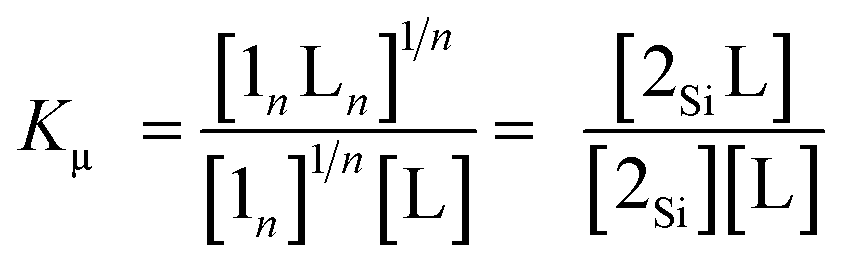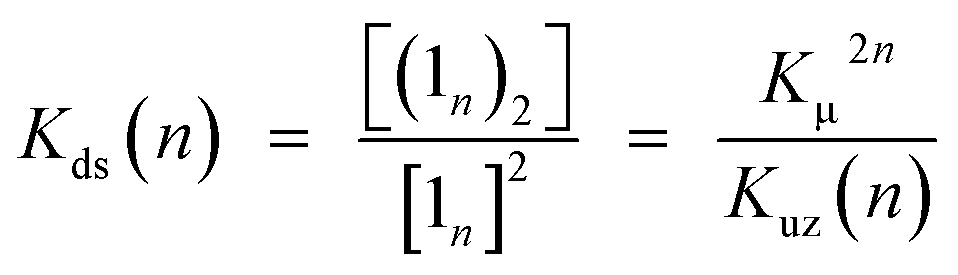DOI:
10.1039/C5SC01101A
(Edge Article)
Chem. Sci., 2015,
6, 6199-6206
Self-complementary double-stranded porphyrin arrays assembled from an alternating pyridyl–porphyrin sequence†
Received
27th March 2015
, Accepted 5th August 2015
First published on 5th August 2015
Abstract
Oligomeric porphyrin arrays with an alternating pyridyl–porphyrin sequence were synthesized to explore double-strand formation through self-complementary pyridyl-to-zinc axial coordination bonds. Competitive titration experiments revealed the thermodynamic aspects involved in the zipper effect within double-strand formation. Multiple axial coordination bonds defined the stacked conformation, despite a marginal contribution to the stability of the double-strands. Thus, the zipper cooperativity was the dominant factor for the remarkable stability. Moreover, the dimeric and trimeric porphyrin arrays were independently assembled into double-strands by self-sorting from a binary mixture. Double-strand formation engineered discretely stacked π-systems. Successive slipped-cofacial stacks of the porphyrin rings progressively extended the π-system via exciton coupling over the double-strand while keeping a relatively high fluorescence quantum yield.
Introduction
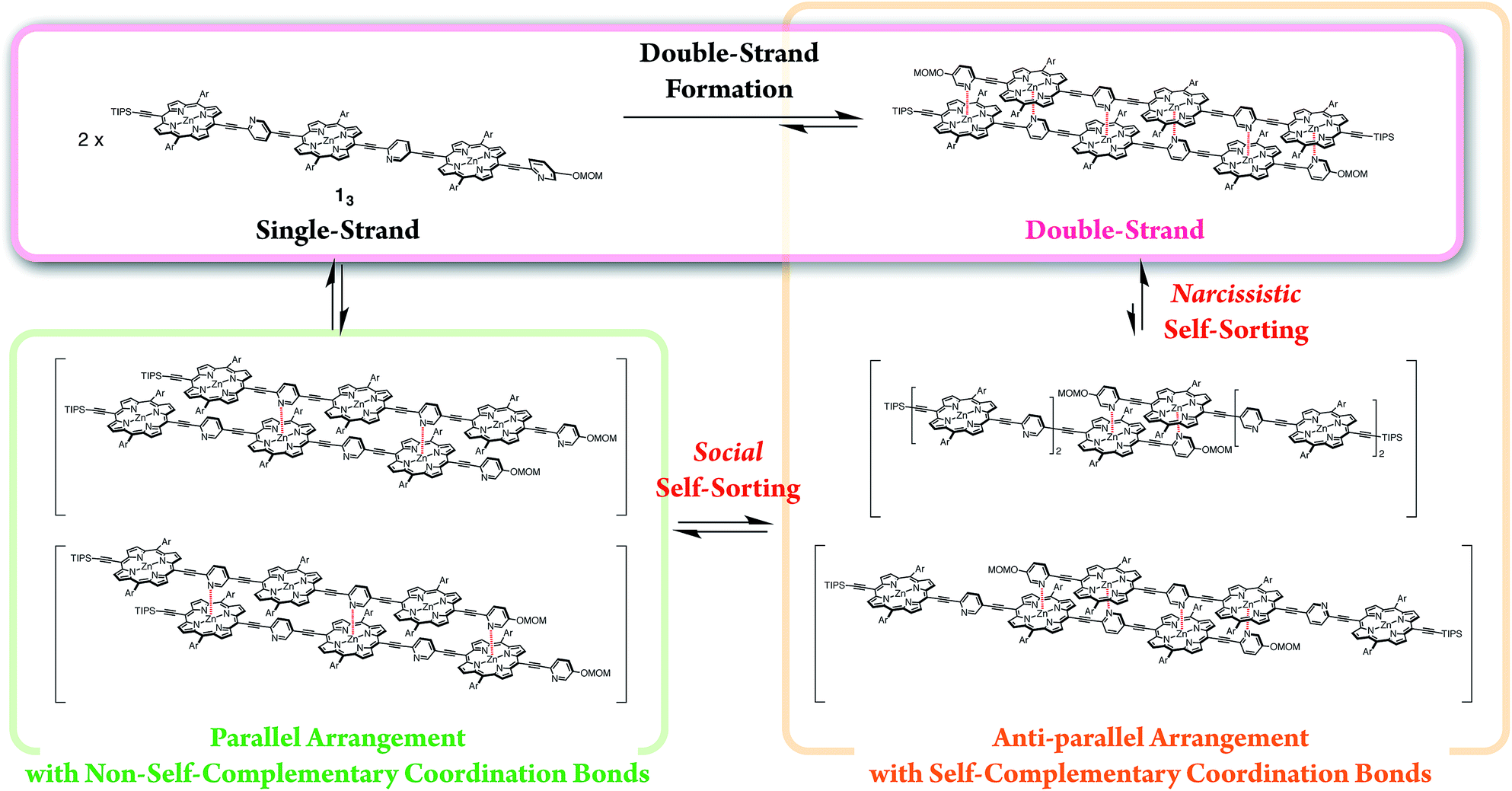
Naturally occurring double-stranded polymers, such as DNA and proteins, display an exquisite molecular organization in biological systems. Double-stranded DNA is currently emerging as a passive scaffold and provides state-of-the-art bottom-up nanotechnologies with a molecular scale precision, as exemplified by DNA-origami.1 This shape-programmable nanotechnology is enabled by both the sequence-specific double-strand formation and shape-persistent double-stranded building units. Therefore, the development of an intelligent double-strand as a building material for sophisticated hierarchical architectures in which functionalities could be integrated should be of significant interest. The present study examines novel double-strand-forming oligomeric porphyrin arrays which may, in a straightforward manner, be used to incorporate photoelectronic functions into structures for artificial photosynthesis.
The high fidelity of Watson–Crick base pairing via complementary hydrogen bonds plays a crucial role in the formation of double-stranded DNA.2 In a similar way, artificial double-strands require the molecular design of complementary pairs through either multiple hydrogen bonds3 or charge-transfer interactions.4 While the majority of research in the field has been devoted to the formation of helical structures,5 almost no synthetic effort has been aimed at the applications of these structures in nanomaterial science, though they represent a promising new paradigm in bottom-up molecular assembly. The molecular design of supramolecular building materials could include not only sequence selectivity but also the shape-persistence of the functional double-stranded structures.
