
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Targeting virulence: salmochelin modification tunes the antibacterial activity spectrum of β-lactams for pathogen-selective killing of Escherichia coli†
Phoom
Chairatana
 ,
Tengfei
Zheng
and
Elizabeth M.
Nolan
*
,
Tengfei
Zheng
and
Elizabeth M.
Nolan
*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, MA 02139, USA. E-mail: lnolan@mit.edu; Fax: +1-617-324-0505; Tel: +1-617-452-2495
First published on 22nd May 2015
Abstract
New antibiotics are required to treat bacterial infections and counteract the emergence of antibiotic resistance. Pathogen-specific antibiotics have several advantages over broad-spectrum drugs, which include minimal perturbation to the commensal microbiota. We present a strategy for targeting antibiotics to bacterial pathogens that utilises the salmochelin-mediated iron uptake machinery of Gram-negative Escherichia coli. Salmochelins are C-glucosylated derivatives of the siderophore enterobactin. The biosynthesis and utilisation of salmochelins are important for virulence because these siderophores allow pathogens to acquire iron and evade the enterobactin-scavenging host-defense protein lipocalin-2. Inspired by the salmochelins, we report the design and chemoenzymatic preparation of glucosylated enterobactin–β-lactam conjugates that harbour the antibiotics ampicillin (Amp) and amoxicillin (Amx), hereafter GlcEnt–Amp/Amx. The GlcEnt scaffolds are based on mono- and diglucosylated Ent where one catechol moiety is functionalized at the C5 position for antibiotic attachment. We demonstrate that GlcEnt–Amp/Amx provide up to 1000-fold enhanced antimicrobial activity against uropathogenic E. coli relative to the parent β-lactams. Moreover, GlcEnt–Amp/Amx based on a diglucosylated Ent (DGE) platform selectively kill uropathogenic E. coli that express the salmochelin receptor IroN in the presence of non-pathogenic E. coli and other bacterial strains that include the commensal microbe Lactobacillus rhamnosus GG. Moreover, GlcEnt–Amp/Amx evade the host-defense protein lipocalin-2, and exhibit low toxicity to mammalian cells. Our work establishes that siderophore–antibiotic conjugates provide a strategy for targeting virulence, narrowing the activity spectrum of antibiotics in clinical use, and achieving selective delivery of antibacterial cargos to pathogenic bacteria on the basis of siderophore receptor expression.
Introduction
Bacterial infections, the rise in antibacterial resistance in hospital and community settings, and the paucity of new antibiotics in the current drug pipeline create a worldwide public health crisis.1,2 New strategies to diagnose and treat bacterial infections as well as counteract the emergence and spread of antibiotic resistance in bacterial pathogens are urgently needed to reduce morbidity and mortality, as well as the economic burden, caused by these infections.3,4 The discovery of narrow-spectrum antibiotics that target select pathogens is one important and necessary facet of this large and complex problem.2,5,6 Pathogen-specific antibiotics that minimally perturb the normal microbial flora are expected to reduce the likelihood of colonisation by pathogenic and drug-resistant microbes during or after antibiotic treatment, and prevent life-threatening secondary infections such as those caused by Clostridium difficile.5,7 Moreover, the availability of narrow-spectrum antibiotics, coupled with rapid diagnostics, is expected to reduce the use of broad-spectrum therapeutics and thereby slow down the evolution of drug resistance.2,5,7 Among current and emerging microbial threats, Gram-negative bacteria, including pathogenic Escherichia coli, Klebsiella pneumoniae, Acinetobacter baumannii, and Salmonella spp., pose a challenge for antibiotic drug discovery.1,2,8 These strains have an outer membrane that serves as a permeability barrier and prevents the cellular entry of many antibiotics.9 In this work, we report a stealth antibiotic delivery strategy that overcomes the outer membrane permeability barrier of Gram-negative E. coli and targets pathogenicity by hijacking the iron import machinery utilised by virulent strains during colonisation in the mammalian host.Iron is an essential nutrient for almost all bacterial pathogens.10,11 Because iron exhibits low solubility in aqueous solutions at physiological pH and enables Fenton chemistry, the levels of “free” iron in mammals (ca. 10−24 M in serum)12 are tightly regulated by homeostatic mechanisms, which include the expression of the iron transport and storage proteins transferrin and ferritin.13 Most bacterial pathogens require micromolar concentrations of iron to colonise and cause disease, and bacterial iron acquisition machineries contribute to virulence.10,14
One way that bacteria scavenge iron in the host environment is to biosynthesize and export siderophores, secondary metabolites that chelate Fe(III) with high affinity.15 The ferric siderophores are recognised and transported into the cell by dedicated uptake machinery. In this work, we consider the catecholate siderophore enterobactin 1 (Ent, Fig. 1a), its glucosylated congeners 2–4 (GlcEnt, Fig. 1a), and the outer membrane receptors for these iron chelators. Ent is biosynthesized by all E. coli and the ferric complex is transported across the outer membrane by the TonB–ExbB–ExbD-dependent outer membrane receptor FepA (Fig. 1b).12 In addition to Ent, many pathogenic E. coli as well as Salmonella spp. biosynthesize salmochelins, C-glucosylated derivatives of Ent (Fig. 1a).16 The iroA gene cluster (iroBCDEN)14,17,18 encodes enzymes that tailor the Ent scaffold to provide the salmochelins (IroBDE), and proteins for salmochelin transport (IroCN). Expression of genes encoded by the iroA locus contributes to virulence by providing Gram-negative pathogens with additional iron acquisition machinery and enabling the pathogens to overcome the host innate immune response.19,20 In the battle against such invading pathogens, the mammalian host mounts a metal-withholding response and secretes lipocalin-2 (Lcn2), a 22 kDa antimicrobial protein that captures ferric Ent.19,21,22 Gram-negative pathogens that utilise salmochelins for iron acquisition readily evade this innate immune mechanism because the salmochelins cannot be sequestered by Lcn2.19
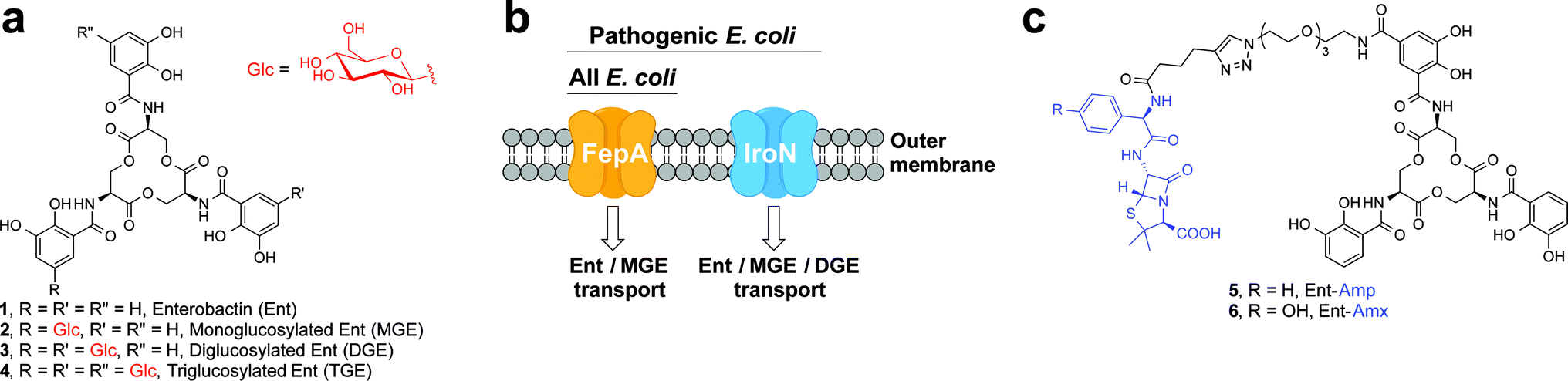 | ||
| Fig. 1 Siderophores and outer membrane siderophore receptors relevant to this work. (a) Chemical structures of enterobactin 1, and its glucosylated derivatives 2–4. (b) A cartoon representation of the outer membrane receptors FepA and IroN considered in this work. (c) Chemical structures of Ent–Amp/Amx 5 and 6. | ||
Because bacteria utilise siderophores to acquire nutrient iron during infection, these molecules, as well as the corresponding biosynthetic and transport machineries, provide opportunities for antibiotic development.10,11,23–27 The notion of using siderophores or siderophore mimics to deliver antibacterial cargo into bacterial cells has garnered attention over several decades.28–44 Our approach to siderophore-based targeting focuses on harnessing native siderophore platforms used by pathogens in the human host for cargo delivery, and we seek to modify these scaffolds in ways that minimally perturb iron binding and receptor recognition. We have designed and utilised a monofunctionalized Ent platform to assemble a variety of Ent–cargo conjugates, and we reported that the Ent uptake machinery (FepABCDG) provides a means to transport small-molecule cargo, including antibiotics in clinical use, into E. coli.44,45 For instance, the Ent–β-lactam conjugates 5 and 6 (Fig. 1c) target and kill E. coli expressing FepA.44 Because all E. coli use Ent for iron acquisition, the Ent–β-lactam conjugates target and kill both non-pathogenic and pathogenic E. coli strains. Some E. coli are commensal microbes, comprising <1% of the total microbial community in the human gut, that biosynthesize vitamin K that is needed by the host.46,47 Thus, the ability to target pathogenic E. coli has utility for minimally perturbing the normal flora. Inspired by prior investigations of native siderophore transport,16,48 we hypothesised that salmochelin–antibiotic conjugates will be specifically recognised by IroN, the outer membrane receptor for the salmochelins, and afford a strategy for overcoming the outer membrane permeability barrier, achieving narrow-spectrum antibacterial activity against pathogenic E. coli, and evading capture by Lcn2.
In this work, we report the design and chemoenzymatic preparation of siderophore–β-lactam conjugates based on salmochelin platforms, and demonstrate targeting of β-lactam antibiotics to pathogenic E. coli that harbour the iroA gene cluster and express IroN. Salmochelin-inspired GlcEnt–β-lactam conjugates based on the diglucosylated Ent (DGE, Fig. 1a) platform provide selective antibacterial activity against pathogenic E. coli and up to 1000-fold enhanced potency relative to the parent β-lactam antibiotics. Moreover, the salmochelin–inspired conjugates remain antibacterial in the presence of Lcn2. These investigations establish a chemoenzymatic route to functionalized salmochelins and provide a new approach for transforming a broad-spectrum antibiotic in clinical use into a narrow-spectrum therapeutic that targets microbial pathogens on the basis of siderophore receptor expression.
