DOI:
10.1039/C5SC01038A
(Edge Article)
Chem. Sci., 2015,
6, 5419-5425
Protein recognition by bivalent, ‘turn-on’ fluorescent molecular probes†
Received
22nd March 2015
, Accepted 7th June 2015
First published on 12th June 2015
Abstract
We show that the conversion of a known intercalating dye (i.e., thiazole orange) into a bivalent protein binder could lead to the realization of a novel class of ‘turn-on’ fluorescent molecular probes that detect proteins with high affinity, selectivity, and a high signal-to-noise (S/N) ratio. The feasibility of the approach is demonstrated with monomolecular probes that light-up in the presence of three different proteins: acetylcholinesterase (AChE), glutathione-s-transferase (GST), or avidin (Av) at low concentrations and with minimal background signal. The way by which such probes can be used to detect individual protein isoforms and be applied in inhibitor screening, cell imaging, and biomarker detection is described.
Introduction
Fluorescent molecular probes that can label, detect, or image specific proteins1 serve as a powerful tools for developing in vitro proteomic assays, for identifying disease biomarkers, as well as for tracking proteins in their native environments. Ideally, such systems should act as ‘turn-on’ fluorescent probes,2 which do not generate any background signal in the absence of the bioanalyte, but emit very strongly in the presence of the protein target. In practice, however, developing fluorescent molecular switches that can recognize their target proteins with high affinity, selectivity, and sensitivity is challenging. Obtaining highly selective probes is complicated by the fact that many protein groups, which can be targeted by small-molecule-based probes, possess well-defined recognition sites that are conserved among structurally similar isoforms of the same family. High sensitivity is also difficult to achieve because common fluorescence signaling mechanisms,3 such as photo-induced electron transfer (PET), charge transfer (CT), or fluorescence resonance energy transfer (FRET) often lead to a background emission signal by the unbound probes. Consequently, an excess of protein is generally required to obtain a sufficient fluorescence response. Finally, a limitation of many molecular probes, when compared with the corresponding antibodies or aptamers, is that they bind their target with lower affinities, which prevents them from detecting proteins at low concentrations. Herein, we describe the operating principles of a novel class of ‘turn-on’ fluorescent molecular probes that were designed to address these limitations. Specifically, we show how thiazole orange (TO) can be modified to detect different proteins, including individual protein isoforms, with high affinity, selectivity, and a high signal-to-noise (S/N) ratio.
Recently, we have shown that structurally similar biomolecules, such as saccharides4 or isoforms of the glutathione-s-transferase (GST) family5 can be discriminated using the “nose/tongue” strategy, in which combinations of non-specific fluorescent probes generate distinct optical “fingerprints” for different analytes.3c Herein, we show that it is also possible to create highly selective turn-on fluorescent molecular probes that can identify specific proteins at low concentrations and with a minimal background signal (Fig. 1).
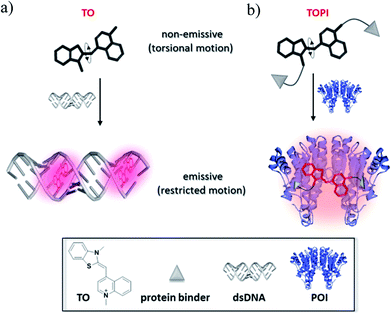 |
| | Fig. 1 Schematic representation of the mechanism responsible for the ‘turn-on’ fluorescence signal generated upon (a) the binding of thiazole orange (TO) to a double-stranded DNA (dsDNA), and (b) the binding of a TO-based protein identifier (TOPI) to the protein of interest (POI). | |
Results and discussion
Design of TO-based protein identifiers (TOPIs)
TO was selected as the signaling unit of these probes because this asymmetrical cyanine dye exhibits a remarkable turn-on fluorescence response once the torsional motion between the benzothiazole and the quinoline rings in the excited-state is restricted,6 for example, upon binding to double-stranded DNA (Fig. 1a). This property has been elegantly used to construct low-noise forced intercalation probes (FIT-probes) for sensitive detection of RNA and DNA.7 Other hybridization probes8 as well as aptamers9 or antibody-based probes2i further demonstrate the versatile use of this dye for sensing applications, and also indicate that fixating of TO in its fluorescent form does not necessarily require intercalation between DNA base pairs.
Accordingly, we anticipated that modifying TO with two protein binders would result in a TO-based protein identifier (TOPI) that is inherently non-fluorescent in the unbound state; however, it becomes highly emissive once its torsional motion is restricted upon binding to the protein of interest (POI, Fig. 1b). The bivalent interaction mode of this probe is another important property that should enable TOPI to bind its target with high affinity.10 Finally, we anticipated that the interaction between the specific protein binders of TOPI (Fig. 2) and the POI would also result in the binding of the TO-core to the protein's surface (Fig. 1b). This assumption is based on a recent study by our group in which we showed that bringing a synthetic agent in the vicinity of a protein surface is likely to enhance its affinity toward the surface of this protein.5 In the case of TOPI, such interactions could also affect the rotational motion of TO, which should enable the system to respond differently to isoforms with distinct surface characteristics.