For decades, porphyrin frameworks have been part of technological advancements at the forefront of material studies because of their large π-systems and outstanding molar extinction coefficients. Their structural relevance in natural photosynthetic systems has been the source of considerable interest and has driven research in supramolecular multiporphyrin architectures.6,7 The rigid porphyrin plane offers an ideal platform for versatile multiporphyrin architectures which can be built using both covalent and supramolecular approaches.8,9 Biomimetic supramolecular porphyrin architectures with slipped-cofacially stacked conformations have been a useful motif for the design of materials with excellent photoelectronic functionalities, as described by Kobuke and coworkers.10,11 A coordination-directed approach is particularly effective in the assembly of ladder complexes that are composed of fully π-conjugated multiporphyrin arrays via the use of bidentate ligands, as demonstrated by Anderson and collaborators.12 A double-strand is an intriguing motif to use to create novel artificial photosynthetic systems and engineer discrete stacked π-systems.
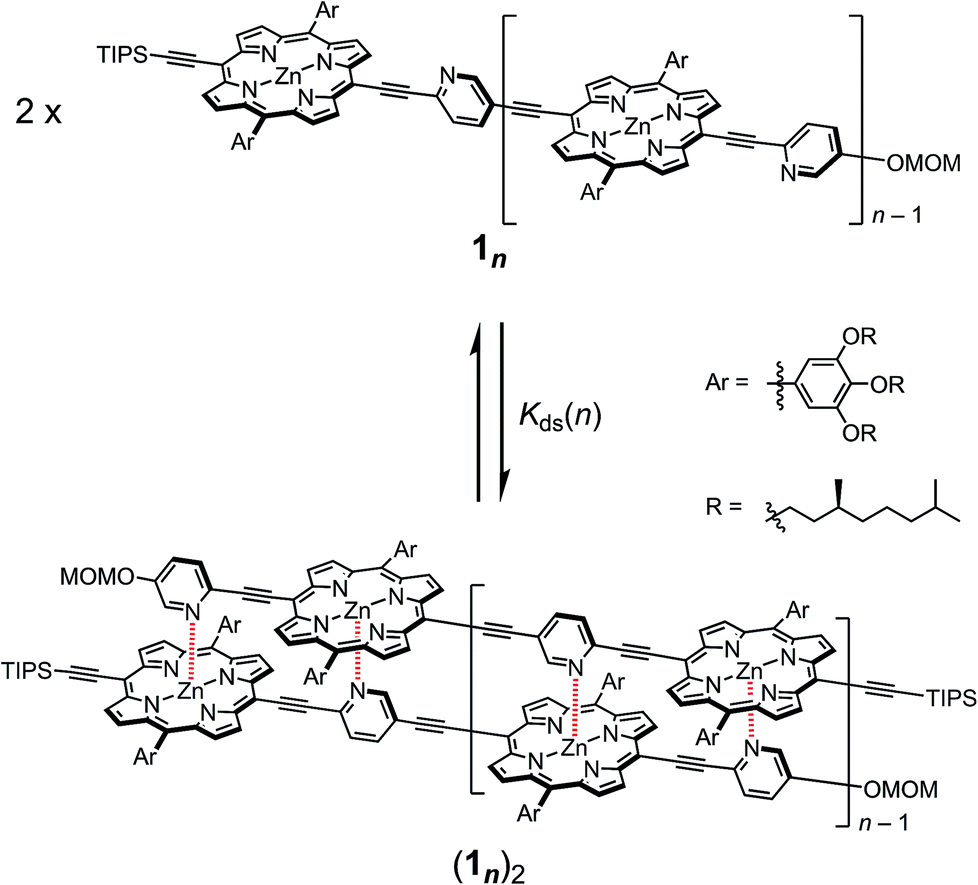
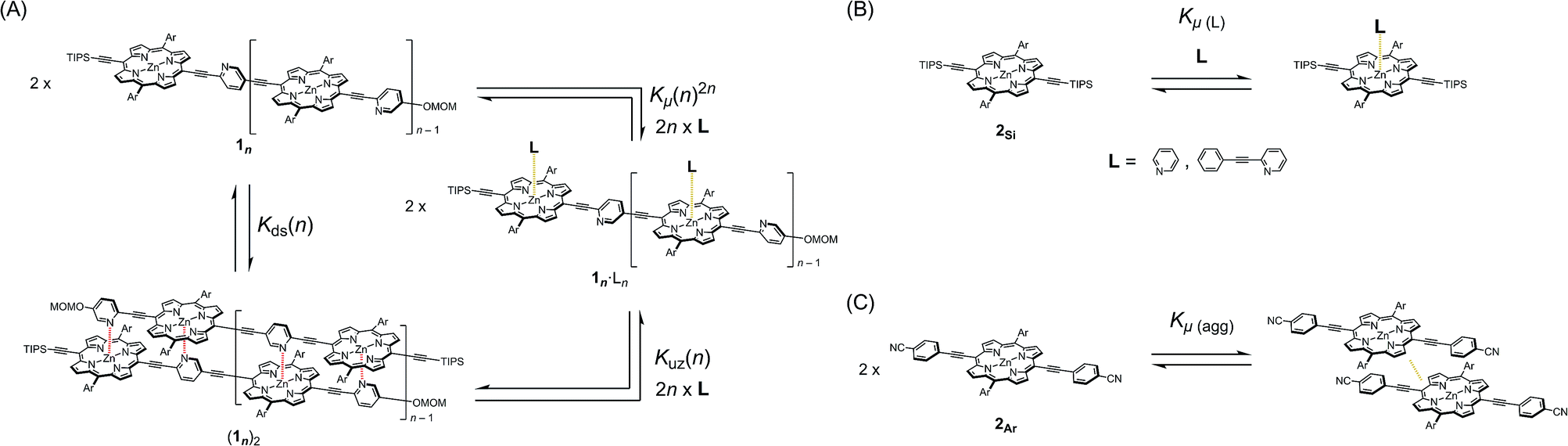
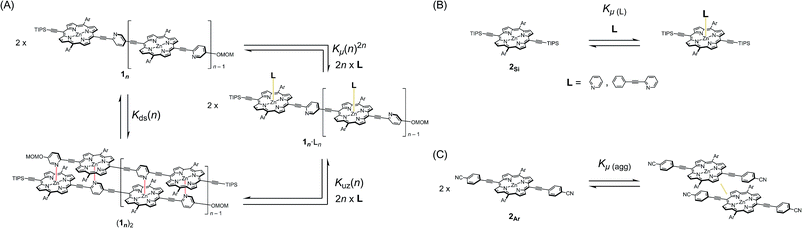
In our previous report, the formation of self-coordinated zinc(2-pyridylethynyl)porphyrin dimers was controlled by the choice of nucleation conditions, where the formation of the initial coordination bond governed how the second coordination bond would form.13 In a non-coordinating solvent, the formation of the second intra-dimer coordination bond was more energetically favorable than the initial binding was, which led to a self-complementary pattern of multiple coordination bonds. According to the same principle, we envisioned that oligomeric zinc(2-pyridylethynyl)porphyrin arrays 1n with an alternating pyridyl–porphyrin sequence could be assembled into a double-strand (1n)2 (Scheme 1). The formation of the initial interstrand coordination bond would induce the spontaneous formation of the second and the subsequent coordination bonds in a self-complementary fashion, because of the increasingly favorable thermodynamics of binding. This is the so-called zipper effect. Here, we present the molecular organization of novel double-stranded porphyrin arrays based on a self-complementary ligand–metalloporphyrin sequence, which provides successively stacked porphyrin arrays. The photophysical properties of these systems are studied.
