
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Generation of 1,2-azaboretidines via reduction of ADC borane adducts†
H.
Braunschweig
*,
A.
Gackstatter
,
T.
Kupfer
,
T.
Scheller
,
F.
Hupp
,
A.
Damme
,
N.
Arnold
and
W. C.
Ewing
Institut für Anorganische Chemie, Julius-Maximilians-Universität Würzburg, Am Hubland, 97074 Würzburg, Germany. E-mail: h.braunschweig@uni-wuerzburg.de; Web: http://www-anorganik.chemie.uni-wuerzburg.de/Braunschweig/
First published on 16th April 2015
Abstract
Reaction of the acyclic (diamino)carbene (ADC) :C(NiPr2)2 (1) with different dihaloboranes of the type RBX2 (R = Mes, Dur; X = Cl, Br) smoothly afforded a novel class of ADC-stabilized borane adducts. For MesBBr2 however, the reaction did not stop at the adduct level, but an uncommon rearrangement process occurred, which eventually resulted in the formation of a 5-membered boracycle after elimination of mesitylene. Chemical reduction of the ADC borane adducts by KC8 selectively yielded air stable 1,2-azaboretidines. Detailed DFT studies suggest a reduction mechanism involving a highly reactive borylene intermediate, which is converted into the boracycles via a rearrangement/C–H activation sequence.
Introduction
Recent years have clearly pointed out the exceptional value of carbenes not only as ligands in catalysis,1 but also in the stabilization of highly reactive transition metal and main group element species.2 Initially, research focused on the use of N-heterocyclic carbenes (A; NHC) as stabilizing Lewis bases, and from 2007 a series of outstanding molecules including diborenes,3 diatomic allotropes (B2,4 Si2,5 Ge2,6 Sn2,7 P2,8 As29), and radical/radical ions (B, Si, pnictogen, organic)2b could be realized. Later on, it was rapidly recognized that the different electronics of cyclic (alkyl)(amino)carbenes (B; CAAC) can also be quite useful in the generation of such species (e.g. B2,10 P2,11 PN,12 Sb2,13 B/Si/P/Sn radicals).2e In many cases however, CAAC stabilization also significantly alters the reactivity of the adduct precursors, thereby granting access to other classes of products.2eThe impact of the subtle electronic differences between NHC and CAAC donor ligands is maybe best illustrated by the reduction chemistry of carbene-stabilized haloboranes. Thus, we have demonstrated that chemical reduction of NHC adducts of the type RBX2·NHC and B2Br4·NHC2 is associated with the generation of boron multiple-bonded species, i.e. diborenes3 and diborynes.4 By contrast, reduction of DurBCl2·CAAC (Dur = 2,3,5,6-tetramethylphenyl) stops at the radical stage enabling the isolation of a neutral boron-containing radical, which is most likely a direct consequence of the stronger π accepting abilities of the CAAC ligand.14 This particular electronic property of the CAAC ligand is also reflected in the cumulenic structure of B2·CAAC2, which shows a decreased B–B bond order in comparison to that of the triply bound B2·NHC2.15 Fascinating results also come from the group of Bertrand, which highlighted that CAACs efficiently stabilize neutral B–H borylene16 and boryl radical species,17 as well as boryl anions.18

With these developments in mind, we wondered why the potential of acyclic (diamino)carbenes (C; ADC) in the stabilization of uncommon low-valent main group element compounds has not been studied in detail so far. This appears particularly surprising given the remarkable structural and electronic properties of ADCs.19 Thus, the wider N–C–N angle in ADCs increases the steric shielding of the carbene carbon atom by the amino substituents, which will also affect the structural environment of any potential bonding partner in providing a larger degree of steric protection to the reactive center.20 In addition, ADCs offer a significantly higher basicity, nucleophilicity, and σ donor capacity than NHCs,19 which might entail other reactivity patterns for the reduction of suitable ADC adducts than those observed for their NHC/CAAC analogs. So far however, ADCs have predominately found application as flexible ligands in transition metal catalysis,19 and the number of publications addressing its main group element chemistry is rather limited. Sporadic studies deal with coupling reactions, decomposition reactions, or small molecule activation,19a,21 while almost nothing is known about their ability to stabilize reactive main group element compounds.