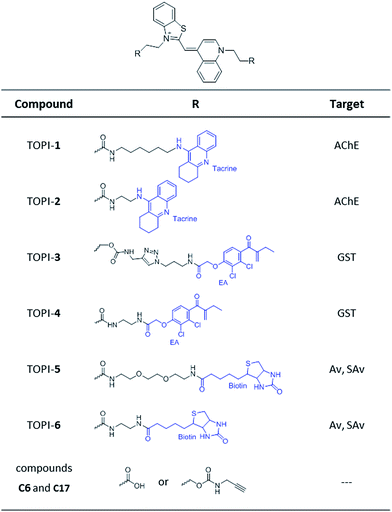 |
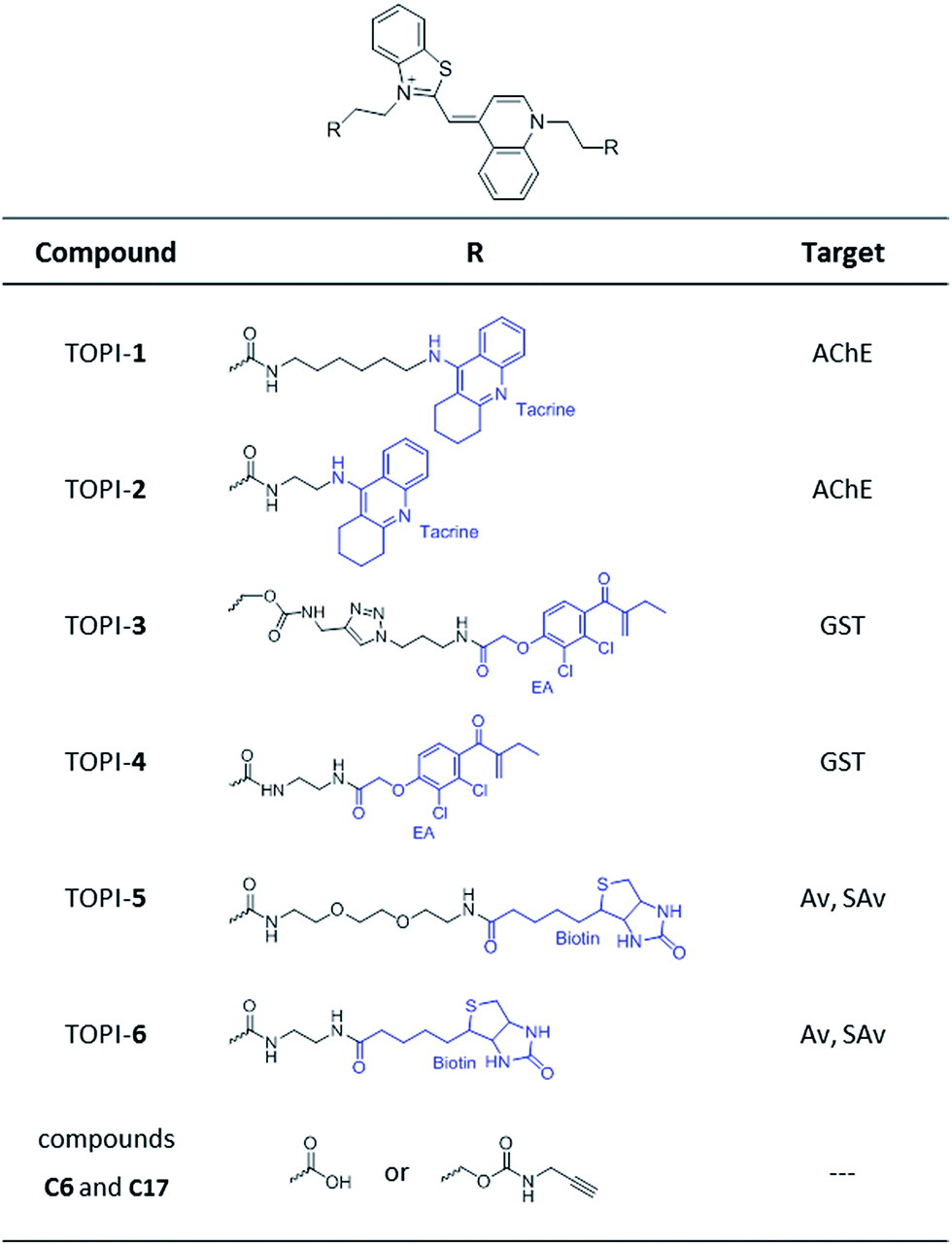
| | Fig. 2 Structures of TO-based probes with selectivity toward acetylcholinesterase (AChE), glutathione-s-transferases (GSTs), and avidin (Av) or streptavidin (SAv). C6 and C17 are control compounds that lack specific protein binders. | |
Based on these principles, we prepared several TOPI probes (Fig. 2) appended with two tacrine (TOPI-1 and TOPI-2), ethacrynic amide (EA) (TOPI-3 and TOPI-4), and biotin (TOPI-5 and TOPI-6) moieties, which were intended to target adjacent binding sites within acetylcholinesterase (AChE), glutathione-s-transferases (GSTs), and avidin (Av) or streptavidin (SAv), respectively. Probes from each group share the same protein binders, but differ in the lengths and/or structure of their linkers. AChE is an acetylcholine hydrolase that regulates the concentration of this transmitter at the synapse.11 The design of TOPI-1 and TOPI-2 is based on the ability of bis-tacrine inhibitors to interact both with the active site and the peripheral site of this enzyme.12 GSTs are a large family of dimeric enzymes that protect the organism from toxic species by conjugating glutathione (GSH) to a variety of electrophilic substrates.13 TOPI-3 and TOPI-4, which are appended with two GST inhibitors (EAs), were expected to detect members of this family by simultaneously binding to identical sites within these dimers.5 GSTs and AChE play a role in a myriad of cellular processes, and overexpression of these enzymes has been associated with various diseases.11,13 Hence, turn-on probes for these proteins could potentially be applied in inhibitor screening, medical diagnosis, and cellular imaging. Avidin (Av) and streptavidin (SAv) are protein tetramers produced from egg whites and bacterium Streptomyces avidinii, respectively.14 These tetramers share a similar biotin binding site but possess very distinct surfaces. Therefore, by measuring the fluorescent response of TOPI-5 and TOPI-6 to Av and SAv, as well as the response of TOPI-3 and TOPI-4 to different GST isozymes, we aimed at understanding whether the local molecular environment of TO plays a role in obtaining ‘turn-on’ fluorescent signals.