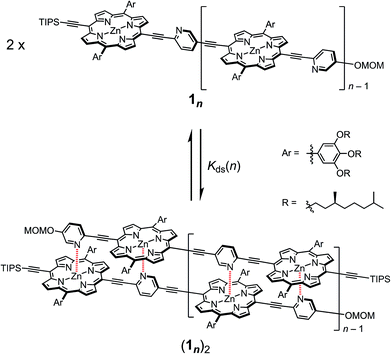 |
| | Scheme 1 Formation of the double-strands (1n)2 (n = 1–3). | |
Results and discussion
Synthesis
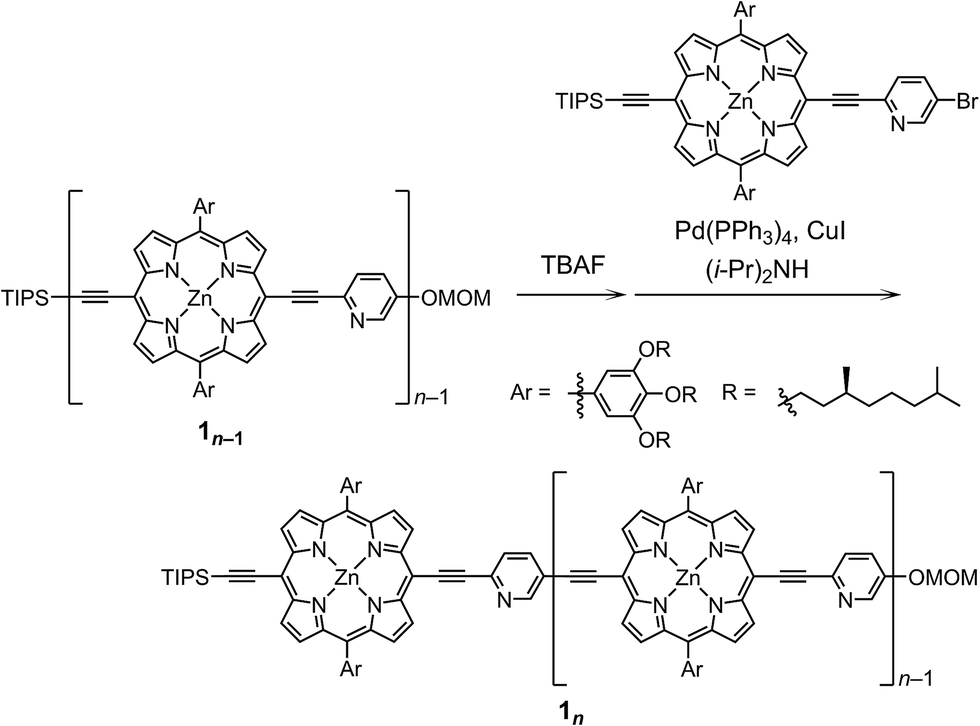
The synthetic route to the monomeric zinc(2-pyridylethynyl)porphyrin has already been established by our group.13 According to the reported procedure, we prepared the monomeric 11 as the precursor for the oligomeric 1n. A systematic series of new oligomers 1n (n = 2–3) were then prepared from 11via repetitive Sonogashira–Hagihara coupling reactions (Scheme 2).14 The details of the synthetic procedures are described in the ESI,† together with the essential thermodynamic and photophysical properties of (11)2 and (12)2. The following sections mainly demonstrate the results representative of (13)2, which is an assembly composed of six porphyrin rings and six pyridyl groups.
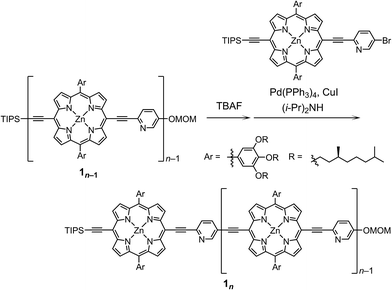 |
| | Scheme 2 Synthesis of the porphyrin arrays 1n (n = 2–3). | |
Structural elucidation of double-strands
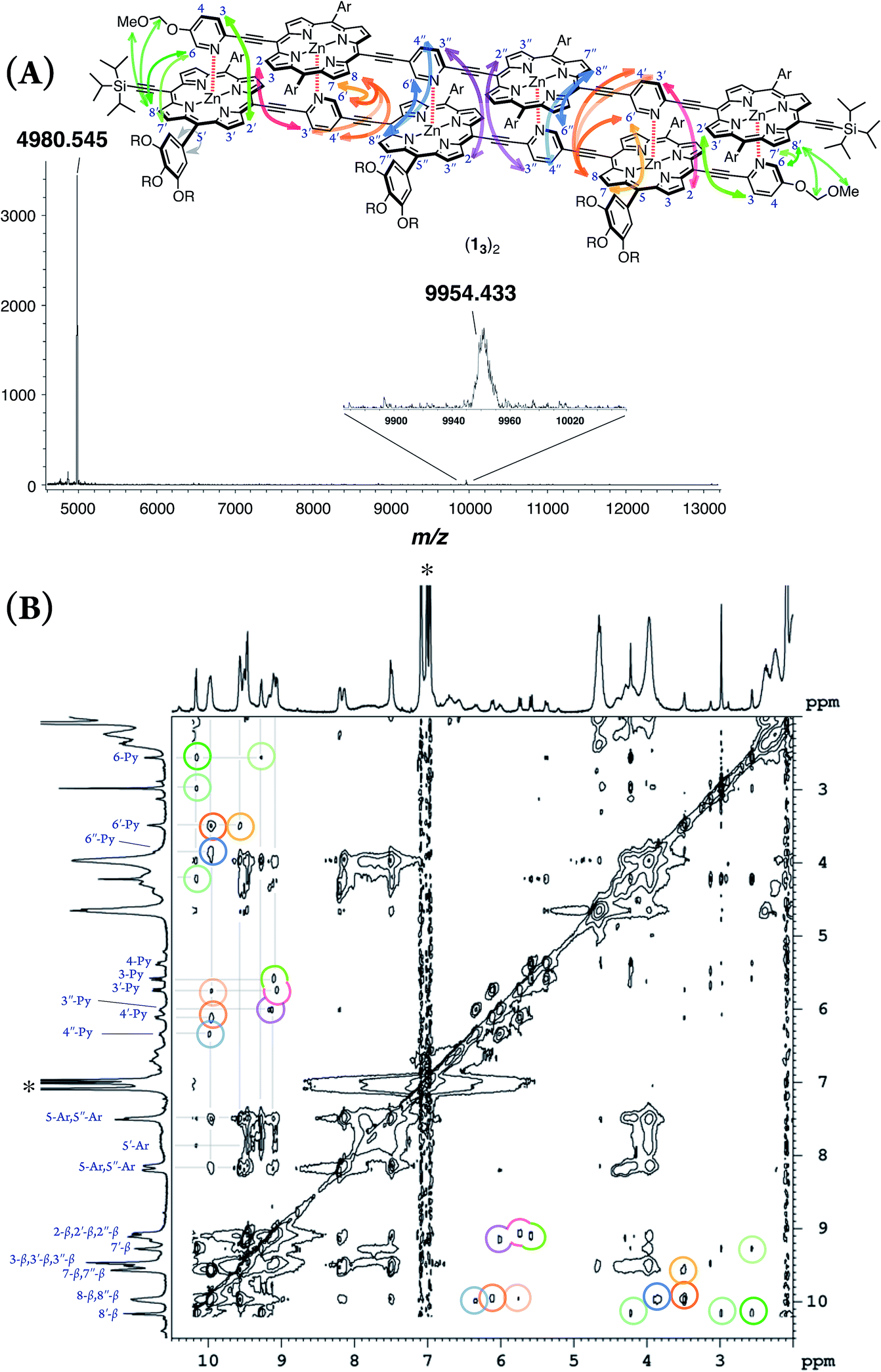
double-strand (13)2 was observed using MALDI-TOF MS measurements (Fig. 1A). (13)2 spontaneously formed in the lone stationary state in toluene, as identified by NMR. Only three sets of protons in the pyridyl and porphyrin-β positions showed unambiguous diagonal correlations via the nuclear Overhauser effect (NOE) (Fig. 1B), which is indicative of two trimeric porphyrin arrays that are cofacially assembled in an antiparallel arrangement (Fig. 1A). NOE correlation with the protons of the MOM group determined one of three porphyrin rings. Subsequently, TOCSY correlations identified two sets of signals corresponding to the 2-, 3- and 6-pyridyl and 2′′-, 3′′- and 6′′-pyridyl protons. An alternative assignment for these two sets was also possible for these pyridyl–porphyrin pairs. However, these pyridyl protons individually showed NOE correlations with the porphyrin rings, which indicated the pairing of the porphyrin rings with the complementary pyridyl groups. This finding is consistent with a symmetrically assembled structure, as indicated by a lack of multiplied signals for the porphyrin array. All of the aromatic resonances of the pyridyl groups were found in the non-aromatic region (6.34–2.56 ppm), which suggested that the axially coordinated pyridyl groups were strongly shielded in the vicinity of the porphyrin ring. The assignment is consistent with all of the observed NMR resonances.