As part of our ongoing efforts to generate uncommon low-valent boron species, we now began to study the chemistry of simple ADC borane adducts of the type RBX2·ADC with an emphasis on their reduction behavior, and the results are presented in this contribution.
Results and discussion
We chose bis(diisopropylamino)carbene 1 as the most suitable ADC for the realization of ADC borane adducts, because of its high stability and its convenient synthetic access. 1 was first prepared by Alders et al. back in 1996 as the first compound of this class, and can be readily isolated in high yields by sublimation.22 The stoichiometric reactions of 1 with different dihaloboranes RBX2 (2a: R = Dur, X = Cl; 2b: R = Mes, X = Cl; 2c: R = Mes, X = Br; Mes = 2,4,6-trimethylphenyl) proceeded spontaneously and with high selectivity to afford ADC borane adducts 3a–c in almost quantitative yields (3a: 92%; 3b: 93%; 3c: 97%; Scheme 1). Identification of 3a–c posed no difficulties, and 11B NMR spectroscopy in solution (3a: δ = 4.8; 3b: δ = 4.5; 3c: δ = −1.3) and X-ray diffraction studies on 3a and 3b (Fig. 1) clearly confirmed adduct formation, and the presence of tetra-coordinate boron centers. The solid-state structures of 3a and 3b revealed no surprises, and all bonding parameters are reminiscent of other known carbene borane adducts. As anticipated, both structures feature N1–C1–N2 bond angles (3a: 117.0(1)°; 3b: 117.1(4)°) that are smaller than in 1 (121.0(5)°).22 | ||
| Scheme 1 Syntheses of ADC borane adducts 3a–c. | ||
 | ||
| Fig. 1 X-ray diffraction structures of 3a (left) and 3b (right). Hydrogen atoms and co-crystallized solvent molecules are omitted for clarity. Selected bond lengths [Å] and angles [°]: 3a: B1–C1 1.681(3), B1–C2 1.622(2), B1–Cl1 1.938(2), B1–Cl2 1.889(2), C1–N1 1.376(2), C1–N2 1.352(2), C1–B1–C2 118.9(1), Cl1–B1–Cl2 103.71(9), N1–C1–N2 117.0(2); 3b: B1–C1 1.676(5), B1–C2 1.623(5), B1–Cl1 1.910(4), B1–Cl2 1.920(4), C1–N1 1.364(5), C1–N2 1.365(4), C1–B1–C2 113.5(3), Cl1–B1–Cl2 101.4(2), N1–B1–N2 116.6(3). | ||
While 3a and 3b were found indefinitely stable under inert conditions, 3c readily undergoes a rearrangement reaction to afford boracycle 4 (Scheme 2). The process occurs both in solution (24 h), and in the solid-state (4–5 days), and is quantitative as judged by 1H NMR and 11B NMR spectroscopy (3c: δ = −1.3; 4: δ = −3.4). From a mechanistic point of view, rearrangement of 3c involves C–H activation of one iPr group with concomitant elimination of mesitylene to generate the 5-membered heterocycle 4, whose identity was eventually verified by an X-ray diffraction study (Fig. 2). As a consequence of ring formation, the N1–C1–N2 angle (122.8(2)°) in 4 is slightly larger than in adducts 3a (117.0(1)°) and 3b (117.1(4)°), while the C1–B1–C2 angle (99.53(1)°) becomes dramatically smaller (3a: 118.9(1)°; 3b: 113.5(3)°). By contrast, the C1–N1 (1.352(2) Å) and C1–N2 (1.340(2) Å) bond lengths remain almost unaffected (cf.3a: 1.376(2) Å, 1.352(2) Å; 3b: 1.364(5) Å, 1.365(4) Å), which suggests that the carbenic character of C1 is retained upon rearrangement. The elimination of mesitylene induced by the coordination of the ADC to the boron center of 3c is highly unexpected and should definitely be emphasized here. To the best of our knowledge, such a behavior has not been observed before in boron chemistry. All cases that report on somekind of rearrangement/elimination processes upon coordination of a σ donor to a haloborane were accompanied exclusively by HX elimination (X = Br, I), which usually seems to be strongly favored over RH elimination.23
 | ||
| Scheme 2 Rearrangement of ADC borane adduct 3c. | ||
 | ||
| Fig. 2 X-ray diffraction structure of 4. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: B1–C1 1.624(2), B1–C2 1.598(3), B1–Br1 2.049(2), B1–Br2 2.067(2), C1–N1 1.352(2), C1–N2 1.340(2), N2–C3 1.507(2), C2–C3 1.532(2), C1–B1–C2 99.53(1), B1–C2–C3 104.3(1), C2–C3–N2 104.5(1), C3–N2–C1 113.3(1), N2–C1–B1 109.0(2), N1–C1–N2 122.8(2). | ||
Next, we studied the reduction chemistry of ADC borane adducts 3a–c. To this end, 3a was treated with an excess of KC8 in benzene solution (Scheme 3). Initially, the reaction mixture turned red in color, which however completely disappeared within one hour to finally leave a colorless solution. 11B NMR spectroscopy indicated quantitative and selective conversion of 3a into a new boron-containing species (5a: δ = 45.0). After work-up, 1,2-azaboretidine 5a was isolated as air stable, colorless crystals by recrystallization from hexanes.