Fluorescence detection of different proteins
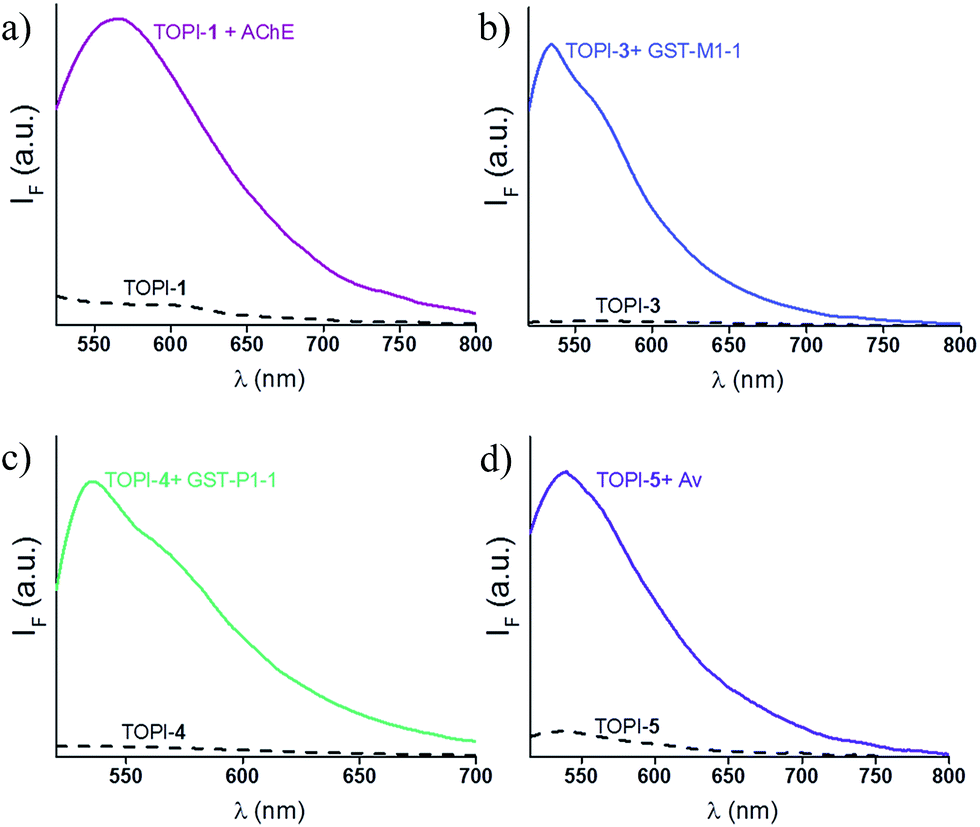
We identified the most efficient TOPIs (Fig. 3) by measuring the fluorescence of the six probes (100 nM) in the absence and presence of their targets (90 nM) (Fig. 4). These measurements revealed that the TOPIs not only can identify their targets at low nanomolar concentrations and with high S/N ratio – their properties can also be adjusted through the systematic modification of the length and type of linkers. TOPI-1, for example, which exhibited 22 fold enhancement in its emission, was found to be more efficient than TOPI-2, although both probes could detect AChE (Fig. 4a). Similarly, the emission of TOPI-5 was more significantly enhanced (16 fold) when compared with TOPI-6 (7 fold) (Fig. 4b); however, both probes responded strongly to Av and weakly to SAv. Whereas TOPI-1 and TOPI-2, or TOPI-5 and TOPI-6 exhibited similar response trends, the emission of TOPI-3 and TOPI-4 was distinctly affected by different GST isozymes. Incubation of TOPI-3 with eight GST isozymes (e.g., GST-M1-1, GST-A1-1, GST-A2-2, GST-P1-1, GST-Z1-1, GST-O1-1, GST-K1-1, and GST-T1-1) resulted in an immediate fluorescence enhancement (55 fold) only in the presence of GST-M1-1 (Fig. 4c), whereas for TOPI-4, a strong (33 fold) turn-on fluorescence signal was observed only when it was incubated with GST-P1-1 (Fig. 4c). Interestingly, in the presence of GST-P1-1 the emission of TOPI-3 was slowly increased over a period of 2 hours (Fig. S1†), which may result from distinct binding kinetics. Fluorescence measurements also show that different TOPIs exhibit different emission maxima (Fig. 3 and S2†), which is expected from differences in the chemical structures of TO-based probes and/or their analyte target.6b The approximate dissociation constants for the TOPI-3–GST-M1-1 (Kd = 16 nM ± 2) and TOPI-4–GST-P1-1 (Kd = 29 nM ± 5) interactions were also obtained by performing fluorescence binding studies, in which we followed the changes in the emission upon incremental addition of each protein (Fig. S3†).