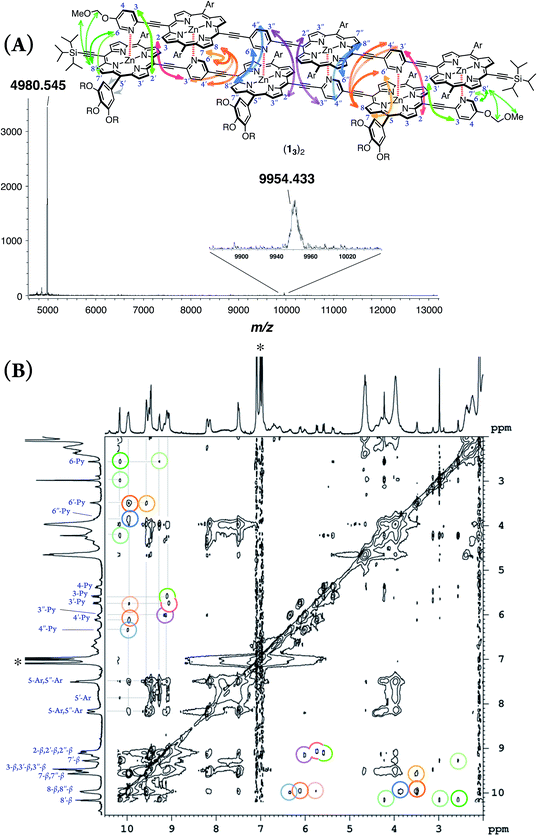 |
| | Fig. 1 (A) MALDI-TOF MS spectrum of (13)2. (B) 1H–1H NOE spectrum of (13)2 in toluene-d8. The asterisk indicates residual toluene. One alternative assignment is shown. | |
In pyridine-d5, which can act as a competitive coordinating ligand, the non-shielded resonances of the pyridyl protons of the disassembled species were observed in the aromatic region with the disappearance of the upfield-signals observed for the double-strand. The comparison of the spectra in the two solvents suggests the assembly of double-strand (13)2 through pyridyl-to-zinc coordination bonds.14 All of the NMR data firmly established that the structural picture of the discrete double-strand (13)2 assembled from an alternating pyridyl–porphyrin sequence via self-complementary coordination bonds.
Thermodynamic behaviors
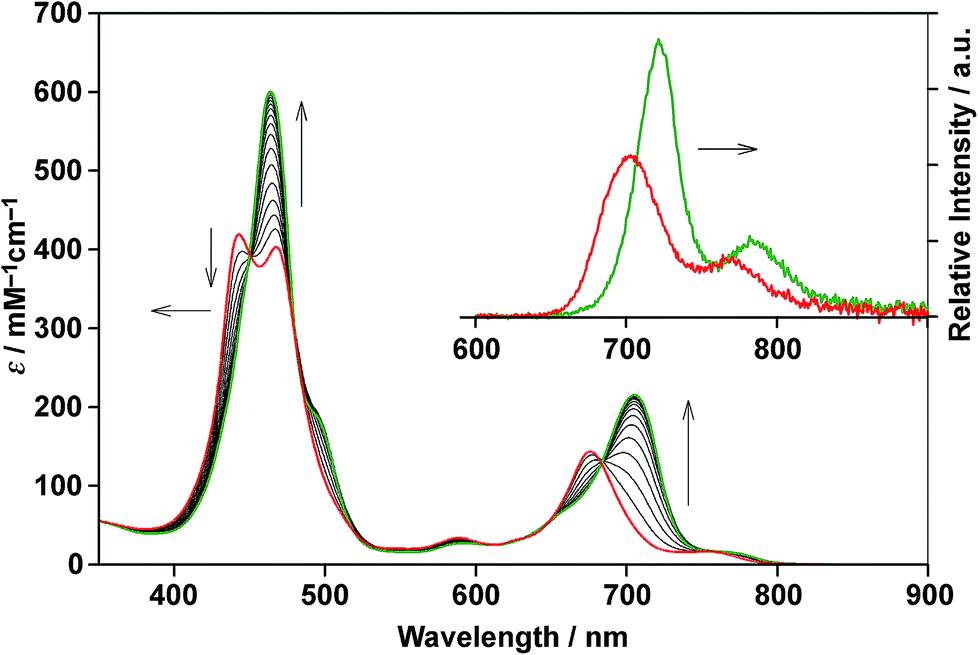
Double-strand (13)2 was sufficiently durable to obey Beer's law over a wide concentration range (10−7 to 10−4 M) in toluene. In contrast, the spectral shape of the electronic absorption spectra of 11 depended on the concentration, which suggested a small association constant for the formation of (11)2 (Kds(1) = 1.3 ± 0.2 × 104 M−1).14 The association constant of (13)2, Kds(3), was found to be too high to directly evaluate the thermodynamic stability of double-strand (13)2. Over the course of competitive titration experiments with pyridine, the spectral changes showed several pseudo-isosbestic points, which suggested that the equilibria involved essentially two stationary states, i.e., the double-strand and the unzipped single-strand (Fig. 2). Competitive titration experiments allowed us to analyze the thermodynamic stability of the double-strands according to the thermodynamic cycle (Scheme 3). We analyzed the unzipping equilibria by employing a tentative one-step unzipping model, which is useful for the description of the simplified overall equilibria. Nonlinear least-square fittings gave reliable binding properties for the overall unzipping equilibria.14 The overall unzipping constant (Kuz) is then described as follows:| | 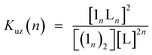 | (1) |
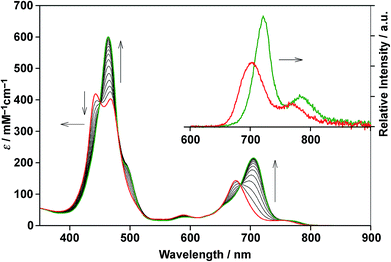 |
| | Fig. 2 Spectrometric titration of (13)2 ([13]0 = 2.9 × 10−6 M) with pyridine (up to 480 equiv., red to green) at 25 °C in toluene. The inset shows the fluorescence spectra of (13)2 and 13 in the presence of excess pyridine (104 equiv.) in toluene (λex = 450 nm, a pseudo-isosbestic point). | |
 |
| | Scheme 3 (A) Generic closed thermodynamic cycle (n = 2–3). (B) Microscopic binding equilibrium of model porphyrin 2Si with axial ligands; L = pyridine and 2-(phenylethynyl)pyridine. (C) Aggregation equilibrium of model porphyrin 2Ar. | |
Assuming that the microscopic binding constant (Kμ) is identical for each ligand-to-zinc axial coordination bond of the single-strand, the microscopic binding constant can be approximated by eqn (2).
| | 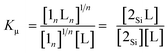 | (2) |
In practice, the titration of model zinc porphyrin 2Si with pyridine to yield the axially coordinated zinc porphyrin gave the experimental Kμ values (Kμ = (3.2 ± 0.1) × 104 M−1) (Scheme 3B). The values, in turn, gave the binding constant for the double-strand formation (Kds(n)) according to eqn (3).
| | 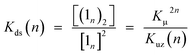 | (3) |
These estimated thermodynamic parameters are useful to the discussion on the durability of double-strand (1n)2 (Table 1). The values of Kds(n), Kds(2) = (2.5 ± 0.3) × 109 M−1 and Kds(3) = (6.5 ± 1.2) × 1011 M−1, were remarkable, considering the small magnitude of the microscopic binding constant as described below.