 | ||
| Scheme 3 Synthesis of 1,2-azaboretidine 5a. | ||
X-ray diffraction served to validate the molecular composition of 5a in the solid-state (Fig. 3). Accordingly, reduction of 3a is followed by a 1,2-shift of one NiPr2 moiety from the carbenic carbon C1 to the boron center, and a subsequent C–H activation/ring closure sequence to afford the 4-membered heterocycle 5a. The structural parameters of 5a are fully consistent with a classification as 1,2-azaboretidine, and relevant bond lengths and angles lie within the same range observed for other 1,2-azaboretidines.24 Thus, the boron center adopts a distorted trigonal planar geometry. The C1–B1–N1 angle of 91.1(2)° indicates the presence of an almost regular BC2N rectangle (B1–N1–C3: 96.0(2)°; N1–C3–C1: 88.5(2)°; C3–C1–B1: 83.8(2)°), which however is not completely planar (torsion angles between −5.0(2)° and 4.9(2)°). Due to the C–H activation event involving the carbene center of 3a, the geometry of C1 changed from trigonal planar in 3a to highly distorted tetrahedral in 5a (B1–C1–N2: 128.0(3)°).
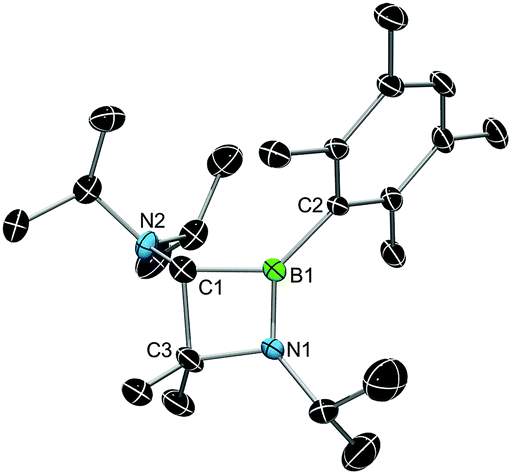 | ||
| Fig. 3 X-ray diffraction structure of 5a. Hydrogen atoms are omitted for clarity. Selected bond lengths [Å] and angles [°]: B1–C1 1.624(4), B1–C2 1.577(4), B1–N1 1.398(4), C1–N2 1.446(4), C1–C3 1.599(5), C3–N1 1.498(4); C1–B1–N1 91.1(2), B1–N1–C3 96.0(2), N1–C3–C1 88.5(2), C3–C1–B1 83.8(2), B1–C1–N2 128.0(3). | ||
The use of ADC 1 has been previously implicated in the formation of 4-membered rings in its reaction with CO, wherein initial complexation of CO and resultant ketene formation leads to the migration of one of the two ADC NiPr2 groups across the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond, initially forming an amide complex that subsequently cyclizes to the final β-lactam product.21b,25 A similar mechanism can be invoked to explain the formation of the 1,2-azaboretidine, since the initial reduction of 3a presumably yields a high-energy borylene (ADC = B–Dur), analogous to the aforementioned ketene (ADC = CO).
C bond, initially forming an amide complex that subsequently cyclizes to the final β-lactam product.21b,25 A similar mechanism can be invoked to explain the formation of the 1,2-azaboretidine, since the initial reduction of 3a presumably yields a high-energy borylene (ADC = B–Dur), analogous to the aforementioned ketene (ADC = CO).