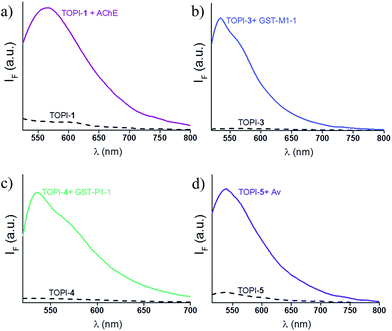 |
| | Fig. 3 Changes in the fluorescence signal of the most efficient TOPIs: (a) TOPI-1, (b) TOPI-3, (c) TOPI-4, and (d) TOPI-5 (100 nM each) upon the addition of 90 nM of AChE, GST-M1-1, GST-P1-1, and Av, respectively. Conditions: (a) λex = 505 nm, PBS buffer (20 mM, pH = 8), (b and c) λex = 500 nm, PBS buffer (5 mM, pH = 6.5) (d) λex = 495 nm, PBS buffer (15 mM, pH = 7.3). | |
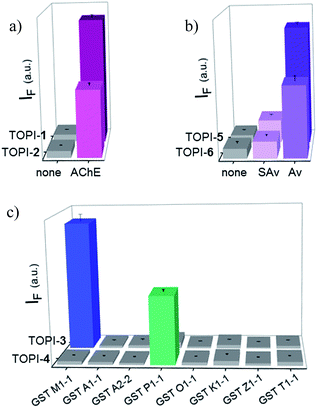 |
| | Fig. 4 Fluorescence response of 100 nM of (a) TOPI-1 and TOPI-2, (b) TOPI-5 and TOPI-6, and (c) TOPI-3 and TOPI-4, to the addition of 90 nM of AChE, Av and SAv, and various GST isozymes, respectively. | |
Enzymatic assays
Enzymatic inhibition assays were performed to elucidate the sensing mechanism underlying TOPI probes (Fig. 5 and S5†). Specifically, these assays were used to determine whether TOPIs bind their targets (a) at the active site, and (b) through a bivalent interaction mode (Fig. 5). In addition, these assays were used to investigate (c) whether the local molecular environment of the TO dye plays a role in obtaining enhanced fluorescence signals (Fig. S5†). As shown in Fig. 5, the catalytic activity of AChE, GST-M1-1, and GST-P1-1 was followed in the absence and presence of the corresponding probe, as well as with the monovalent inhibitor (tacrine or EA) or a control TO derivative (C6 or C17) that lacks the inhibitors. The fact that the three enzymes were hardly inhibited by the control compounds and that TOPIs were found to be considerably more potent than the monovalent inhibitors confirms the manifestation of the “multivalency effect”, which results from the simultaneous binding of each probe at two binding sites within the enzyme. The same enzymatic assays were used to determine the approximate inhibition constant for the TOPI-1-AChE (Ki = 0.32 nM ± 0.04), TOPI-3–GST-M1-1 (Ki = 4 nM ± 0.6), and TOPI-4–GST-P1-1 (Ki = 28.09 nM ± 3.81) interactions (Fig. S4†). The Ki values for TOPI-3 and TOPI-4 are in the same range of the Kd values derived from the fluorescence assay (Fig. S3†), which further validates that these probes bind at the active site, and that non-specific binding of the TO moiety is not the cause of the enhanced emission.
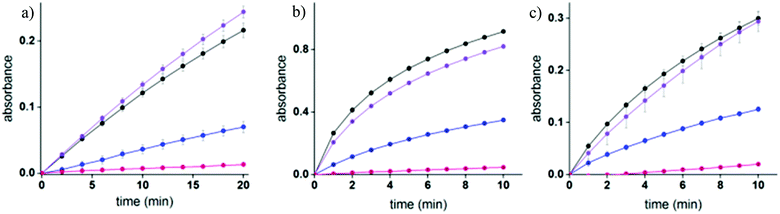 |
| | Fig. 5 (a) Enzymatic activity of AChE (8 pM) ( ) in the presence of 50 nM tacrine ( ) in the presence of 50 nM tacrine ( ), 5 μM C6 ( ), 5 μM C6 ( ), and 50 nM TOPI-1 ( ), and 50 nM TOPI-1 ( ). (b and c) Enzymatic activity of 20 nM of GST-M1-1 (b) or GST-P1-1 (c) in the absence ( ). (b and c) Enzymatic activity of 20 nM of GST-M1-1 (b) or GST-P1-1 (c) in the absence ( ) and the presence of 500 nM of TOPI-3 or TOPI-4 ( ) and the presence of 500 nM of TOPI-3 or TOPI-4 ( ), respectively, as well as with 5 μM EA ( ), respectively, as well as with 5 μM EA ( ) or control compounds (C6 or C17) ( ) or control compounds (C6 or C17) ( ). ). | |
Next, enzymatic assays were used to investigate the reason for the selective response of some of the TOPI probes toward specific protein isoforms (Fig. 4c). This selectivity may result from enhanced affinity toward these isoforms. Alternatively, these probes might also bind to the other (non-detected) isoforms, which would indicate that this unique isoform discrimination results from differences in the molecular environment of the protein-bound TO. Fig. S5† shows the catalytic activity of GST-A1-1 and GST-A2-2 in the presence and absence of TOPI-3. Although these isozymes could not be detected by TOPI-3, both of them were strongly inhibited by this probe with a Ki value of 20 nM and 450 nM, respectively (Fig. S6†), indicating a bivalent probe–protein interaction. Considering that SAv was also hardly detected by TOPI-5 and TOPI-6, despite its remarkable affinity to biotin (Kd = 10−14 M), we can conclude that the selective detection of isoforms does not result from isozyme-specific binding. Instead, the interaction of TO with amino acid side chains in its surroundings must play an important role in achieving a turn-on emission signal. Inspection of the crystal structures and electrostatic maps of the different GSTs (Fig. S7†) reveals that they possess very similar structures. In GST-M1-1 and GST-P1-1, in particular, even the crevices between the EA binding sites have very similar dimensions and are negatively charged. This structural similarity indicates that the TOPI probes are sensitive to subtle changes in the protein structure, a property that could be either advantageous or limiting (see the conclusion section).