Results and discussion
Design and syntheses of GlcEnt–β-lactam conjugates
We present a family of salmochelin–inspired GlcEnt–β-lactam conjugates 7–10 that exhibit ampicillin (Amp) or amoxicillin (Amx) attached to either monoglucosylated Ent (MGE, 2) or diglucosylated Ent (DGE, 3) by a stable polyethylene glycol (PEG3) linker. The design of the GlcEnt–β-lactam conjugates 7–10 builds upon Ent–Amp/Amx 5 and 6 (Fig. 1C).44 These conjugates are based on a monosubstituted Ent platform where one catechol moiety is modified at the C5 position for cargo attachment. We sought to install glucose moieties at the C5 position of one or both of the unfunctionalized catechol rings to afford MGE–Amp/Amx 7 and 8 and DGE–Amp/Amx 9 and 10, respectively. Although the total chemical syntheses of salmochelins have been reported, nine steps are required to achieve the requisite glucosylated 2,3-dihydroxybenzoic acid building block.49 We therefore established a chemoenzymatic approach that employs enzymes involved in salmochelin biosynthesis, which affords the desired glucosylated conjugates and requires only one additional step compared to the reported preparation of Ent–Amp/Amx.IroB and MceC are C-glucosyltransferases that catalyse C-glucosylation of Ent at the C5 positions of the catechol rings. MceC is encoded by the MccE492 gene cluster of K. pneumoniae RYC492, and has 75% amino acid sequence identity with IroB.50 IroB catalyses up to three C-glucosylation events, affording MGE, DGE and TGE as products (Fig. 1a).51 MceC, in contrast, produces only MGE and DGE.52 On the basis of these observations, we hypothesised that both IroB and MceC would accept monofunctionalized Ent as a substrate, providing a preparative route to 7–10. Initial activity assays where either IroB or MceC was incubated with Ent-PEG3-N311, UDP-Glc, and Mg(II) revealed that both enzymes accept Ent-PEG3-N311 as a substrate and afford MGE-PEG3-N312 and DGE-PEG3-N313 as products (Fig. S1 and S2†). Accumulation of 12 was observed in the MceC-catalysed reactions, whereas 13 accumulated in reactions catalysed by IroB. When Ent–Amp/Amx 5 and 6 were employed as substrates, complex product mixtures were obtained. LC/MS analysis of the mixtures revealed the desired products as well as multiple byproducts, including products of β-lactam decomposition. We therefore performed large-scale C-glucosylation reactions employing Ent-PEG3-N311 as a substrate to afford milligram quantities of MGE-PEG3-N312, and DGE-PEG3-N313 (Scheme 1). We subsequently employed copper-catalysed azide/alkyne cycloaddition to install the β-lactam moieties (Scheme 1).44 This route achieved the mono- and diglucosylated conjugates 7–10 in high purity and in yields of 26–59% from 7 following HPLC purification. As expected, the GlcEnt–β-lactam conjugates 7–10 bind iron.53 Each Fe(III) complex exhibits a broad absorption band (ca. 400–700 nm, MeOH) characteristic of ferric Ent and its derivatives (Fig. S3†).44,45,54
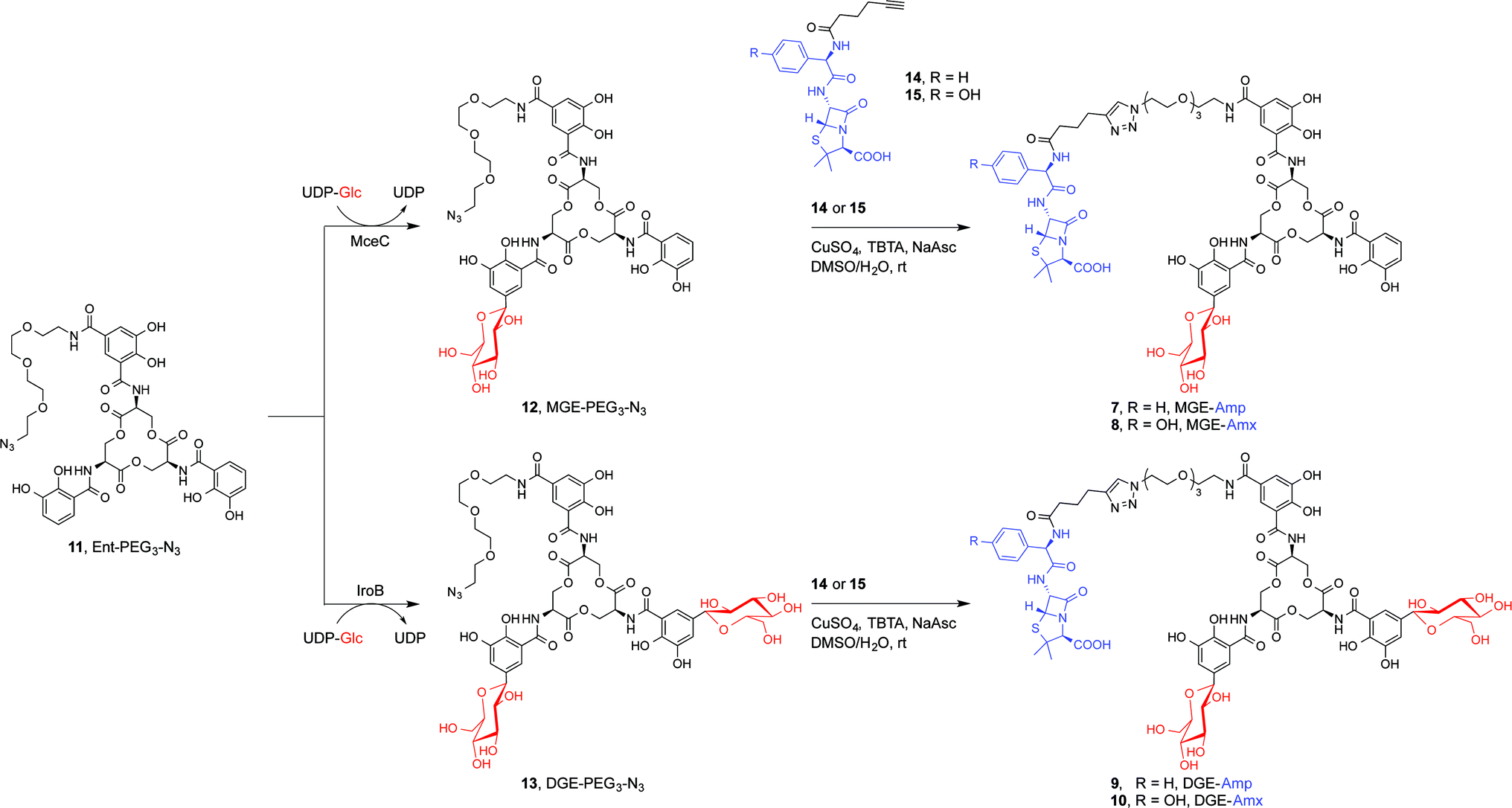 | ||
| Scheme 1 Chemoenzymatic syntheses of GlcEnt–Amp/Amx 7–10. The synthetic route consists of MceC- or IroB-catalysed glucosylation of Ent-PGE3-N311 followed by a copper-catalysed click reaction to achieve the GlcEnt–β-lactam conjugates 7–10. We abbreviate the siderophore family 1–4 (Fig. 1a) as (Glc)Ent and the siderophore–β-lactam conjugates 5–10 as (Glc)Ent–Amp/Amx. | ||
DGE–β-lactam conjugates target pathogenic E. coli expressing IroN
To evaluate whether GlcEnt–Amp/Amx 7–10 target pathogenic E. coli expressing IroN, we compared the antibacterial activities of the parent antibiotics Amp/Amx, Ent–Amp/Amx 5 and 6, and GlcEnt–Amp/Amx 7–10. We selected five E. coli strains on the basis of siderophore receptor expression (Table S1†). E. coli CFT073![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 55 and UTI8956 harbour the iroA gene cluster, biosynthesize and utilise salmochelins for iron acquisition in the host, and cause urinary tract infections.57,58 In contrast, E. coli H9049 is a clinical isolate that does not have the iroA cluster.22E. coli K-1259 and E. coli B60 are non-pathogenic laboratory strains that also lack the iroA cluster. To ascertain the effect of iron limitation on antibacterial activity, we performed antibacterial activity assays in the absence or presence of the metal-ion chelator 2,2′-dipyridyl (DP, 200 μM). This concentration of DP inhibits E. coli growth (Fig. S4†). These assays revealed that DGE–Amp/Amx 9 and 10 target pathogenic E. coli that express IroN.
55 and UTI8956 harbour the iroA gene cluster, biosynthesize and utilise salmochelins for iron acquisition in the host, and cause urinary tract infections.57,58 In contrast, E. coli H9049 is a clinical isolate that does not have the iroA cluster.22E. coli K-1259 and E. coli B60 are non-pathogenic laboratory strains that also lack the iroA cluster. To ascertain the effect of iron limitation on antibacterial activity, we performed antibacterial activity assays in the absence or presence of the metal-ion chelator 2,2′-dipyridyl (DP, 200 μM). This concentration of DP inhibits E. coli growth (Fig. S4†). These assays revealed that DGE–Amp/Amx 9 and 10 target pathogenic E. coli that express IroN.