Table 1 Thermodynamic parameters of 1n at 25 °C in toluene
|
|
n
|
K
uz(n)a/M1−2n |
K
ds(n)/M−1 (ΔG°ds(n)/kJ mol−1) |
ΔΔG°(n)d/kJ mol−1 (EMe/M) |
|
Estimated from the competitive titration experiments with pyridine.
Directly estimated from variations in concentration.
Estimated using eqn (3), wherein Kμ = (3.2 ± 0.1) × 104 M−1.
Estimated by employing ΔG°μ = −5.5 ± 0.3 kJ mol−1, wherein Kμ = Kμ(L)1/2Kμ(agg)1/2 = 9.4 ± 1 M−1.
Effective molarity (EM) was evaluated from eqn (6).
|
|
11
|
1 |
— |
(1.3 ± 0.2) × 104 (−23 ± 1)b |
−12 ± 0.4 (147 ± 9) |
|
12
|
2 |
(4.1 ± 0.2) × 108 |
(2.5 ± 0.3) × 109 (−53 ± 1)c |
−31 ± 0.2 (68 ± 8) |
|
13
|
3 |
(6.5 ± 1.2) × 1015 |
(1.7 ± 0.3) × 1011 (−64 ± 1)c |
−31 ± 0.2 (12 ± 1) |
The zipper effect was quantified by the synergetic free energy change (ΔΔG(n)), the excess energy beyond the sum of the independent free energy changes induced by pyridyl-to-zinc axial coordinating and π-stacked microscopic binding.15
| | | ΔΔG(n) = ΔGds(n) − 2nΔGμ | (4) |
The ΔΔG(n) values substantially dominated the stability of the double-strands, with multiple axial coordination bonds defining the discrete conformation of the double-strand despite their minimal thermodynamic contribution. To quantify a reliable microscopic binding constant, we employed model compounds. The binding constant for the axial coordination of 2-(phenylethynyl)pyridine to 2Si as the model for each ligand-to-zinc axial coordination bond is Kμ(L) = 7.8 ± 1.1 M−1 (Scheme 3B) and that for the self-aggregation of 2Ar as the model for π-stacked interactions is Kμ(agg) = 11 ± 1 M−1(Scheme 3C). The microscopic binding constant for single coordination together with π-stacked interactions was, then, determined to be 9.4 ± 1 M−1 (Kμ = Kμ(L)1/2Kμ(agg)1/2, ΔG°μ = −5.5 ± 0.3 kJ mol−1). The dramatic change from ΔΔG°(1) to ΔΔG°(2) suggested that the zipper effect, as a consequence of chelate cooperativity including interactions such as π-stacking, van der Waals interactions, and desolvation entropy, became greater with the addition of the repeating units. In contrast, the ΔΔG°(3) value was similar to that of ΔΔG°(2), which indicated that the compensatory effects gave rise to structural strain, such as the distortion of the porphyrin planes, and a loss of structural entropy. The analyses elucidated that the increasing number of repeat units increased the durability of (1n)2, although the cooperativity per interaction decreased. The nucleation step of multiple coordination bonds is dominant when 1n self-assembles, and synergetic effects govern the durability of (1n)2 for n ≤ 3. The self-complementary coordination bonds were significantly stabilized by the zipper effect, thereby thermodynamically funnelling the self-assembled structures into the most stable form without kinetic entrapment by metastable states. This allowed for the realization of the specific double-strand formation.
The situations are alternatively examined in terms of “effective molarity (EM)”, an empirical parameter used to describe the effectiveness of intramolecular interactions, to index the upper limit of concentration for the double-strand formation. The EM value is defined in eqn (5) and (6).16,17
| | | EM = (Kds(n)/Kμ2n)1/(2n−1) | (5) |
| | | = exp{−ΔΔG(n)/(2n − 1)RT} | (6) |
The EM values of (11)2 and (12)2 (Table 1) were in the high end range of the typical values for supramolecular systems,17 inferring that the model systems were, in the most precise sense, imperfect for the formation of discrete double-strands. However, the EM values predicted a significant selectivity of the double-strand formation even at very concentrated conditions.
Self-sorting behaviors
The exclusive formation of the self-complementary double-strand (1n)2 intrinsically involves self-sorting behaviors due to the possibility of several binding patterns for the self-association of 1n (Scheme 4). Briefly, social self-sorting favors self-complementary binding patterns, whereas narcissistic self-sorting distinguishes differences in the numbers of the repeating units of the self-complementary binding patterns.12b,18 Based on the lack of hysteresis, the self-sorting assembly of the double-strands occurred during the heating/cooling processes (25–70 °C) of a binary mixture of (12)2 and (13)2.14 The electronic absorption spectra appeared as a superimposition of the spectra of (12)2 and (13)2 at 25 °C before and after heating, although thermally dissociated 12 and 13 mutually interacted to a degree at 70 °C. The mutually orthogonal self-assembly from a thermally unzipped mixture perfectly restored the initial fluorescence properties, as indicated by the superimposition of (12)2 and (13)2 at 25 °C. The self-sorting capacity of double-strands (1n)2 makes them promising in the construction of units that are capable of simultaneous multiple molecular assembly within bottom-up nanotechnology applications.