Selectivity studies
To test the selectivity of the best TOPI probes (Fig. 3, TOPI-1, TOPI-3, TOPI-4, and TOPI-5) toward their targets, three additional fluorescence experiments were performed (Fig. 6 and 7). In the first, we confirmed that these probes do not respond to the addition of a large excess of other proteins (1–2 μM) (Fig. 6). These include common serum proteins such as human serum albumin (HSA), which are notorious for forming non-specific interactions. In a second experiment we showed that the control TO derivatives, which lack the specific protein binders (Fig. 2), do not fluoresce in the presence of the detected proteins (Fig. 7a–d). Hence, this experiment further shows that it is the strong interaction of the specific protein binders (Fig. 2) that enhances a weak and non-specific interaction between the TO-core of these probes and the surface of the POIs. Finally, we tested whether the emission of these probes (in the absence of POI) is triggered by the addition of double-stranded DNA (dsDNA) (Fig. 7e).
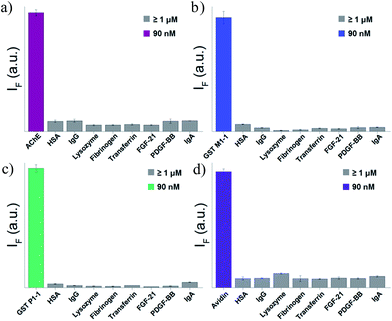 |
| | Fig. 6 Fluorescence response of (a) TOPI-1, (b) TOPI-3, (c) TOPI-4, and (d) TOPI-5 to the addition of the corresponding protein target (90 nM) (colored) or an excess (≥1 μM) of serum proteins (grey). | |
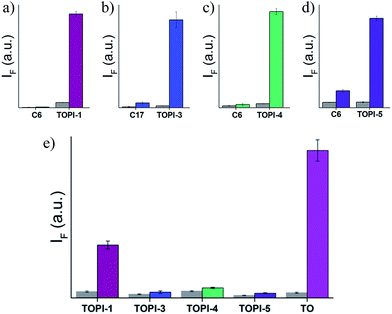 |
| | Fig. 7 Fluorescence emission of (a) TOPI-1, (b) TOPI-3, (c) TOPI-4, and (d) TOPI-5 or control compounds C6 or C17 before (grey) and after (colored) the addition of AChE, GST-M1-1, GST-P1-1, and Av, respectively (90 nM). (e) Fluorescence of TOPIs and TO (100 nM each) before (grey) and after (colored) the addition of dsDNA (400 nM). | |
Our probes rely on an intercalating dye as the main sensing element and therefore, the goal of this experiment was to test the sensitivity of TOPIs to the presence of nucleic acids, which are prevalent in various biological samples. The strong fluorescence signal generated by the known intercalator (i.e., TO) and the weaker emission generated by TOPIs under the same conditions indicate that the bulky substituents disrupt the intercalation of the probe with dsDNA. TOPIs 3–5, in particular, exhibited negligible fluorescence responses to the addition of dsDNA, which demonstrates the feasibility of converting the known DNA intercalator (i.e., TO) into a highly specific turn-on protein probe.
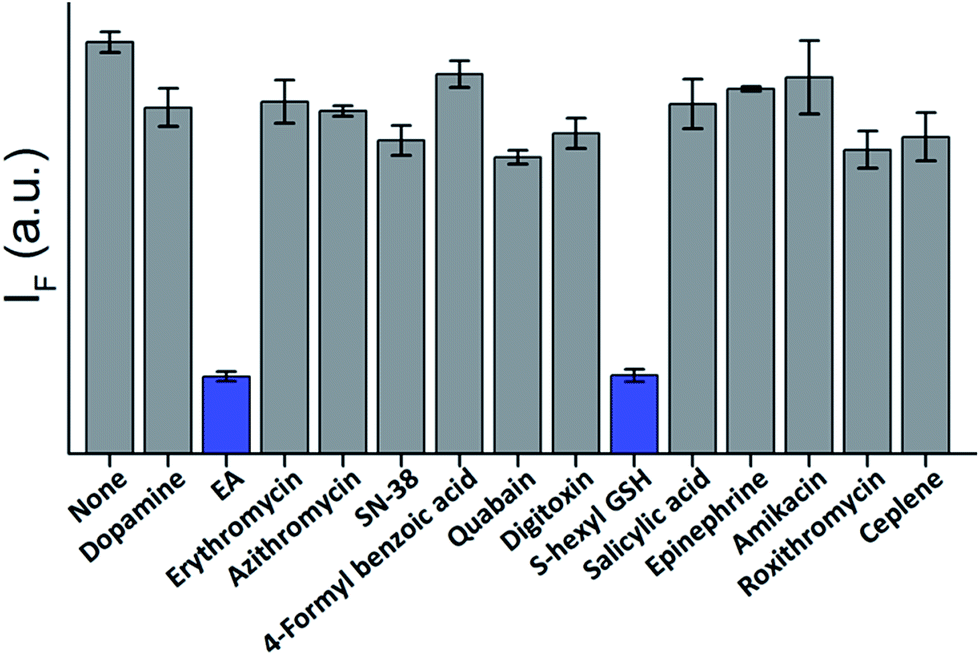
The high affinity and specificity of TOPIs toward their targets were also demonstrated by using them to identify inhibitors. In the following experiment (Fig. 8) TOPI-3–GST-M1-1 complex was subjected to different compounds and the displacement of the probes by “hit” compounds was identified by observing a reduction in the emission intensity. To demonstrate the relevance of such systems to drug discovery applications, the TOPI–protein complex was treated with a random library of drugs (grey), as well as with known inhibitors (blue). The selective identification of EA and S-hexyl GSH (Fig. 8), as well as the displacement of AChE-bound TOPI-1 by tacrine and avidin-bound TOPI-5 by biotin (Fig. S8†) provides another evidence that the probes' response originates from selective and reversible binding at the enzymes' active sites.