Amp/Amx exhibit minimum inhibitory concentration (MIC) values of 10 μM against the five E. coli strains (±DP, Fig. 2 and S5–S9†). Under conditions of iron limitation, Ent–Amp/Amx provide 100- to 1000-fold enhanced activity against all five strains (50% MHB, +DP). These results are in agreement with our prior studies of Ent–Amp/Amx killing of E. coli.44 Glucosylation affords strain-dependent antimicrobial activity that correlates with IroN expression (Fig. 2a and b, S5 and S6†). Like Ent–Amp/Amx 5 and 6, GlcEnt–Amp/Amx 7–10 provide 100- and 1000-fold enhanced antimicrobial activity against E. coli UTI89 and E. coli CFT073, respectively (+DP). The susceptibility of E. coli CFT073 to GlcEnt–Amp/Amx remains enhanced in the absence of DP, as observed previously for Ent–Amp/Amx.44 The antibacterial activity of GlcEnt–Amp/Amx 7–10 against E. coli H9049, K-12, and B is attenuated relative to that of Ent–Amp/Amx 5 and 6 (+DP, Fig. 2c–e and S7–S9†). Moreover, for these non-pathogenic strains, the MIC values of (Glc)Ent–Amp/Amx follow the trend Ent–Amp/Amx < MGE–Amp/Amx < DGE–Amp/Amx. The MGE modification provides enhanced potency relative to Amp/Amx because growth reduction (K-12) or complete growth inhibition (H9049 and B) occurs at 1 μM MGE–Amp/Amx (+DP). In contrast, the DGE–β-lactam conjugates exhibit negligible antibacterial activity against the three strains that lack IroN (MIC > 10 μM). The growth medium contains ≈4 μM iron (Table S2†) and we attribute the growth inhibition observed at 10 μM DGE–Amp/Amx to iron deprivation that results from DGE–Amp/Amx sequestering the iron in the growth medium (Table S2†).
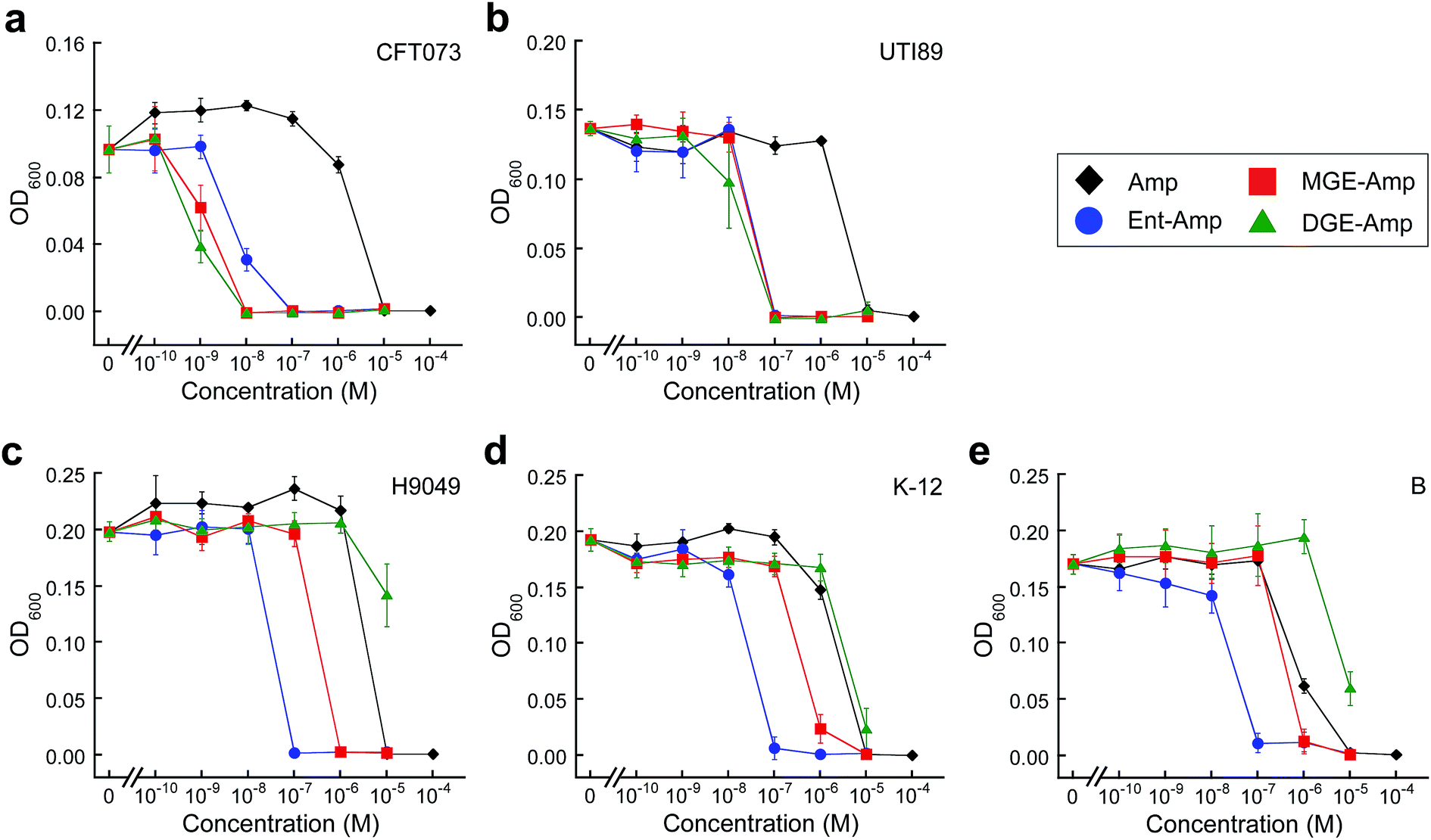 | ||
| Fig. 2 Antibacterial activity of (Glc)Ent–Amp against five E. coli strains. (a)–(e) Antibacterial activity of (Glc)Ent–Amp 5/7/9 against (a) uropathogenic E. coli CFT073, (b) uropathogenic E. coli UTI89, (c) non-pathogenic clinical isolate E. coli H9049, (d) laboratory strain E. coli K-12, (e) laboratory strain E. coli B. All assays were performed in 50% MHB medium supplemented with 200 μM DP (t = 19 h, T = 30 °C) (mean ± standard deviation, n ≥ 3). The data for (Glc)Ent–Amx 6/8/10 and for the assays performed in the absence of DP are presented in Fig. S5–S9.† | ||
In the antibacterial activity assays described above, we treated the bacterial cultures with the apo conjugates and expected that the siderophore moieties chelate iron from the growth medium, allowing for recognition of the ferric–siderophore complexes by FepA and IroN. We previously reported that preloading of Ent–Amp/Amx with Fe(III) prior to antibacterial activity assays against E. coli K-12 had negligible effect on the MIC value.44 Here we report that preloading of MGE–Amp/Amx and DGE–Amp/Amx also has a negligible effect on the growth inhibitory properties (Fig. S10†). This result is expected given that the concentration of iron in the growth medium far exceeds the MIC values obtained for the conjugates under conditions where FepA and IroN are expressed. Lastly, mixtures of unmodified Amp/Amx and (Glc)Ent 1–3 against E. coli CFT073 and UTI89 provide the same MIC values as Amp/Amx alone and confirm that the enhanced antibacterial activity of (Glc)Ent–Amp/Amx 5–10 requires the covalent attachment of β-lactams to the siderophore scaffolds (Fig. S11 and S12†).
Siderophore modification accelerates killing of pathogenic E. coli CFT073
E. coli CFT073 is rapidly killed by conjugates 5–10 (Fig. 3a and S13†). The OD600 value of E. coli CFT073 culture (108 CFU mL−1) treated with 5 μM (Glc)Ent–β-lactam is reduced to almost the baseline value (≈0.04) after 1 h, which corresponds to a ≈2-fold log reduction in CFU mL−1, whereas the change in OD600 and CFU mL−1 for E. coli CFT073 treated with 50 μM Amp/Amx is negligible over this time period. In contrast, siderophore modification has negligible effect on the time-kill kinetics observed for E. coli UTI89 (Fig. 3b and S14†); the (Glc)Ent–β-lactam conjugates provide similar profiles as observed for Amp/Amx. This result is reminiscent of our prior observations for E. coli K-12 where attachment of Ent to Amp/Amx provided only a modest increase in the time-kill kinetics compared to the parent antibiotics.44 The origin of this strain-dependence is unclear and warrants further investigation. Nevertheless, these data show that glucosylation of Ent–Amp/Amx does not alter the time-kill kinetics of Ent–Amp/Amx for either E. coli CFT073 or UTI89, and (Glc)Ent–Amp/Amx 5–10 kill CFT073 more rapidly than UTI89.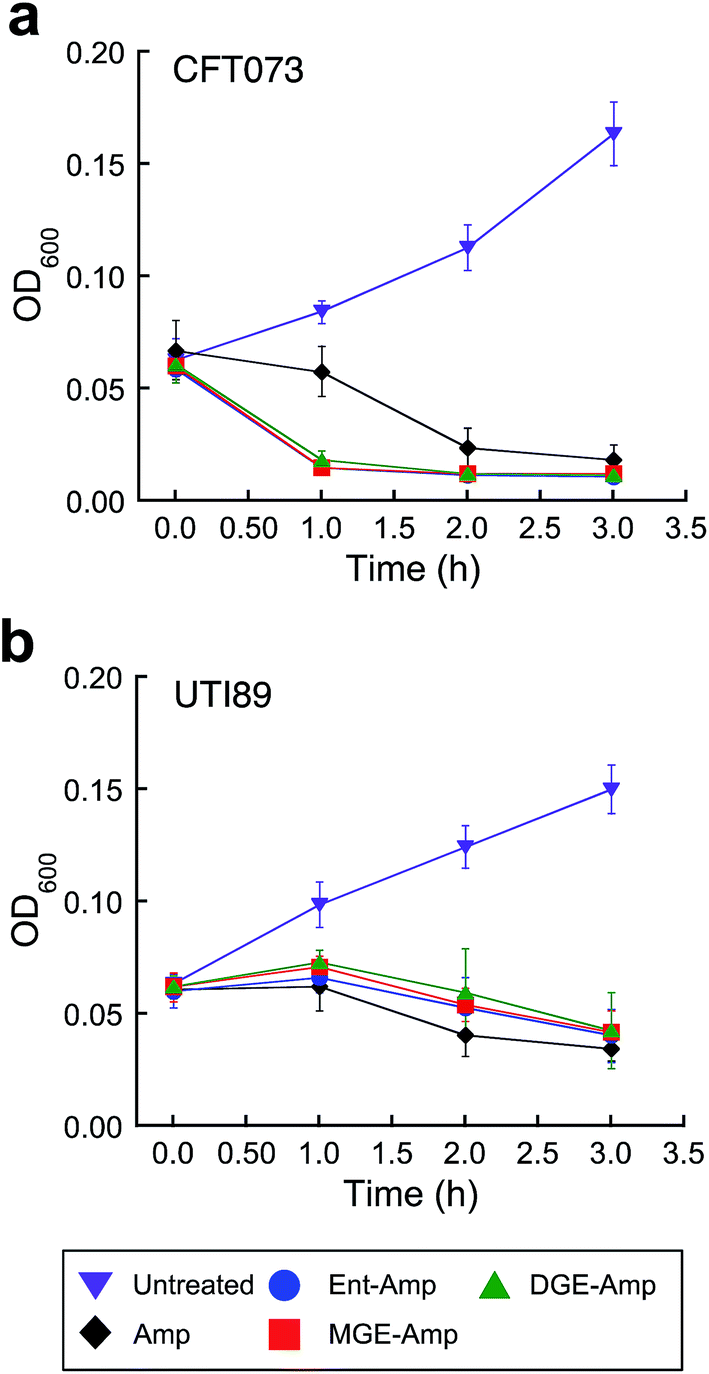 | ||