Through DFT calculations (B3LYP/6-311G(d)) we were able to investigate such a mechanism (Fig. 4). A structure for the post-reduction high energy borylene was successfully identified (INT1), showing a linear CA–B–CDur unit (175.0°), as well as one wide and one more acute N–C–B angle (NA–CA–B: 106.8°; NB–CA–B: 134.3°). The transition states in the migration of the diisopropylamine could not be located, likely as a result of the shallow potential energy surface in this region, but an intermediate was found with NA bridging CA and B (INT2) only 1.2 kcal mol−1 lower in energy than INT1. Completion of the migration of the diisopropylamine group from CA to B (INT3) results in a large decrease in energy (−37.5 kcal mol−1), giving a structure with a planar boron (Σangles = 359.5°), but a pronounced bend at CA (B–CA–NB: 143.4°) and a short CA–NB bond (1.282 Å). Activation of the central C–H bond on one of the two iPr groups on NA presumably leads to TS1, 18.7 kcal mol−1 higher in energy than INT3, but still 20.0 kcal mol−1 lower in energy than the initial borylene. Given the high activation barrier for this rearrangement process however, a mechanism involving boron-based radical intermediates might also be effective at this stage, even though we did not obtain any definite experimental evidence for the presence of such radical species. Completion of the C–H activation gives compound 5a, 54.4 kcal mol−1 lower in energy than the borylene product of the reduction of 3a.
 | ||
| Fig. 4 Proposed reaction pathway from the initial borylene product of the reduction of 3a to 5a, as calculated by DFT (B3LYP/6-311G(d)). Electronic energies are given in kcal mol−1 and are ZPE-corrected. | ||
Chemical reduction of ADC borane adducts 3b and 3c pursues an identical reaction pathway as observed for 3a (Scheme 4), and in both cases 1,2-azaboretidine 5b is formed with high selectivity as suggested by 11B NMR spectroscopy of the reaction mixtures (δ = 44.9). 5b can only be isolated as an colorless oil after the standard work-up procedure, which precluded a determination of its molecular structure by X-ray diffraction. The results of NMR spectroscopic studies and an elemental analysis however clearly legitimate the assigned composition.
 | ||
| Scheme 4 Synthesis of 1,2-azaboretidine 5a. | ||
Conclusions
In summary, we have demonstrated that the ADC borane adducts 3a–c are conveniently accessible by stoichiometric reaction of dihaloboranes RBX2 with bis(diisopropylamino)-carbene 1. While the chloro-substituted species 3a and 3b were found indefinitely stable under ambient conditions, the bromo-substituted derivative 3c was prone to undergo a rearrangement process both in solution, and in the solid-state to afford the 5-membered heterocycle 4via C–H activation and mesitylene elimination steps. Subsequent chemical reduction of the ADC borane adducts 3a–c led to the formation of air stable 1,2-azaboretidines 5a and 5b. According to DFT calculations, this transformation most likely involves the initial formation of a reactive borylene species, which is converted into the heterocyclic products by a sequence of 1,2-NiPr2-shift and C–H activation/ring closure.Acknowledgements
This work was supported by the Deutsche Forschungs-gemeinschaft.Notes and references
- For special issues on this topic, see for instance: (a) T. Rovis and S. P. Nolan, Synlett, 2013, 24, 188 Search PubMed; (b) A. J. Arduengo and G. Bertrand, Chem. Rev., 2009, 109, 3209 CrossRef CAS PubMed; (c) X. Bugaut and D. Glorius, Chem. Soc. Rev., 2012, 41, 3511 RSC.
- See for instance: (a) Y. Wang and G. H. Robinson, Dalton Trans., 2012, 41, 337 RSC; (b) C. D. Martin, M. Soleilhavoup and G. Betrand, Chem. Sci., 2013, 4, 3020 RSC; (c) H. Braunschweig and R. D. Dewhurst, Organometallics, 2014, 33, 6271 CrossRef CAS; (d) Y. Wang and G. H. Robinson, Inorg. Chem., 2014, 53, 11815 CrossRef CAS PubMed; (e) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed.