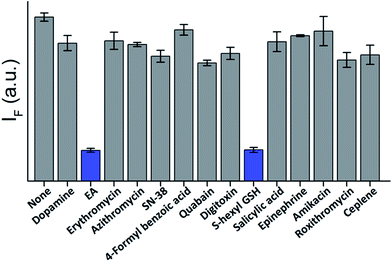 |
| | Fig. 8 Displacement assay in which the TOPI-3–GST-M1-1 complex (90 nM) is treated with 50 μM of randomly selected drugs (grey) as well as with the known GST inhibitors, EA and S-hexyl GSH (blue). | |
Protein detection in biofluids and in live cells
GST-P1-1 is a cytosolic protein, whereas DNA is located in the nucleus. Hence, the differences between TOPI-4 and the known DNA intercalator (i.e., TO) (Fig. 7e) could also be observed by monitoring their localization in live cells (Fig. 9). Breast cancer cells (MDA-MB-231), known to overexpress GST-P1-1,15 were incubated with TO or TOPI-4, as well as with TOPI-4 and an excess of EA. Fluorescent imaging shows a difference in the localization of these probes according to their biomolecule targets. Whereas treatment with the TO intercalator led to a strong green emission mainly from the nucleus (Fig. 9b), the fluorescence generated by TOPI-4 was distributed within the cell (Fig. 9a). This emission was eliminated in the presence of EA (Fig. 9c), which is expected from the displacement of the probe by the GST inhibitor (Fig. 8). A much weaker emission signal was observed when TOPI-4 was incubated with healthy MCF-10A cells (Fig. 9d), which further demonstrates the compatibility of TOPIs with live cell imaging applications.
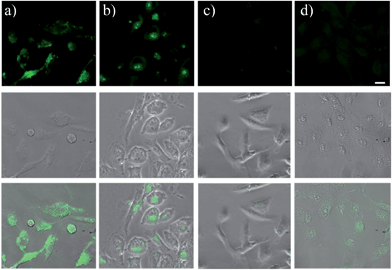 |
| | Fig. 9 (Top) Fluorescent, (middle) bright field and (down) overlay images of MDA-MB-231 cancer cells, overexpressing GST-P1-1, after incubation with (a) TOPI-4 (2 μM), (b) TO (2 μM), and (c) TOPI-4 (2 μM) and EA (50 μM). (d) Images of healthy MCF-10A cells incubated with TOPI-4 (2 μM). The scale bar is 20 μm. | |
Finally, we tested the suitability of our probes to be applied in biomarker detection. In the following experiments, we demonstrate how the selective response of some of the TOPI probes to particular protein isoforms could be used to circumvent the challenge of identifying specific isoforms within mixtures. We first tested the ability of TOPI-3 (100 nM) to detect GST-M1-1 (20 nM) in an isozyme mixture (mixture 1) containing seven additional GSTs (20 nM each) and compared the resulting emission to that generated in a solution containing the same seven isozymes without GST-M1-1 (mixture 2) (Fig. 10a). The selective detection of GST-M1-1 in a mixture of isoforms could be achieved not only due to the strong TOPI-3–GST-M1-1 interaction and excellent fluorescence response of the probe (55 fold), but also owing to the low background emission (Fig. 3b), which enabled us to use an excess of TOPI-3 (100 nM) and thus, to ensure that the other isozymes will not compete with the probe–GST-M1-1 interaction.
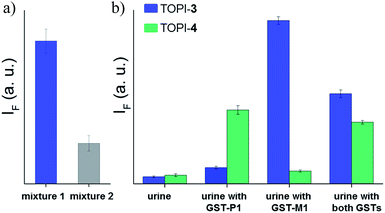 |
| | Fig. 10 (a) Detection of GST-M1-1 by TOPI-3 in a mixture (mixture 1) of multiple GST isozymes (GST A1-1, GST A2-2, GST P1-1, GST T1-1, GST K1-1, GST O1-1, and GST Z1-1, 20 nM each). Mixture 2 contains the same isozymes without GST-M1-1. (b) Analyzing the content of GST-M1-1 and GST-P1-1 (800 ng mL−1 each) in human urine by measuring the response of TOPI-3 ( ) and TOPI-4 ( ) and TOPI-4 ( ) to each sample. ) to each sample. | |
In the next step, TOPI-3 and TOPI-4 were used to sense specific GST isozymes in human urine (Fig. 10b). Elevated levels of specific GST isozymes, such as GST-P1-1, have been detected in several kidney-related diseases.16 Hence, by discriminating among isozymes in urine (Fig. 10b) we aimed at demonstrating the applicability of this approach to biomarker detection. Human urine samples were spiked with medicinally relevant concentrations of GST-P1-1 (800 ng mL−1),16 as well as GST-M1-1, and their combination. Isozyme analysis was achieved by enriching the GST content of each sample using a GSH column subjecting it to TOPI-3 and TOPI-4. As shown in Fig. 10b, the resulting fluorescence intensities provided a clear-cut analysis of the isozyme composition within each sample. This experiment also highlights the differences between our system and common techniques used in biomarker detection. Enzymatic assays,17 for example, which can straightforwardly detect high enzyme concentrations, are often unsuitable for distinguishing among isozymes, whereas isozyme detection by antibody-based techniques generally requires stepwise incubation and labelling steps.