| Fig. 3 Time-kill kinetics of (Glc)Ent–Amp against E. coli CFT073 and UTI89. (a and b) Time-kill kinetics of (Glc)Ent–Amp 5/7/9 against (a) uropathogenic E. coli CFT073 (≈108 CFU mL−1) treated with 50 μM Amp or 5 μM (Glc)Ent–Amp 5/7/9 and (b) uropathogenic E. coli UIT89 (≈108 CFU mL−1) treated with 50 μM Amp or 50 μM (Glc)Ent–Amp 5/7/9. All assays were performed in 50% MHB medium supplemented with 200 μM DP (T = 37 °C) (mean ± standard deviation, n ≥ 3). The data for (Glc)Ent–Amx 6/8/10 and for the assays performed in the absence of DP are presented in Fig. S13–S14.† | ||
Siderophore competition supports recognition of (Glc)Ent–β-lactam conjugates by IroN
To investigate the interactions between (Glc)Ent–β-lactam conjugates 5–10 and the siderophore receptors FepA and IroN of E. coli CFT073 and UTI89, we performed modified antimicrobial activity assays where varying concentrations (0–10 μM) of (Glc)Ent 1–3 were combined with 100 nM (Glc)Ent–Amp/Amx 5–10 (Fig. 4 and S15†). These mixtures provide a means to probe competition between exogenous native siderophores and the conjugates for receptor recognition because siderophore uptake of the former molecules results in growth promotion whereas the latter afford growth inhibition. The competition assays establish that Ent and MGE attenuate the antibacterial activity of all (Glc)Ent–β-lactam conjugates 5–10 to varying degrees, whereas DGE only inhibits the activity of the glucosylated congeners 7–10. Moreover, DGE fully attenuates DGE–Amp/Amx 9–10 but not MGE–Amp/Amx 7 and 8. These conclusions are drawn from the following observations: (i) a 100-fold molar excess of Ent recovers the growth of E. coli CFT073 treated with Ent/MGE–Amp/Amx 5–8 to levels comparable to that of the untreated control (Fig. 4a and S15a†). In contrast, a 100-fold excess of Ent provides only partial growth recovery of E. coli CFT073 treated with DGE–Amp/Amx 9 and 10. (ii) A 100-fold excess of MGE fully recovers the growth of E. coli CFT073 treated with (Glc)Ent–Amp/Amx 5–10 (Fig. 4b and S15b†). (iii) A 100-fold molar excess of DGE does not recover the growth of E. coli CFT073 treated with Ent–Amp/Amx 5 and 6, whereas it provides partial and complete growth recovery of E. coli CFT073 treated with MGE–Amp/Amx 7 and 8 and DGE–Amp/Amx 9 and 10, respectively (Fig. 4c and S15c†). In total, this work indicates that FepA recognises and delivers Ent/MGE–Amp/Amx 5–8 but not DGE–Amp/Amx 9 and 10, whereas IroN binds and transports all conjugates based on the three siderophore scaffolds. Competition assays employing E. coli UTI89 afford overall trends that are similar to those observed for E. coli CFT073 except that lower concentrations of exogenous siderophores effectively block the antibacterial action of (Glc)Ent–Amp (Fig. 4d–f and S15d–f†).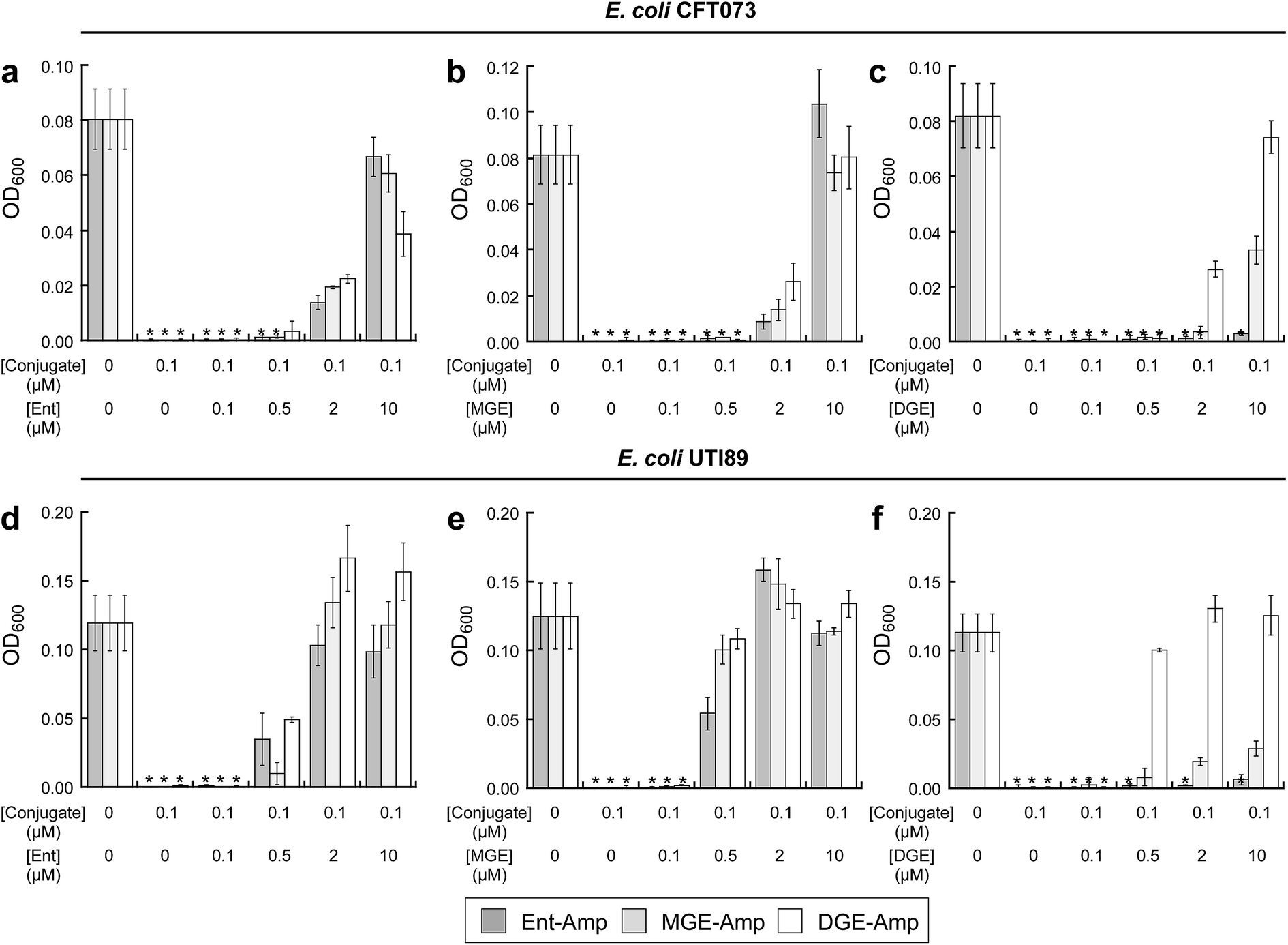 | ||
| Fig. 4 Exogenous (Glc)Ent compete with (Glc)Ent–Amp conjugates for FepA and IroN recognition. (a)–(c) Growth of E. coli CFT073 in the presence of 100 nM (Glc)Ent–Amp 5/7/9 and mixtures of 100 nM (Glc)Ent–Amp 5/7/9 and 1, 5, 20, or 100 equiv of exogenous (a) Ent 1, (b) MGE 2, or (c) DGE 3 in the presence of 200 μM DP. (d)–(f) Growth of E. coli UTI89 in the presence of 100 nM (Glc)Ent–Amp 5/7/9 and mixtures of 100 nM (Glc)Ent–Amp 5/7/9 and 1, 5, 20, or 100 equiv of exogenous (d) Ent 1, (e) MGE 2, or (f) DGE 3 in the presence of 200 μM DP. All assays were performed in 50% MHB medium (t = 19 h, T = 30 °C) (mean ± standard deviation, n = 3). An asterisk indicates OD600 < 0.01. The data for (Glc)Ent–Amx 6/8/10 are presented in Fig. S15.† | ||
GlcEnt–Amp/Amx kill pathogenic E. coli in the presence of other microbes that include non-pathogenic E. coli and commensal Lactobacilli