- (a) P. Bissinger, H. Braunschweig, A. Damme, T. Kupfer and A. Vargas, Angew. Chem., Int. Ed., 2012, 51, 9931 CrossRef CAS PubMed; (b) P. Bissinger, H. Braunschweig, A. Damme, T. Kupfer, I. Krummenacher and A. Vargas, Angew. Chem., Int. Ed., 2014, 53, 5689 CrossRef CAS PubMed; (c) H. Braunschweig, R. D. Dewhurst, C. Hörl, A. K. Phukan, F. Pinzner and S. Ullrich, Angew. Chem., Int. Ed., 2014, 53, 3241 CrossRef CAS PubMed; (d) P. Bissinger, H. Braunschweig, A. Damme, C. Hörl, I. Krummenacher and T. Kupfer, Angew. Chem., Int. Ed., 2015, 54, 359 CrossRef CAS PubMed; (e) P. Bissinger, A. Steffen, A. Vargas, R. D. Dewhurst, A. Damme and H. Braunschweig, Angew. Chem., Int. Ed., 2015, 54, 4362 CrossRef CAS PubMed.
- (a) H. Braunschweig, R. D. Dewhurst, K. Hammond, J. Mies, K. Radacki and A. Vargas, Science, 2012, 336, 1420 CrossRef CAS PubMed; (b) H. Braunschweig, T. Dellermann, R. D. Dewhurst, W. C. Ewing, K. Hammond, J. O. C. Jimenez-Halla, T. Kramer, I. Krummenacher, J. Mies, A. K. Phukan and A. Vargas, Nat. Chem., 2013, 5, 1025 CrossRef CAS PubMed.
- Y. Wang, Y. Xie, R. B. King, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Science, 2008, 321, 1069 CrossRef CAS PubMed.
- A. Sidiropoulos, C. Jones, A. Stasch, S. Klein and G. Frenking, Angew. Chem., Int. Ed., 2009, 48, 9701 CrossRef CAS PubMed.
- C. Jones, A. Sisiropoulos, N. Holzmann, G. Frenking and A. Stasch, Chem. Commun., 2012, 48, 9855 RSC.
- Y. Wang, Y. Xie, P. Wei, R. B. King, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, J. Am. Chem. Soc., 2008, 130, 14970 CrossRef CAS PubMed.
- M. Y. Abraham, Y. Wang, Y. Xie, P. Wei, H. F. Schaefer III, P. v. R. Schleyer and G. H. Robinson, Chem.–Eur. J., 2010, 16, 432 CrossRef CAS PubMed.
- (a) J. Böhnke, H. Braunschweig, W. C. Ewing, C. Hörl, T. Kramer, I. Krummenacher, J. Mies and A. Vargas, Angew. Chem., Int. Ed., 2014, 53, 9082 CrossRef PubMed; (b) J. Böhnke, H. Braunschweig, P. Constantinidis, T. Dellermann, W. C. Ewing, I. Fischer, K. Hammond, F. Hupp, J. Mies, H.-C. Schmitt and A. Vargas, J. Am. Chem. Soc., 2015, 137, 1766 CrossRef PubMed; (c) J. Böhnke, H. Braunschweig, T. Dellermann, W. C. Ewing, T. Kramer, I. Krummenacher and A. Vargas, Angew. Chem., Int. Ed., 2015, 54, 4469 CrossRef PubMed.
- O. Back, G. Kuchenbeiser, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2009, 48, 5530 CrossRef CAS PubMed.
- R. Kinjo, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 5930 CrossRef CAS PubMed.
- R. Kretschmer, D. A. Ruiz, C. E. Moore, A. L. Rheingold and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 8176 CrossRef CAS PubMed.
- P. Bissinger, H. Braunschweig, A. Damme, I. Krummenacher, A. K. Phukan, K. Radacki and S. Sugawara, Angew. Chem., Int. Ed., 2014, 53, 7360 CrossRef CAS PubMed.
- F. A. Perras, W. C. Ewing, T. Dellermann, J. Böhnke, S. Ulrich, T. Schäfer, H. Braunschweig and D. L. Bryce, Chem. Sci., 2015, 6 10.1039/c5sc00644a.
- R. Kinjo, B. Donnadieu, M. Ali Celik, G. Frenking and G. Bertrand, Science, 2011, 333, 610 CrossRef CAS PubMed.