Conclusions
In summary, we have presented a new class of turn-on fluorescent molecular probes for the sensitive and selective detection of different proteins. The generality of this approach was demonstrated by the ability of TOPIs to detect members within all three protein groups that were selected for this study. The ways by which these probes can be applied in inhibitor screening, cellular imaging, and biomarker detection have also been demonstrated. A unique property of these systems is their ability to distinguish among proteins that have very similar binding sites. We have shown that although one probe can bind several proteins with similar affinities, these interactions lead to markedly distinct fluorescence responses. This property thus indicates that the emission of TOPIs is largely affected by the local molecular environment that is created for TO dye upon forming the probe–protein complex. A weakness of this sensing mechanism lies in the general difficulty of predicting the way synthetic molecules interact with protein surfaces,18 which could complicate the rational design of isoform-specific probes. On the other hand, our results show that this challenge can be addressed by systematically modifying the structure of these probes and screening for their fluorescence responses. What distinguishes the TOPI technology from various other fluorescent molecular probes for proteins2 is the ability to use a single molecular sensing platform, which integrates the strong ‘turn-on’ emission of TO and the high binding affinity of bivalent inhibitors for detecting various different proteins including individual protein isoforms. Given that about 30% of human proteins are homodimers,19 and that a general and easily applicable procedure for preparing such systems is in hand, we expect that various other TOPIs would be developed and contribute to the ability to detect and image proteins with fluorescent molecular probes.
Acknowledgements
This work was supported by the European Research Council Starting Grant 338265. We are grateful to Prof. Israel Silman for kindly providing us with Torpedo californica AChE.
Notes and references
- For selected reviews, see:
(a) H. Kobayashi, M. Ogawa, R. Alford, P. L. Choyke and Y. Urano, Chem. Rev., 2010, 110, 2620–2640 CrossRef CAS PubMed;
(b) R. W. Sinkeldam, N. J. Greco and Y. Tor, Chem. Rev., 2010, 110, 2579–2619 CrossRef CAS PubMed;
(c) A. T. Krueger and B. Imperiali, ChemBioChem, 2013, 14, 788–799 CrossRef CAS PubMed;
(d) L. D. Lavis and R. T. Raines, ACS Chem. Biol., 2008, 3, 142–155 CrossRef CAS PubMed;
(e) Y. Takaoka, A. Ojida and I. Hamachi, Angew. Chem. Int. Ed., 2013, 52, 4088–4106 CrossRef CAS PubMed;
(f) G. Lukinavičius and K. Johnsson, Curr. Opin. Chem. Biol., 2011, 15, 768–774 CrossRef PubMed.
- Examples of 'turn-on' fluorescent probes for proteins. For disassembly driven probes, see:
(a) K. Mizusawa, Y. Ishida, Y. Takaoka, M. Miyagawa, S. Tsukiji and I. Hamachi, J. Am. Chem. Soc., 2010, 132, 7291–7293 CrossRef CAS PubMed;
(b) K. Mizusawa, Y. Takaoka and I. Hamachi, J. Am. Chem. Soc., 2012, 134, 13386–13395 CrossRef CAS PubMed. For solvatochromic probes, see:
(c) M. Sainlos, W. S. Iskenderian and B. Imperiali, J. Am. Chem. Soc., 2009, 131, 6680–6682 CrossRef CAS PubMed;
(d) Y.-D. Zhuang, P.-Y. Chiang, C.-W. Wang and K.-T. Tan, Angew. Chem. Int. Ed., 2013, 52, 8124–8128 CrossRef CAS PubMed. For peptide beacon-based probes, see:
(e) K. J. Oh, K. J. Cash and K. W. Plaxco, J. Am. Chem. Soc., 2006, 128, 14018–14019 CrossRef CAS PubMed;
(f) S. Thurley, L. Röglin and O. Seitz, J. Am. Chem. Soc., 2007, 129, 12693–12695 CrossRef CAS PubMed. For genetically targeted probes, see:
(g) A. B. Griffin, S. R. Adams and R. Y. Tsien, Science, 1998, 281, 269–272 CrossRef PubMed;
(h) T. L. Halo, J. Appelbaum, E. M. Hobert, D. M. Balkin and A. Schepartz, J. Am. Chem. Soc., 2008, 131, 438–439 CrossRef PubMed;
(i) C. Szent-Gyorgyi, B. F. Schmidt, Y. Creeger, G. W. Fisher, K. L. Zakel, S. Adler, J. A. J. Fitzpatrick, C. A. Woolford, Q. Yan, K. V. Vasilev, P. B. Berget, M. P. Bruchez, J. W. Jarvik and A. Waggoner, Nat. Biotechnol., 2008, 26, 235–240 CrossRef CAS PubMed;
(j) K. Honda, S.-H. Fujishima, A. Ojida and I. Hamachi, ChemBioChem, 2007, 8, 1370–1372 CrossRef CAS PubMed;
(k) M. Kamoto, N. Umezawa, N. Kato and T. Higuchi, Chem.–Eur. J., 2008, 14, 8004–8012 CrossRef CAS PubMed;
(l) A. Rutkowska, C. H. Haering and C. Schultz, Angew. Chem. Int. Ed., 2011, 50, 12655–12658 CrossRef CAS PubMed;
(m) E. G. Guignet, R. Hovius and H. Vogel, Nat. Biotechnol., 2004, 22, 440–444 CrossRef CAS PubMed.