To further probe the activity spectrum and investigate strain selectivity of GlcEnt–Amp/Amx, we performed mixed-species assays to determine whether these conjugates will selectively kill pathogenic E. coli that express IroN cultured in the presence of other organisms. These assays confirmed that GlcEnt–Amp/Amx 7–10 selectively kill pathogenic E. coli that express IroN in the presence of E. coli strains that do not express this receptor. Treatment of co-cultures of pathogenic E. coli (CFT073 or UTI89, transformed with the chloramphenicol resistance plasmid pSG398) and non-pathogenic E. coli K-12 (transformed with the kanamycin resistance plasmid pET29a) with 100 nM Ent–Amp/Amx 5 and 6 results in complete killing of both strains (Fig. 5a–d and S16a–d†). In contrast, treatment of the co-cultures with 100 nM GlcEnt–Amp/Amx 7–10 affords killing of the uropathogenic E. coli concomitant with E. coli K-12 survival (Fig. 5a–d and S16a–d†). Taken together, these results demonstrate that GlcEnt–β-lactam conjugates 7–10 provide strain-specific targeting of the antibacterial cargo to virulent E. coli that express IroN.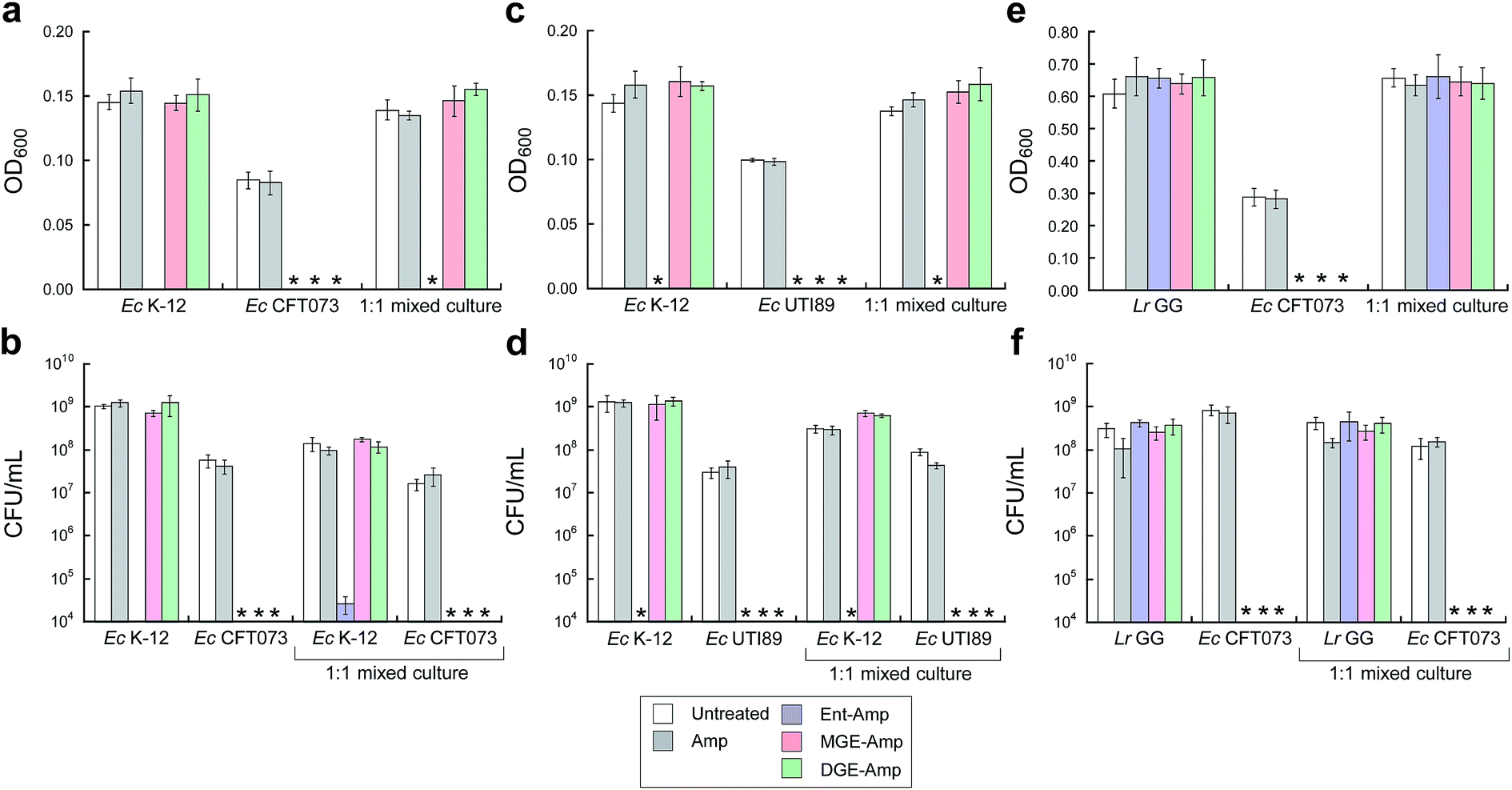 | ||
| Fig. 5 MGE/DGE–Amp selectively kill uropathogenic E. coli in the presence of non-pathogenic E. coli K-12 and the probiotic L. rhamnosus GG. (a and b) Bacterial growth monitored by (a) OD600 and (b) CFU mL−1 for cultures of E. coli K-12 only, CFT073 only, and 1:1 K-12/CFT073 mixtures treated with 100 nM Amp or 100 nM (Glc)Ent–Amp 5/7/9 in the presence of 200 μM DP. (c and d) Bacterial growth monitored by (c) OD600 and (d) CFU mL−1 for cultures of E. coli K-12 only, UTI89 only, and 1:1 K-12/UTI89 mixtures treated with 100 nM Amp or 100 nM (Glc)Ent–Amp 5/7/9 in the presence of 200 μM DP. (e and f) Bacterial growth monitored by (e) OD600 and (f) CFU mL−1 for cultures of L. rhamnosus GG only, E. coli CFT073 only, and 1:1 L. rhamnosus GG/E. coli CFT073 mixtures treated with 1 μM Amp or 1 μM (Glc)Ent–Amp 5/7/9 in the presence of 200 μM DP. All mixed-E. coli antimicrobial assays were performed in 50% MHB medium and all mixed-species antimicrobial assays were conducted in 1:1 MRS/MHB medium (t = 19 h, T = 30 °C) (mean ± standard deviation, n = 3). An asterisk indicates OD600 < 0.01 or no colony formation. The data for (Glc)Ent–Amx 6/8/10 are presented in Fig. S16.† | ||
(Glc)Ent–Amp/Amx 5–10 also target pathogenic E. coli in the presence of commensal microbes. Lactobacilli are Gram-positive commensal bacteria of the human gastrointestinal tract, and are also found in the urinary and genital tracts.61 Some Lactobacilli reduce recurrent urinary tract infections in women.62 Lactobacilli have little-to-no minimal metabolic iron requirement, and do not employ enterobactin or salmochelins for iron acquisition.63,64Lactobacillus rhamnosus GG (ATCC 53103), a human commensal that is considered to be a probiotic, is susceptible to β-lactam antibiotics, and we obtained a MIC value of 10 μM for Amp/Amx against this strain (1:1 MRS/MHB medium, ±DP) (Fig. S17†). In contrast, 10 μM (Glc)Ent–Amp/Amx 5–10 have negligible effect on L. rhamnosus GG growth (Fig. S17†). Treatment of E. coli CFT073 and L. rhamnosus GG co-cultures with (Glc)Ent–Amp/Amx 5–10 affords selective killing of E. coli CFT073 (Fig. 5e and f, S16e and f†).
We previously reported that modification of Amp/Amx with Ent attenuated the activity of the β-lactam against Staphylococcus aureus ATCC 25923.44 In the current work we obtained a similar result with GlcEnt–Amp/Amx, and found that the salmochelin modification lowers the antibacterial activity of Amp/Amx against S. aureus by 10-fold (Fig. S18†). Moreover, treatment of E. coli CFT073 and S. aureus co-cultures with DGE–Amp/Amx 9,10 affords selective killing of E. coli CFT073 (Fig. S19a and b, S20a and b†). Selective killing of E. coli CFT073 co-cultured with Acinetobacter baumannii ATCC 17961 also occurred (Fig. S19c and d, S20c and d, S21†). Substitution of E. coli CFT073 with UTI89 in these assays afforded similar selectivity trends (Fig. S22 and S23†). In total, the mixed-species assays provide support for DGE-based targeting of the antibacterial cargo to IroN-expressing strains.