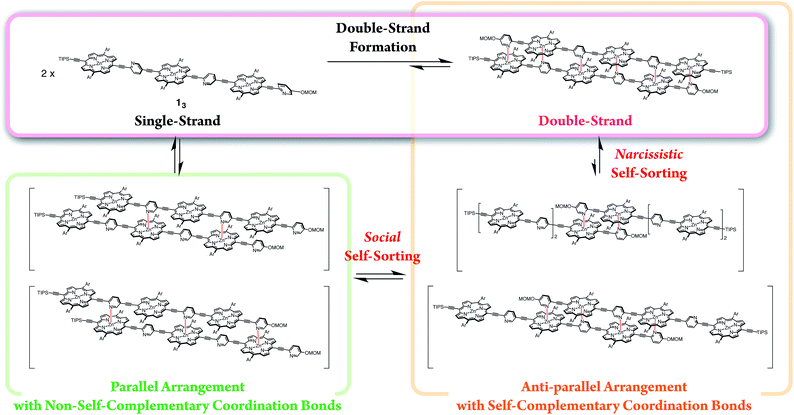 |
| | Scheme 4 Double-strand formation of 13 self-sorted from possible self-assembled patterns. | |
Engineering discrete stacked π-systems
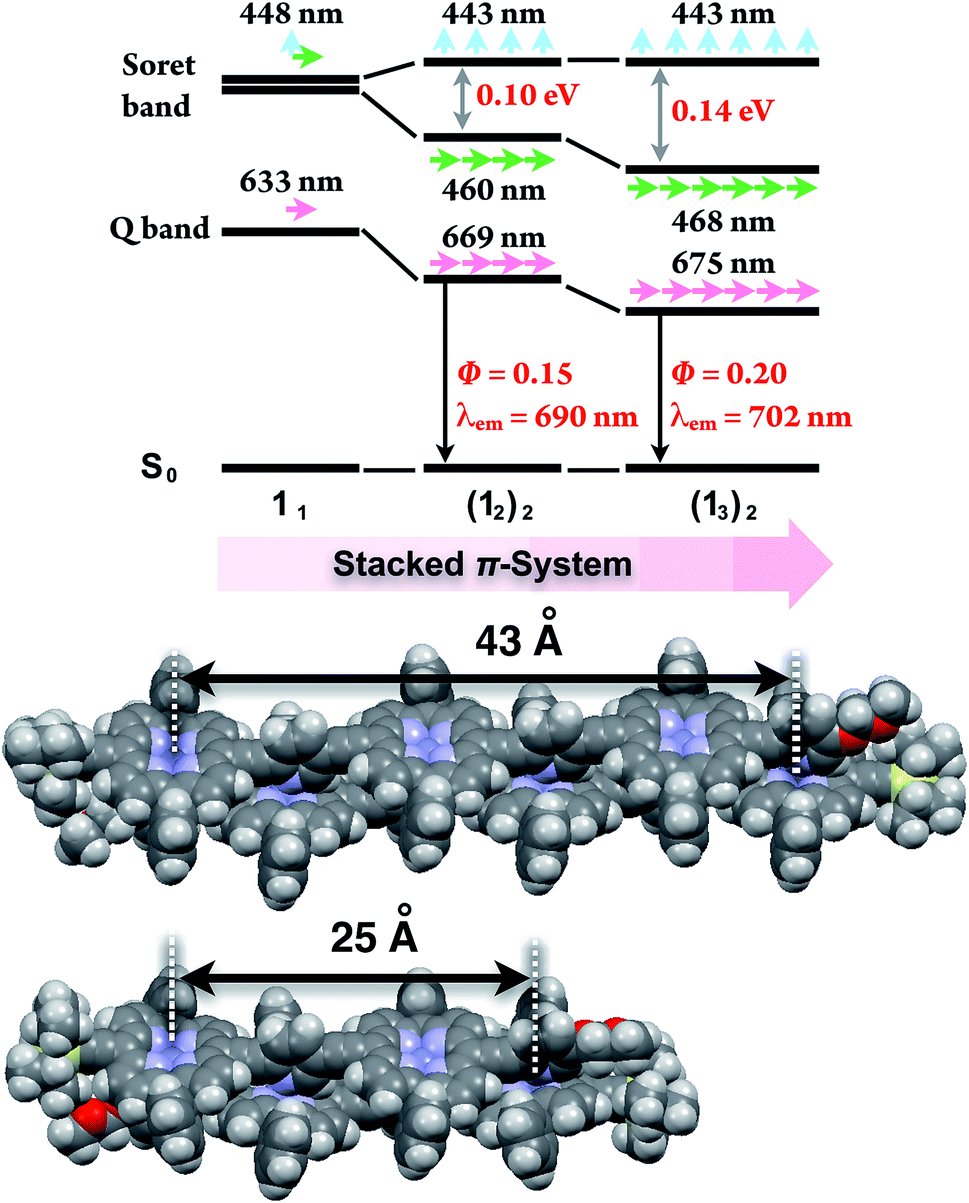
In the last stage, our attention turned to the electronic properties of the double-strands (12)2 and (13)2. The double-strand formation of 1n resulted in a successively slipped-cofacial stack of porphyrin arrays. The double-strands (1n)2 showed a splitting of the Soret band with a bathochromic shift of the lower band due to exciton coupling in the electronic absorption spectra.10,19 The split width of the Soret band of (13)2 (0.14 eV) was wider than that of (12)2 (0.10 eV), where the lowest exciton band was red-shifted and the highest exciton band remained unchanged. At the same time, the Q band of (13)2 displayed a larger bathochromic shift to 675 nm (1.83 eV) than that of (12)2 at 669 nm (1.85 eV), and the longest Zn⋯Zn distances were estimated to be 43 Å for (13)2 and 25 Å for (12)2 based on the geometry-optimized structures (Fig. 3). The comparison of the electronic structures of (13)2 and (12)2 unveiled that the double-stranded multiporphyrin arrays exhibited exciton coupling due to successive slipped-cofacial stacks, leading to long-range π-electronic interactions.
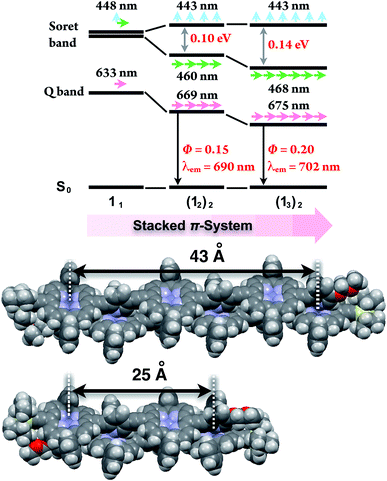 |
| | Fig. 3 Energy diagrams of the double-strands (1n)2, and their geometry-optimized structures calculated using the MM + force field (HyperChem Ver. 8.0 software). The alkoxy side chains are omitted for visual clarity. The wavelengths denote the absorption maxima. | |
The emission properties are noteworthy because the double-strands (1n)2 did not dissipate a photoexcited singlet. The absolute fluorescence quantum yields (Φ) of (12)2 and of (13)2 were 0.15 and 0.20, respectively (Table 2). Increasing the number of porphyrin units raised the Φ values by extending the π-systems. The relatively high fluorescence efficiency of (1n)2 suggested that the double-stranded structure was effective in circumventing nonradiative deactivation pathways, even in the near-infrared wavelength region. The successive slipped-cofacial stacks of the porphyrin planes in the double-strand serve as structural and functional mimics of bacterial light-harvesting antenna complexes which display an efficient capture of sunlight and photoexcited energy transfer.7
Table 2 Photophysical properties of double-strands (1n)2, 1n accommodated with pyridine (1n·Pyn) and 2 in toluene
|
|
λ
em
/nm (Φ) |
τ/ns (α) |
k
em
/s−1 |
|
Emission maximum (λem) and fluorescence quantum yield (Φ) obtained using an integration sphere (excitation at 452 nm for (12)2, 450 nm for (13)2, and 405 nm for 2Si and 2Si·Py).
Fluorescence lifetime (τ) and the normalized amplitude (α) determined from the fluorescence decay profiles in the range of 623–773 nm upon excitation at 483 nm (Fig. 4).
Emission at 635 nm upon excitation at 405 nm.
Emission at 650 nm upon excitation at 405 nm.
Radiative rate constant defined as kem = Φ/τ. The single-strand 2Si·Py was observed in the presence of excess pyridine (104 equiv.).