- For selected reviews, see:
(a) A. P. de Silva, H. Q. N. Gunaratne, T. Gunnlaugsson, A. J. M. Huxley, C. P. McCoy, J. T. Rademacher and T. E. Rice, Chem. Rev., 1997, 97, 1515–1566 CrossRef CAS PubMed;
(b) J. F. Callan, A. P. de Silva and D. C. Magri, Tetrahedron, 2005, 61, 8551–8588 CrossRef CAS;
(c) B. Rout, L. Motiei and D. Margulies, Synlett, 2014, 25, 1050–1054 CrossRef.
-
(a) B. Rout, L. Unger, G. Armony, M. A. Iron and D. Margulies, Angew. Chem. Int. Ed., 2012, 21, 12477–12481 CrossRef PubMed;
(b) B. Rout, P. Milko, M. A. Iron, L. Motiei and D. Margulies, J. Am. Chem. Soc., 2013, 135, 15330–15333 CrossRef CAS PubMed.
-
(a) L. Motiei, Z. Pode, A. Koganitsky and D. Margulies, Angew. Chem. Int. Ed., 2014, 53, 9289–9293 CrossRef CAS PubMed;
(b) S. Karuthapandi, L. Motiei and D. Margulies, J. Am. Chem. Soc., 2015, 137, 4892–4895 CrossRef PubMed.
-
(a) V. Karunakaran, J. L. Pérez Lustres, L. Zhao, N. P. Ernsting and O. Seitz, J. Am. Chem. Soc., 2006, 128, 2954–2962 CrossRef CAS PubMed;
(b) I. I. Timcheva, V. A. Maximova, T. G. Deligeorgiev, N. I. Gadjev, R. W. Sabnis and I. G. Ivanov, FEBS Lett., 1997, 405, 141–144 CrossRef CAS PubMed.
-
(a) E. Socher, L. Bethge, A. Knoll, N. Jungnick, A. Herrmann and O. Seitz, Angew. Chem. Int. Ed., 2008, 47, 9555–9559 CrossRef CAS PubMed;
(b) F. Hövelmann, L. Bethge and O. Seitz, ChemBioChem, 2012, 13, 2072–2081 CrossRef PubMed;
(c) F. Hövelmann, I. Gaspar, A. Ephrussi and O. Seitz, J. Am. Chem. Soc., 2013, 135, 19025–19032 CrossRef PubMed.
- Y. Kam, A. Rubinstein, A. Nissan, D. Halle and E. Yavin, Mol. Pharm., 2012, 9, 685–693 CrossRef CAS PubMed.
- R. Pei, J. Rothman, Y. Xie and M. N. Stojanovic, Nucleic Acids Res., 2009, 37, e59 CrossRef PubMed.
-
(a) C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E.-W. Knapp and R. Haag, Angew. Chem. Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed;
(b) C. A. Hunter and H. L. Anderson, Angew. Chem. Int. Ed., 2009, 48, 7488–7499 CrossRef CAS PubMed.
- I. Silman and J. L. Sussman, Curr. Opin. Pharmacol., 2005, 5, 293–302 CrossRef CAS PubMed.
- E. H. Rydberg, B. Brumshtein, H. M. Greenblatt, D. M. Wong, D. Shaya, L. D. Williams, P. R. Carlier, Y.-P. Pang, I. Silman and J. L. Sussman, J. Med. Chem., 2006, 49, 5491–5500 CrossRef CAS PubMed.
- R. N. Armstrong, Chem. Res. Toxicol., 1997, 10, 2–18 CrossRef CAS PubMed.
- G. Gitlin, E. A. Bayer and M. Wilchek, Biochem. J., 1990, 269, 527–530 CrossRef CAS PubMed.
- J. Ge, A.-X. Tian, Q.-S. Wang, P.-Z. Kong, Y. Yu, X.-Q. Li, X.-C. Cao and Y.-M. Feng, PLoS One, 2013, 8, e67589 CAS.
-
(a) T. J. Cawood, M. Bashir, J. Brady, B. Murray, P. T. Murray and D. O'Shea, Am. J. Nephrol., 2010, 32, 219–225 CrossRef CAS PubMed;
(b) J. Westhuyzen, Z. H. Endre, G. Reece, D. M. Reith, D. Saltissi and T. J. Morgan, Nephrol., Dial., Transplant., 2003, 18, 543–551 CrossRef CAS.
- Y. Fujikawa, Y. Urano, T. Komatsu, K. Hanaoka, H. Kojima, T. Terai, H. Inoue and T. Nagano, J. Am. Chem. Soc., 2008, 130, 14533–14543 CrossRef CAS PubMed.
- H. Yin and A. D. Hamilton, Angew. Chem. Int. Ed., 2005, 44, 4130–4163 CrossRef CAS PubMed.
- E. D. Levy, J. B. Pereira-Leal, C. Chothia and S. A. Teichmann, PLoS Comput. Biol., 2006, 2, e155 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc01038a |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence


 ) in the presence of 50 nM tacrine (
) in the presence of 50 nM tacrine ( ), 5 μM C6 (
), 5 μM C6 ( ), and 50 nM TOPI-1 (
), and 50 nM TOPI-1 ( ). (b and c) Enzymatic activity of 20 nM of GST-M1-1 (b) or GST-P1-1 (c) in the absence (
). (b and c) Enzymatic activity of 20 nM of GST-M1-1 (b) or GST-P1-1 (c) in the absence ( ) and the presence of 500 nM of TOPI-3 or TOPI-4 (
) and the presence of 500 nM of TOPI-3 or TOPI-4 ( ), respectively, as well as with 5 μM EA (
), respectively, as well as with 5 μM EA ( ) or control compounds (C6 or C17) (
) or control compounds (C6 or C17) ( ).
).



 ) and TOPI-4 (
) and TOPI-4 ( ) to each sample.
) to each sample.