GlcEnt–Amp/Amx kill E. coli in the presence of lipocalin-2
To ascertain whether GlcEnt–Amp/Amx 7–10 overcome Lcn2 sequestration, in analogy to Lcn2 evasion by the salmochelins,14,19 we conducted antibacterial assays with E. coli CFT073 in the absence or presence of Lcn2 or bovine serum albumin (BSA, control). These assays were conducted in modified M9 medium,65 and 100 nM (Glc)Ent–Amp/Amx 5–10 provide complete growth inhibition of E. coli CFT073 in this medium (Fig. 6). A 10-fold excess of Lcn2 attenuates the antibacterial activity of Ent–Amp/Amx 5 and 6, in agreement with prior work.44 In contrast, Lcn2 has negligible effect on the antimicrobial activity of GlcEnt–Amp/Amx 7–10 against E. coli CFT073 (Fig. 6 and S24†).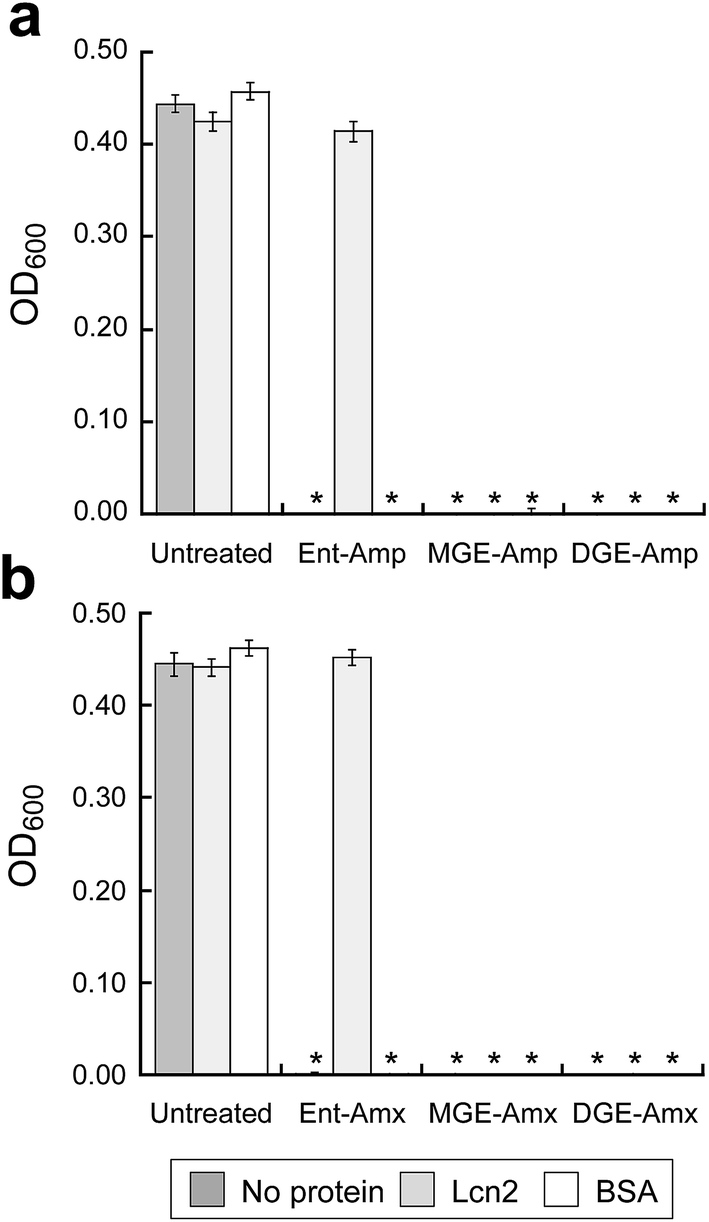 | ||
| Fig. 6 Antibacterial activity of (Glc)Ent–Amp/Amx against E. coli CFT073 in the presence of Lcn2 or bovine serum albumin (BSA). E. coli CFT073 was treated with (a) 100 nM (Glc)Ent–Amp 5/7/9 or (b) 100 nM (Glc)Ent–Amx 6/8/10 in the absence (control) and presence of 1 μM Lcn2 or 1 μM BSA. The assays were conducted in modified M9 medium (t = 24 h, T = 37 °C) (mean ± standard deviation, n = 3). An asterisk indicates OD600 < 0.01. The data for the conjugates preloaded with Fe(III) are presented in Fig. S24.† | ||
GlcEnt–Amp/Amx exhibit low cytotoxicity to human T84 cells
The cytotoxicity of apo and iron-bound GlcEnt–Amp/Amx 7–10 (≤10 μM) against human T84 colon epithelial cells was evaluated by the MTT assay. In all cases, the cell viability was ≥80% of that of the untreated control, indicating that the conjugates exhibit negligible cytotoxicity to T84 cells over a 24 h period (Fig. S25†).Conclusions
In this work, inspired by the siderophore recognition strategies utilised by E. coli for iron acquisition in the host, we report a siderophore-based approach for antibiotic delivery that targets strains that express IroN, a siderophore receptor that contributes to virulence. First, we establish that the tailoring enzymes IroB and MceC can C-glucosylate monofunctionalized Ent and therefore be employed in chemoenzymatic synthesis to afford functionalized salmochelins. Next, we demonstrate that GlcEnt–β-lactam conjugates are recognised by siderophore transport machinery, target IroN, provide ≥100-fold enhanced antibacterial activity against uropathogenic E. coli relative to the parent β-lactams, afford killing of virulent E. coli in the presence of non-pathogenic E. coli and other commensal strains, and overcome the enterobactin–sequestering host-defense protein Lcn2. Our results establish that conjugation of a broad-spectrum antibiotic to a siderophore tunes the activity profile of the parent antibiotic. With the appropriate choice of siderophore, the antibacterial activity spectrum can be modulated to afford species- and strain-specific targeting. In broad terms, targeting pathogens is important for pharmaceutical development, which will ultimately afford treatment options that minimally perturb the commensal microbiota.66,67IroN was first discovered in Salmonella18 and subsequently identified in other Enterobacteriaceae. Our current work focuses on antibiotic delivery to uropathogenic E. coli that harbour the iroA gene cluster, and we expect that this strategy will be applicable to other pathogens that employ salmochelins for iron acquisition. At present, 121 completely sequenced E. coli genomes are available, which include 46 human pathogens. A BLAST search using iroN from E. coli CFT073 afforded hits with ≥99% sequence identity for three uropathogenic E. coli (UTI89, 536, and 83792), adherent invasive E. coli UM146, the meningitis isolate E. coli IHE3034, and a carbapenemase-producing isolate E. coli ECONIH1 (Table S4†). The probiotic E. coli Nissle 1917 and the laboratory reference strain for antimicrobial testing E. coli ATCC 25922 were the only other E. coli revealed as hits. Studies of the distribution of siderophore biosynthetic machinery in E. coli isolated from feces of healthy mammals indicate that ≈20% of the commensal isolates produce salmochelins.68 This observation suggests that one potential limitation of GlcEnt-based antibiotic delivery is that a fraction of commensal E. coli harbour the iroA cluster are susceptible and, conversely, that some pathogenic E. coli do not. Regarding the former possibility, the healthy gut is considered to be a reservoir for E. coli that cause infections of the urinary tract,58,69–71 and the ability to target such pathobionts using siderophores may be advantageous in certain cases. In addition to Salmonella and E. coli, BLAST revealed that the genomes of the human pathogens Shigella dysenteriae 1617 and Sd197, Enterobacter cloacae, Klebsiella pneumoniae, and Enterobacter aerogenes encode iroN (Table S4†). Thus, it will be informative to determine whether DGE also provides targeted antibiotic delivery to these problematic strains.
Our current investigations also provide fundamental insights into siderophore recognition and transport. Prior studies of siderophore uptake in Salmonella revealed that both FepA and IroN recognise and transport Ent.72 Our competition assays employing uropathogenic E. coli are in agreement with this observation, and indicate that both receptors deliver Ent–Amp/Amx 5 and 6 into E. coli. Moreover, our competition data suggest that MGE 2 and MGE–Amp/Amx 7 and 8 are recognised and transported by FepA as well as IroN of E. coli. In contrast, DGE 3 only competes with GlcEnt–Amp/Amx 7–10 and most effectively blocks the activity of DGE–Amp/Amx 9 and 10. These observations support exclusive transport of DGE–Amp/Amx 9 and 10 through IroN. Indeed, prior studies demonstrated that IroN is required for transporting salmochelin extracts isolated from several S. enterica strains,16 and in vitro activity assays reveal that IroB accumulates DGE 3.51
We previously reported that E. coli CFT073 exhibits greater sensitivity to Ent–Amp/Amx 5 and 6 than E. coli UTI89,44 and we observe the same trend with GlcEnt–Amp/Amx 7–10. The physiological origins of this observation remain unclear. One possible explanation may be differences in the siderophore biosynthetic and uptake machineries employed by these two uropathogens. E. coli CFT073 expresses a third catecholate siderophore receptor, IhA,73 whereas E. coli UTI89 biosynthesizes yersiniabactin, a siderophore mainly used by Yersinia spp.74 Alternatively, as-yet unidentified factors may account for these trends, and further studies are warranted to understand these observations.
In closing, this investigation establishes that siderophores and the siderophore uptake machinery employed by virulent bacteria provide a powerful approach for targeting pathogenesis in the context of antibacterial drug discovery. Narrow-spectrum and species-specific antibiotics are needed for treating infections where the causative agent is known and, when coupled with rapid diagnostics, will ultimately reduce the onset of secondary infections and evolution of antibiotic resistance.2,5,7 The current study focuses on targeting broad-spectrum β-lactam antibiotics to pathogenic E. coli on the basis of iron acquisition machinery that is employed by these pathogens during colonisation in the host. We establish that native salmochelins can be used as scaffolds for “Trojan horse” antibiotic delivery to hijack the iron acquisition machinery that contributes to pathogenicity. It will be important to ascertain whether this salmochelin–inspired strategy is applicable to other Gram-negatives, such as Salmonella and K. pneumoniae, which cause human disease and utilise salmochelins for iron acquisition. Leveraging this strategy to target other antibacterial cargos and thereby modulate activity and mitigate off-target effects is another important avenue of future chemical and biological investigation.
Experimental section
Synthetic reagents
Anhydrous dimethyl sulfoxide (DMSO) was purchased from Sigma-Aldrich and used as received. All other chemicals and solvents were purchased from Sigma-Aldrich or Alfa Aesar in the highest available purity and used as received. The syntheses of Ent 1,75 MGE 2,52 DGE 3,51 Ent–Amp 5,44 Ent–Amx 6,44 Ent-PEG3-N311,44 Amp–alkyne 14,44 and Amx–alkyne 1544 are reported elsewhere.
Instrumentation
Analytical and semi-preparative high-performance liquid chromatography (HPLC) were performed using an Agilent 1200 series HPLC system outfitted with a Clipeus reverse-phase C18 column (5 μm pore size, 4.6 × 250 mm; Higgins Analytical, Inc.) at a flow rate of 1 mL min−1 and an Agilent Zorbax reverse-phase C18 column (5 μm pore size, 9.4 × 250 mm) at a flow rate of 4 mL min−1, respectively. The multi-wavelength detector was set to read the absorbance at 220, 280, and 316 (catecholate absorption) nm. HPLC-grade acetonitrile (MeCN) and trifluoroacetic acid (TFA) were purchased from EMD and Alfa Aesar, respectively. For HPLC analyses, solvent A was 0.1% TFA/H2O and solvent B was 0.1% TFA/MeCN, unless stated otherwise. The HPLC solvents were prepared with HPLC-grade MeCN and TFA, and Milli-Q water (18.2 mΩ cm), and filtered through a 0.2 μm filter before use. For analytical HPLC to evaluate conjugate purity, the entire portion of each HPLC-purified compound was dissolved in a mixture of 1:1 MeCN/H2O and an aliquot was taken for HPLC analysis. The remaining solution was subsequently lyophilized.
High-resolution mass spectrometry was performed by using an Agilent LC-MS system comprised of an Agilent 1260 series LC system outfitted with an Agilent Poroshell 120 EC-C18 column (2.7 μm pore size) and an Agilent 6230 TOF system housing an Agilent Jetstream ESI source. For all LC-MS analyses, solvent A was 0.1% formic acid/H2O and solvent B was 0.1% formic acid/MeCN (LC-MS grade, Sigma-Aldrich). The samples were analysed using a solvent gradient of 5–95% B over 10 min with a flow rate of 0.4 mL min−1. The MS profiles were analysed and deconvoluted by using Agilent Technologies Quantitative Analysis 2009 software version B.03.02.