|
| (12)2 |
690 (0.15) |
0.65 (0.51), 1.15 (0.49)b |
1.7 × 108 |
| (13)2 |
702 (0.20) |
0.56 (0.32), 1.03 (0.68)b |
2.3 × 108 |
|
2Si
|
635 (0.07) |
1.61c |
4.3 × 107 |
|
2Si·Py |
650 (0.08) |
1.45d |
5.6 × 107 |
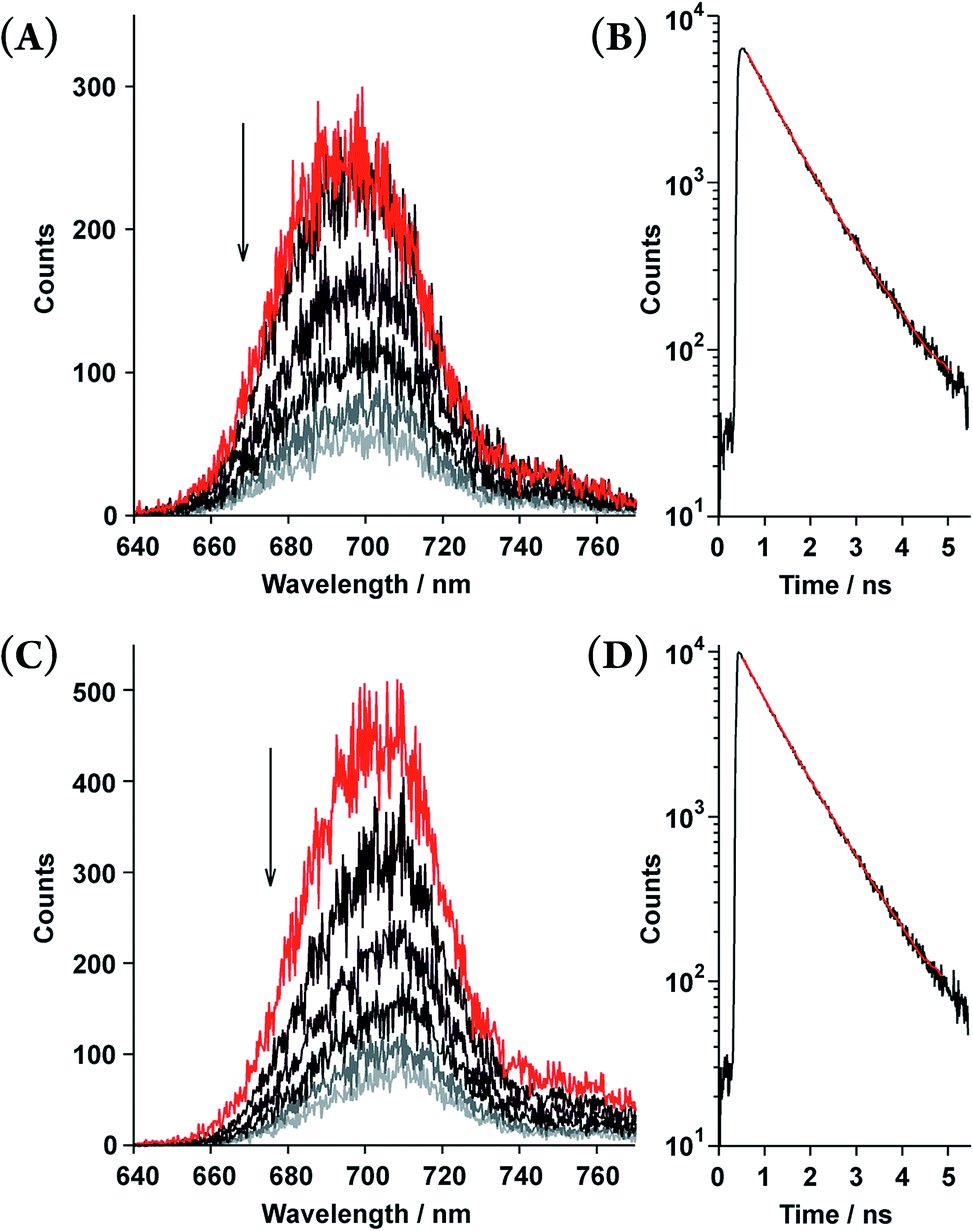
Time-resolved fluorescence spectroscopy gave further insight into the photophysical dynamics of the excited states (Fig. 4). The biexponential fluorescence decay profiles of both double-strands indicated the existence of dual fluorescent states (Table 2). Over time, double-strand (13)2 showed a small red-shift in its emission wavelength. In contrast, the spectral shift of (12)2 was smaller than that of (13)2. It is intriguing to consider that the photophysical properties are relevant to the thermodynamic aspect of the double-strands; (13)2 was much more strained than (12)2. Structural relaxation could provide an energy sink that would be capable of trapping photoexcited singlets. For instance, it is known that porphyrin planes are ruffled in the photoexcited singlet state.20,21 This interpretation is a likely reason as to why the slow fluorescence decay manifests as a red shift.
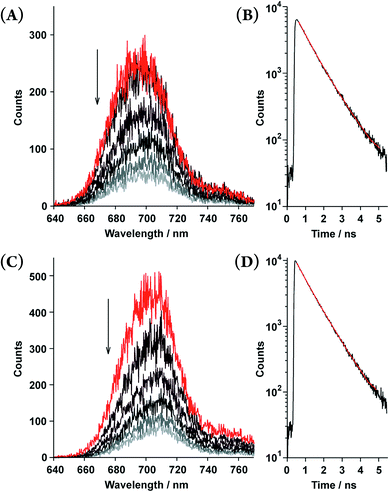 |
| | Fig. 4 Time-resolved fluorescence spectra recorded every 0.3 ns (upper to lower; 0.45–0.55, 0.75–0.85, 1.05–1.15, 1.35–1.45, 1.65–1.75, and 1.95–2.05 ns) and fluorescence decay profiles of the double-strands (12)2 at [12] = 5.5 × 10−6 M (A and B) and (13)2 at [13] = 6.9 × 10−6 M (C and D) in toluene. The fluorescence decay profiles (black lines) are shown with fitted curves based on the biexponential decay constants (red lines, τ shown in Table 2) in the range of 623–773 nm. | |
Conclusions
In summary, we have synthesized double-stranded porphyrin arrays that yielded successive slipped-cofacial stacks with strong exciton coupling. Titration experiments revealed the thermodynamic aspects underlying the specific double-strand formation, and are used to describe a process in which self-complementary coordination bonds define the discrete structure. The zipper effect dominated the stability of the double-stranded structure. The remarkable selectivity for double-strand formation may serve as a powerful building tool for sophisticated bottom-up molecular assemblies with molecular scale precision, similar to those found in DNA nanotechnology. Moreover, the double-strands extended the π-electron network without any dissipation of the fluorescence properties due to the assembly of successive slipped-cofacial stacks. The shape-persistent double-stranded porphyrin arrays provide new options for artificial photosynthetic systems based on shape-programmable, and bottom-up molecular architectures.
Acknowledgements
This work was partly supported by a Grant-in-Aid for Scientific Research (No. 25102525) on the Innovative Area “New Polymeric Materials Based on Element-Blocks (No. 2401)” from the Ministry of Education, Culture, Sports, Science and Technology, Government of Japan. The authors thank Prof. Shinjiro Machida (Kyoto Institute of Technology) for time-resolved fluorescence spectroscopy. We thank the referees for their helpful suggestions concerning the thermodynamic analyses.
Notes and references
- DNA nanotechnology:
(a) P. W. K. Rothemund, Nature, 2006, 440, 297–302 CrossRef CAS PubMed;
(b) F. A. Aldaye, A. L. Palmer and H. F. Sleiman, Science, 2008, 321, 1795–1799 CrossRef CAS PubMed;
(c) M. S. Strano, Science, 2012, 338, 890–891 CrossRef CAS PubMed;
(d) B. Sacca and C. M. Niemeyer, Angew. Chem., Int. Ed., 2012, 51, 58–66 CrossRef CAS PubMed;
(e) A. Rajendran, M. Endo and H. Sugiyama, Angew. Chem., Int. Ed., 2012, 51, 874–890 CrossRef CAS PubMed;
(f) F. Zhang, J. Nangreave, Y. Liu and H. Yan, J. Am. Chem. Soc., 2014, 136, 11198–11211 CrossRef CAS PubMed;
(g) M. R. Jones, N. C. Seeman and C. A. Mirkin, Science, 2015, 347, 1260901 CrossRef PubMed.
-
(a)
W. Saenger, Principle of Nucleic Acid Structure, Springer-Verlag, New York, 1984 Search PubMed;
(b) J. SantaLucia Jr, H. T. Allawi and P. A. Seneviratne, Biochemistry, 1996, 35, 3555–3562 CrossRef CAS PubMed.