- F. Dahcheh, D. Martin, D. W. Stephan and G. Bertrand, Angew. Chem., Int. Ed., 2014, 53, 13159 CrossRef CAS PubMed.
- D. A. Ruiz, G. Ung, M. Melaimi and G. Bertrand, Angew. Chem., Int. Ed., 2013, 52, 7590 CrossRef CAS PubMed.
- (a) J. Vignolle, X. Cattöen and D. Bourissou, Chem. Rev., 2009, 109, 3333 CrossRef CAS PubMed; (b) D. Kremzow, G. Seidel, C. W. Lehmann and A. Fürstner, Chem.–Eur. J., 2005, 11, 1833 CrossRef CAS PubMed; (c) B. Dhudshia and A. N. Thadani, Chem. Commun., 2006, 6, 668 RSC; (d) A. G. Tskhovrebov, K. V. Luzyanin, M. L. Kuznetsov, V. N. Sorokoumov, I. A. Balova, M. Haukka and V. Y. Kukushin, Organometallics, 2011, 30, 863 CrossRef CAS; (e) C. Bartolomé, Z. Ramiro, P. Pérez-Galán, C. Bour, M. Raducan, A. M. Echavarren and P. Espinet, Inorg. Chem., 2008, 47, 11391 CrossRef PubMed; (f) C. Bartolomé, Z. Ramiro, D. García-Cuadrado, P. Pérez-Galán, M. Raducan, C. Bour, A. M. Echavarren and P. Espinet, Organometallics, 2010, 29, 951 CrossRef; (g) H. Seo, B. P. Roberts, K. A. Abboud, K. M. Merz Jr and S. Hong, Org. Lett., 2010, 21, 4860 CrossRef PubMed; (h) V. P. Boyarskiy, K. V. Luzyanin and V. Y. Kukushkin, Coord. Chem. Rev., 2012, 256, 2029 CrossRef CAS PubMed; (i) L. M. Slaughter, ACS Catal., 2012, 2, 1802 CrossRef CAS.
- (a) W. A. Herrmann, K. Öfele, D. v. Preysing and E. Herdtweck, J. Organomet. Chem., 2003, 684, 235 CrossRef CAS; (b) E. L. Rosen, M. D. Sanderson, S. Saravanakumar and C. W. Bielawski, Organometallics, 2007, 26, 5774 CrossRef CAS; (c) K. Denk, P. Sirsch and W. A. Herrmann, J. Organomet. Chem., 2002, 649, 219 CrossRef CAS; (d) G. D. Frey, E. Herdtweck and A. Herrmann, J. Organomet. Chem., 2006, 691, 2465 CrossRef CAS PubMed.
- (a) T. Schulz, M. Leibold, C. Färber, M. Maurer, T. Porsch, M. C. Holthausen and U. Siemeling, Chem. Commun., 2012, 48, 9123 RSC; (b) T. Schulz, C. Färber, M. Leibold, C. Bruhn, W. Baumann, D. Selent, T. Porsch, M. C. Holthausen and U. Siemeling, Chem. Commun., 2013, 49, 6834 RSC.
- R. W. Alder, P. R. Allen, M. Murray and A. G. Orpen, Angew. Chem., Int. Ed., 1996, 35, 1121 CrossRef CAS PubMed.
- (a) P. Bissinger, H. Braunschweig, A. Damme, R. D. Dewhurst, T. Kupfer, K. Radacki and K. Wagner, J. Am. Chem. Soc., 2011, 133, 19044 CrossRef CAS PubMed; (b) H. Braunschweig, A. Damme and T. Kupfer, Chem. Commun., 2013, 49, 2774 RSC.
- (a) S. Channareddy, B. Glaser, E. P. Mayer, H. Nöth and S. W. Helm, Chem. Ber., 1993, 126, 1119 CrossRef CAS PubMed; (b) M. Suginome, T. Fukuda and Y. Iko, J. Organomet. Chem., 2002, 643–644, 508 CrossRef CAS.
- U. Siemeling, C. Färber, C. Bruhn, M. Leibold, D. Selent, W. Baumann, M. v. Hopffgarten, C. Goedecke and G. Frenking, Chem. Sci., 2010, 1, 697 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental, crystallographic, and computational details. CCDC 1042591–1042594. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc01077b |
| This journal is © The Royal Society of Chemistry 2015 |