Optical absorption spectra were recorded on a Beckman Coulter DU800 spectrophotometer (1 cm quartz cuvettes, Starna). A BioTek Synergy HT plate reader was used to record absorbance at 600 nm (OD600) for antimicrobial activity assays and absorbance at 550 nm for cytotoxicity assays.
Enzymatic activity assays for IroB and MceC
The enzymes MceC and IroB were overexpressed as N-terminal His6-fusion proteins in E. coli BL21(DE3) and purified as reported.51,52 To a 405 μL solution containing Ent-PEG3-N311 (100 μM), uridine diphosphoglucose (UDP-Glc, 3 mM), and MgCl2 (5 mM) prepared in 75 mM Tris–HCl buffer, pH 8.0, MceC (10 μM, 45 μL) or IroB (10 μM, 45 μL) was added to afford a final enzyme concentration of 1 μM. The reaction was incubated at room temperature and an aliquot (100 μL) was quenched by adding 10 μL of 6% TFA (aq) after 0, 15, 30, 60 min. The quenched reaction aliquots were immediately vortexed, centrifuged (13000 rpm × 10 min, 4 °C), and analysed by HPLC (0–100% B over 30 min, 1 mL min−1). The results are shown in Fig. S1 and S2.†
Synthesis of MGE/DGE-PEG3-N3 (12 and 13)
A 6.3 mL solution containing Ent-PEG3-N311 (500 μM), uridine diphosphoglucose (UDP-Glc, 3 mM), and MgCl2 (5 mM) was prepared in 75 mM Tris–HCl buffer, pH 8.0 and divided into seven 900 μL aliquots. MceC (50 μM, 100 μL) or IroB (50 μM, 100 μL) was added to each aliquot to afford a final enzyme concentration of 5 μM. The 1 mL reactions were incubated at room temperature and quenched by addition of 100 μL of 6% TFA (aq) after 15 min (MceC reaction) or 2 h (IroB reaction). The quenched reaction aliquots were immediately vortexed, combined, and lyophilized to dryness. The resulting powder was dissolved in 3 mL of 1:1 MeCN/water and centrifuged (13000 rpm × 10 min, 4 °C). MGE-PEG3-N312 and DGE-PEG3-N313 were purified from the supernatants of MceC- and IroB-catalysed reactions, respectively, by using semi-preparative HPLC (20–45% B over 8.5 min, 4 mL min−1). Both compounds were obtained as white powders (MGE-PEG3-N312, 0.66 mg, 41% from MceC-catalyzed reaction; DGE-PEG3-N313, 0.85 mg, 45% from IroB-catalysed reaction). HRMS (ESI): MGE-PEG3-N312, [M + H]+m/z calcd. 1076.3215, found 1076.3214; DGE-PEG3-N313, [M + H]+m/z calcd. 1238.3743, found 1238.3744. The analytical HPLC traces of the purified compounds are reported in Fig. S26 and S27.†
Synthesis of MGE–Amp (7)
Amp–alkyne 14 (50 μL of a 50 mM solution in DMSO, 2.5 μmol) and MGE-PEG3-N312 (73 μL of an 11.3 mM solution in DMSO, 0.825 μmol) were combined and 100 μL of DMSO was added. An aliquot of aqueous CuSO4 (50 μL of a 90 mM solution in water, 4.5 μmol) and tris[(1-benzyl-1H-1,2,3-triazol-4-yl)methyl]amine (TBTA, 100 μL of a 50 mM solution in DMSO, 5 μmol) were combined to give a blue-green solution, to which sodium ascorbate (NaAsc, 100 μL of a 180 mM solution in water, 18.0 μmol) was added. This solution became light yellow and was immediately added to the alkyne/azide solution. The reaction was gently mixed on a bench-top rotator for 2 h at room temperature and conjugate 7 was purified by semi-preparative HPLC (20% B for 5 min and 20–50% B over 11 min, 4 mL min−1; 0.005% TFA was used in solvents A and B because of the acid-sensitive β-lactam moiety). Conjugate 7 was obtained as white powder (0.75 mg, 59%). HRMS (ESI): [M + H]+m/z calcd, 1519.4730; found, 1519.4639. The analytical HPLC trace of the purified product is reported in Fig. S28.†Synthesis of MGE–Amx (8)
As described for MGE–Amp with the exception that Amx–alkyne 15 was used instead of Amp–alkyne 14. Conjugate 8 was purified by semi-preparative HPLC (20% B for 5 min and 20–42% B over 11 min, 4 mL min−1) and obtained as white powder (0.49 mg, 31%). HRMS (ESI): [M + H]+m/z calcd, 1535.4679; found, 1535.4685. The analytical HPLC trace of the purified product is reported in Fig. S29.†Synthesis of DGE–Amp (9)
As described for MGE–Amp with the exception that DGE-PEG3-N313 was used instead of MGE-PEG3-N312. Conjugate 9 was purified by semi-preparative HPLC (0% B for 5 min and 0–50% B over 13 min, 4 mL min−1) and obtained as white powder (0.67 mg, 48%). HRMS (ESI): [M + Na]+m/z calcd, 1703.5077; found, 1703.5069. The analytical HPLC trace of the purified product is reported in Fig. S30.†Synthesis of DGE–Amx (10)
As described for MGE–Amp with the exception that Amx–alkyne 15 and DGE-PEG3-N313 were used instead of Amp–alkyne 14 and MGE-PEG3-N312. Conjugate 10 was purified by semi-preparative HPLC (0% B for 5 min and 0–50% B over 13 min, 4 mL min−1) and obtained as white powder (0.36 mg, 26%). HRMS (ESI): [M + H]+m/z calcd, 1697.5207; found, 1697.5235. The analytical HPLC trace of the purified product is reported in Fig. S31.†Storage and handling of siderophores and siderophore–antibiotic conjugates
All (Glc)Ent 1–3 and siderophore–antibiotic conjugates 5–10 were stored as DMSO stock solutions at −20 °C. The stock solution concentrations for (Glc)Ent–Amp/Amx 5–10 ranged from 2 to 5 mM. These values were determined by diluting the DMSO stock solution in MeOH and using the reported extinction coefficient for enterobactin in MeOH (316 nm, 9500 M−1 cm−1).76 To minimize multiple freeze–thaw cycles, the resulting solutions were divided into 50 μL aliquots and stored at −20 °C. The β-lactam moieties and enterobactin trilactone are susceptible to hydrolysis, and aliquots were routinely analysed by HPLC to confirm the integrity of the samples.General microbiology materials and methods
Information pertaining to all bacterial strains used in this study is listed in Table S1.† Freezer stocks of all Escherichia coli strains (E. coli K-12, B, H9049, CFT073, and UTI89), Staphylcoccus aureus ATCC 25923, and Acinetobacter baumannii ATCC 17961 were prepared from single colonies in 25% glycerol/Luria Broth (LB) medium. Freezer stocks of Lactobacillus rhamnosus GG ATCC 53103 were prepared from single colonies in 25% glycerol/de Man, Rogosa, and Sharpe (MRS) medium.LB, MRS, 5× M9 minimal medium and agar were purchased from BD. Mueller Hinton Broth (MHB) was purchased from Fluka. Recombinant human Lcn2 was purchased from R&D System (Minneapolis, MN). The iron chelator 2,2′-dipyridyl (DP) was purchased from Sigma-Aldrich. All growth medium and Milli-Q water used for bacterial cultures or for preparing solutions of the enterobactin–antibiotic conjugates were sterilised by using an autoclave. A DP stock solution (200 mM) was prepared in DMSO and used in the bacteria growth assays requiring iron-depleted conditions. Working dilutions of the siderophore and siderophore–antibiotic conjugate stock solutions were prepared in 10% DMSO/H2O. For all assays, the final cultures contained 1% v/v DMSO. Sterile polypropylene culture tubes and sterile polystyrene 96-well plates used for culturing were purchased from VWR and Corning Incorporated, respectively. The optical density at 600 nm (OD600) was recorded on a Beckman Coulter DU800 spectrophotometer or by using a BioTek Synergy HT plate reader.
Growth studies of E. coli in the presence of DP
Overnight cultures of E. coli were prepared by inoculating 5 mL of Luria Broth (LB) medium with bacterial freezer stocks. The cultures were incubated at 37 °C in a tabletop incubator with shaking at 150 rpm for 16–18 h. The overnight culture was diluted 1:100 into 5 mL of fresh LB medium containing DP (200 μM) and incubated at 37 °C with shaking at 150 rpm until OD600 reached 0.6. The cultures were subsequently diluted to an OD600 value of 0.001 using 50% MHB medium (11.5 g L−1). A 90 μL aliquot of the diluted culture was combined with a 10 μL aliquot of a 10× solution of DP (0, 0.25, 0.5, 1, 2, 4, and 8 mM) in a 96-well plate, which was wrapped in Parafilm and incubated at 30 °C with shaking at 150 rpm. Bacterial growth was determined at t = 0, 2, 4, 6, 8, 10, and 20 h by measuring the OD600 using a BioTek Synergy HT plate reader. Each well condition was prepared in duplicate and at least three independent replicates were conducted on different days and using two different DP stock solutions. The resulting mean OD600 values are reported and the error bars represent the standard deviation.
General procedure for antimicrobial activity assays
Overnight cultures of E. coli, S. aureus, and A. baumannii were prepared by inoculating 5 mL of Luria Broth (LB) medium with bacterial freezer stocks. The cultures were incubated at 37 °C in a tabletop incubator with shaking at 150 rpm for 16–18 h. The overnight culture was diluted 1:100 into 5 mL of fresh LB medium containing DP (200 μM) and incubated at 37 °C with shaking at 150 rpm until OD600 reached 0.6. The cultures were subsequently diluted to an OD600 value of 0.001 using 50% MHB medium (11.5 g L−1) with or without DP (200 μM). A 90 μL aliquot of the diluted culture was combined with a 10 μL aliquot of a 10× solution of Amp/Amx or (Glc)Ent–Amp/Amx 5–10 in a 96-well plate, which was wrapped in Parafilm and incubated at 30 °C with shaking at 150 rpm for 19 h. Bacterial growth was determined by measuring the OD600 using a BioTek Synergy HT plate reader. Each well condition was prepared in duplicate and at least three independent replicates were conducted on different days and using different synthetic batches of each conjugate. The resulting mean OD600 values are reported and the error bars represent the standard deviation.