- Hydrogen-bonding:
(a) A. P. Bisson, F. J. Carver, C. A. Hunter and J. P. Waltho, J. Am. Chem. Soc., 1994, 116, 10292–10293 CrossRef CAS;
(b) A. P. Bisson, F. J. Carver, D. S. Eggleston, R. C. Haltiwanger, C. A. Hunter, D. L. Livingstone, J. F. McCabe, C. Rotger and A. E. Rowan, J. Am. Chem. Soc., 2000, 122, 8856–8868 CrossRef CAS;
(c) V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720–723 CrossRef CAS PubMed;
(d) T. Maeda, Y. Furusho, S.-I. Sakurai, J. Kumaki and E. Yashima, J. Am. Chem. Soc., 2008, 130, 7938–7945 CrossRef CAS PubMed;
(e) H. Goto, Y. Furusho, K. Miwa and E. Yashima, J. Am. Chem. Soc., 2009, 131, 4710–4719 CrossRef CAS PubMed;
(f) H. Yamada, Z.-Q. Wu, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2012, 134, 9506–9520 CrossRef CAS PubMed.
- Charge-transfer interactions:
(a) G. J. Gabriel and B. L. Iverson, J. Am. Chem. Soc., 2002, 124, 15174–15175 CrossRef CAS PubMed;
(b) Q.-Z. Zhou, X.-K. Jiang, X.-B. Shao, G.-J. Chen, M.-X. Jia and Z.-T. Li, Org. Lett., 2003, 5, 1955–1958 CrossRef CAS PubMed;
(c) Q.-Z. Zhou, Q. M.-X. Jia, X.-B. Shao, L.-Z. Wu, X.-K. Jiang, Z.-T. Li and G.-J. Chen, Tetrahedron, 2005, 61, 7117–7124 CrossRef CAS PubMed.
- Reviews:
(a) D. Haldar and C. Schmuck, Chem. Soc. Rev., 2009, 38, 363–371 RSC;
(b) Y. Furusho and E. Yashima, Macromol. Rapid Commun., 2011, 32, 136–146 CrossRef CAS PubMed , and the references cited therein.
-
(a) G. McDermott, S. M. Prince, A. A. Freer, A. M. Hawthornthwaite-Lawless, M. Z. Papiz, R. J. Cogdell and N. W. Isaacs, Nature, 1995, 374, 517–521 CrossRef CAS PubMed;
(b) A. W. Roszak, T. D. Howard, J. Southall, A. T. Gardiner, C. J. Law, N. W. Isaacs and R. J. Cogdell, Science, 2003, 302, 1969–1972 CrossRef CAS PubMed;
(c) S. Niwa, L.-J. Yu, K. Takeda, Y. Hirao, T. Kawakami, Z.-Y. Wang-Otomo and K. Miki, Nature, 2014, 508, 228–232 CrossRef CAS PubMed.
-
(a) R. J. Cogdell, A. Gall and J. Köhler, Q. Rev. Biophys., 2006, 39, 227–324 CrossRef CAS PubMed;
(b) V. Sundström, Annu. Rev. Phys. Chem., 2008, 59, 53–77 CrossRef PubMed.
- Covalent multiporphyrin architectures:
(a) A. K. Burrell, D. L. Officer, P. G. Plieger and D. C. W. Reid, Chem. Rev., 2001, 101, 2751–2796 CrossRef CAS PubMed;
(b) D. Kim and A. Osuka, Acc. Chem. Res., 2004, 37, 735–745 CrossRef CAS PubMed;
(c)
N. Aratani and A. Osuka, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2010, vol. 1, ch. 1 Search PubMed.
- Supramolecular multiporphyrin architectures:
(a) J. Wojaczynski and L. Latos-Grazynski, Coord. Chem. Rev., 2000, 204, 113–171 CrossRef CAS;
(b) I. Beletskaya, V. S. Tyurin, V. A. Y. Tsivadze, R. Guilard and C. Stern, Chem. Rev., 2009, 109, 1659–1713 CrossRef CAS PubMed;
(c) J. S. A. W. Elemans, R. van Hameren, R. J. M. Nolte and A. E. Rowan, Adv. Mater., 2006, 18, 1251–1266 CrossRef CAS PubMed;
(d) C. Zou and C.-D. Wu, Dalton Trans., 2012, 3879–3888 RSC;
(e) S. Durot, J. Taesch and V. Heitz, Chem. Rev., 2014, 114, 8542–8578 CrossRef CAS PubMed.
-
M. Morisue and Y. Kobuke, in Handbook of Porphyrin Science, ed. K. M. Kadish, K. M. Smith and R. Guilard, World Scientific, Singapore, 2014, vol. 32, ch. 166 Search PubMed.
-
(a) Y. Kobuke and H. Miyaji, J. Am. Chem. Soc., 1994, 116, 4111–4112 CrossRef CAS;
(b) K. Kameyama, M. Morisue, A. Satake and Y. Kobuke, Angew. Chem., Int. Ed., 2005, 44, 4763–4766 CrossRef CAS PubMed;
(c) M. Morisue and Y. Kobuke, Chem.–Eur. J., 2008, 14, 4993–5000 CrossRef CAS PubMed.
-
(a) H. L. Anderson, Inorg. Chem., 1994, 33, 972–981 CrossRef CAS;
(b) P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538–11545 CrossRef CAS;
(c) J. K. Sprafke, B. Odell, T. D. W. Claridge and H. L. Anderson, Angew. Chem., Int. Ed., 2011, 50, 5572–5575 CrossRef CAS PubMed.
- M. Morisue, T. Morita and Y. Kuroda, Org. Biomol. Chem., 2010, 8, 3457–3463 CAS.
- See the ESI.†.
-
(a) S. L. Cockroft and C. A. Hunter, Chem. Soc. Rev., 2007, 36, 172–188 RSC;
(b) A. Camara-Campos, D. Musumeci, C. Hunter and S. Turega, J. Am. Chem. Soc., 2009, 131, 18518–18524 CrossRef CAS PubMed;
(c) H. J. Hogben, J. K. Sprafke, M. Hoffmann, M. Pawlicki and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 20962–20969 CrossRef CAS PubMed.
-
(a) C. Galli and L. Mandolini, Eur. J. Org. Chem., 2000, 3117–3125 CrossRef CAS;
(b) A. Mulder, J. Huskens and D. N. Reinhoudt, Org. Biomol. Chem., 2004, 2, 3409–3424 RSC;
(c) C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed.
- M. C. Misuraca, T. Grecu, Z. Freixa, V. Garanini, C. A. Hunter, P. W. N. M. van Leeuwen, M. D. Segarra-Maset and S. M. Turega, J. Org. Chem., 2011, 76, 2723–2732 CrossRef CAS PubMed.
-
(a) A. Wu and L. Isaacs, J. Am. Chem. Soc., 2003, 125, 4831–4835 CrossRef CAS PubMed;
(b) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed.
-
(a) M. Kasha, Radiat. Res., 1963, 20, 55–71 CrossRef CAS;
(b) M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 CrossRef CAS.
- S. Gentemann, N. Y. Nelson, L. Jaquinod, D. J. Nurco, S. H. Leung, C. J. Medforth, K. M. Smith, J. Fajer and D. Holten, J. Phys. Chem. B, 1997, 101, 1247–1254 CrossRef CAS.
-
(a) R. A. Freitag, J. A. Mercer-Smith and D. G. Whitten, J. Am. Chem. Soc., 1981, 103, 1226–1228 CrossRef CAS;
(b) R. A. Freitag and D. G. Whitten, J. Phys. Chem., 1983, 87, 3918–3925 CrossRef CAS;
(c) K. Konishi, K. Miyazaki, T. Aida and S. Inoue, J. Am. Chem. Soc., 1990, 112, 5639–5640 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthesis and full characterization of new compounds, electronic absorption spectra from titration experiments, and full characterization of (11)2 and (12)2. See DOI: 10.1039/c5sc01101a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a