For L. rhamnosus GG ATCC 53103, the bacterial culture was grown in MRS medium overnight. The resulting culture was diluted 1:50 into 5 mL of fresh MRS medium containing DP (200 μM) and incubated at 37 °C with shaking at 150 rpm until OD600 reached 1.0. The culture was subsequently diluted to an OD600 value of 0.004 in 1:1 MRS/MHB medium with or without DP (200 μM). The antibacterial activity assays were performed as described above for E. coli.
Time-kill kinetic assays
A 5 mL overnight culture of E. coli CFT073 or UTI89 grown in LB medium was diluted 1:100 into 5 mL of fresh LB medium with 200 μM DP and incubated at 37 °C with shaking at 150 rpm until OD600 reached ≈0.3. The culture was centrifuged (3000 rpm × 10 min, rt) and the resulting pellet was resuspended in 50% MHB and centrifuged (3000 rpm × 10 min, rt). The resulting pellet was resuspended in 50% MHB with or without DP (200 μM) and the OD600 was adjusted to 0.3. A 90 μL aliquot of the resulting culture was combined with a 10 μL aliquot of a 10× solution of Amp/Amx or (Glc)Ent–Amp/Amx 5–10 in a 96-well plate, which was wrapped in Parafilm and incubated at 37 °C with shaking at 150 rpm. The OD600 values were recorded at t = 0, 1, 2, and 3 h. In a parallel experiment, a 10 μL aliquot of the culture was taken at t = 0, 1, 2, and 3 h and serially diluted by using sterile phosphate-buffered saline (PBS) and plated on LB agar to obtain colony forming units (CFU mL−1). Each well condition was repeated at least three times independently on different days. The resulting mean OD600 or CFU mL−1 is reported and the error bars are the standard deviation.
Antimicrobial activity assays in the presence of unmodified (Glc)Ent
These assays were performed following the general procedure described above except that varying concentrations of Ent, MGE, or DGE were mixed with Ent–Amp/Amx 5 and 6, MGE–Amp/Amx 7–8, or DGE–Amp/Amx 9 and 10. Ent was synthesized following a literature procedure,75 MGE 2 and DGE 3 were prepared from Ent using MceC and IroB as described for MGE-PEG3-N312 and DGE-PEG3-N313. Stock solutions of (Glc)Ent 1–3 were prepared in DMSO and stored at −20 °C.Mixed-E. coli assays
The pET29a plasmid (kanamycin resistance) was transformed into E. coli K-12, and the pHSG398 plasmid (chloramphenicol resistance) was transformed into E. coli CFT073 and UTI89, by electroporation. Overnight cultures of the bacterial strains were prepared by inoculating 5 mL of LB medium containing the appropriate antibiotic (kanamycin, 50 μg mL−1; chloramphenicol, 34 μg mL−1) with bacterial freezer stocks, and the cultures were incubated at 37 °C in a tabletop incubator shaker set at 150 rpm for 16–18 h. Each overnight culture was diluted 1:100 into 5 mL of fresh LB medium containing 200 μM DP, but no antibiotics, and incubated at 37 °C with shaking at 150 rpm until OD600 reached 0.6. The cultures were diluted to an OD600 value of 0.001 in 50% MHB separately or in a 1:1 mixture (106 CFU mL−1 for each strain), with or without 200 μM DP. No antibiotic marker was included in these cultures. Aliquots of these cultures were serially diluted by using sterile PBS and plated on LB agar plates with or without corresponding antibiotic to confirm the CFU of the starter cultures. A 90 μL aliquot of each culture was combined with a 10 μL aliquot of a 1 μM solution of Amp/Amx or (Glc)Ent–Amp/Amx 5–10 in a 96-well plate. The plate was then wrapped in Parafilm and incubated at 30 °C with shaking at 150 rpm for 19 h. Bacterial growth was evaluated by measuring OD600 as well as counting colonies formed on LB agar with or without kanamycin/chloramphenicol after serial dilution with sterile PBS. Each well condition was repeated at least three times independently on different days. The resulting mean OD600 and CFU mL−1 values are reported and the error bars are the standard deviation.
Mixed-species assays
These assays were performed following the mixed-E. coli assay procedure except that E. coli CFT073 and L. rhamnosus GG ATCC 53103 were used. A 5 mL culture of E. coli CFT073 or L. rhamnosus GG was grown for 16–18 h in LB or MRS medium, respectively. The overnight culture was diluted 1:100 (E. coli) or 1:50 (L. rhamnosus GG) into 5 mL of fresh LB or MRS medium with 200 μM DP and incubated at 37 °C with shaking at 150 rpm until OD600 reached 0.6 (E. coli) or 1.0 (L. rhamnosus GG). The cultures were diluted to an OD600 value of 0.001 (E. coli) or 0.004 (L. rhamnosus GG) in 1:1 MRS/MHB containing 200 μM DP separately or in a 1:1 mixture (106 CFU mL−1 for each strain). Aliquots of these cultures were serially diluted by using sterile PBS and plated on LB and MRS agar plates to confirm the CFU of the starter culture. A 90 μL aliquot of each culture was combined with a 10 μL aliquot of a 10 μM solution of Amp/Amx or (Glc)Ent–Amp/Amx 5–10 in a 96-well plate, which was wrapped in Parafilm and incubated at 30 °C with shaking at 150 rpm for 19 h. Bacterial growth was assayed by both measuring OD600 and counting colonies formed on LB and MRS agar plates after serial dilution with sterile PBS. Each well condition was repeated at least three times independently on different days. The resulting mean OD600 and CFU mL−1 values are reported and the error bars are the standard deviation. Comment: E. coli CFT073 forms colonies more quickly than L. rhamnosus GG on LB agar plates, whereas L. rhamnosus GG colonies appear more quickly than those of E. coli CFT073 on MRS agar plates, and these behaviours allow for each strain to be monitored independently over a 24 h period.
The assays were also performed by co-culturing E. coli CFT073 or UTI89 with S. aureus ATCC 25923 or A. baumannii ATCC 17961. A 5 mL culture of each individual bacterial strain was grown for 16–18 h in LB. The overnight culture was diluted 1:100 into 5 mL of fresh LB with 200 μM DP and incubated at 37 °C with shaking at 150 rpm until OD600 reached 0.6. The cultures were diluted to an OD600 value of 0.001 in 50% MHB containing 200 μM DP separately or in a 1:1 mixture (106 CFU mL−1 for each strain). A 90 μL aliquot of each culture was combined with a 10 μL aliquot of a 10 μM solution of Amp/Amx or (Glc)Ent–Amp/Amx 5–10 in a 96-well plate, which was wrapped in Parafilm and incubated at 30 °C with shaking at 150 rpm for 19 h. Bacterial growth was assayed by both measuring OD600 and counting colonies formed on HardyCHROM UTI plates after serial dilution with sterile PBS. Plating E. coli strains on these plates results in pink colonies, whereas S. aureus and A. baumannii provide white colonies. Each well condition was repeated at least three times independently on different days. The resulting mean OD600 and CFU mL−1 values are reported and the error bars are the standard deviation.
Antimicrobial activity assays in the presence of lipocalin 2
Cultures of E. coli CFT073 were grown in modified M9 minimal medium65 (Na2HPO4 6.8 g L−1, KH2PO4 3 g L−1, NaCl 0.5 g L−1, NH4Cl 1 g L−1, 0.4% glucose, 2 mM MgSO4, 0.1 mM CaCl2, 0.2% casein amino acids, and 16.5 μg mL−1 of thiamine) for 16–18 h. The overnight culture grew to saturation and was diluted 1:100 into 5 mL of fresh modified M9 minimal medium and incubated at 37 °C with shaking at 150 rpm until OD600 reached 0.6. The OD600 of the culture was adjusted to 0.001, and the culture was further diluted 1:100 with the M9 medium in two steps (1:10 × 1:10). The corresponding CFU was determined to be ≈104 CFU mL−1 by plating on LB agar plates. Lipocalin 2 (Lcn2, R&D Systems) was diluted into PBS, pH 7.4 to a concentration of 20 μM and frozen at −20 °C until use. Bovine serum albumin (BSA, Sigma-Aldrich) was prepared in PBS, pH 7.4 to achieve a concentration of 20 μM. A 90 μL aliquot of the diluted culture was combined with a 5 μL aliquot of a 20× solution of (Glc)Ent–Amp/Amx 5–10 and a 5 μL aliquot of Lcn2 or BSA in a 96-well plate, which was wrapped in Parafilm and incubated at 37 °C with shaking at 150 rpm for 24 h. Bacterial growth was determined by OD600. Each well condition was repeated at least three times independently on different days. The resulting mean OD600 is reported and the error bars are the standard deviation.
Cytotoxicity assays
The human colon epithelial T84 cell line was purchased from ATCC and cultured in 1:1 DMEM/F12 medium with 10% fetal bovine serum, and 1% penicillin and streptomycin (v/v, ATCC). The cells were grown to approximately 95% confluency and treated with 3 mL of trypsin-EDTA (Corning). A 12 mL portion of fresh medium was added to the detached cells, and the T84 cell suspension was centrifuged (600 rpm × 5 min, 37 °C). The supernatant was discarded and the cell pellet was resuspended in 6 mL of the fresh culture medium. The concentration of cells was quantified by using a manual hemocytometer (VWR International) and adjusted to 1 × 105 cells per mL. A 90 μL aliquot of T84 cells were then added to 96-well plates and incubated at 37 °C and 5% CO2 for 24 h. Stock solutions (10×) of Amp/Amx or (Glc)Ent–Amp/Amx 5–10 were prepared in sterile-filtered 10% DMSO/H2O and 10 μL of each solution was added to the appropriate well. The plate was incubated at 37 °C and 5% CO2 for another 24 h. 3-[4,5-Dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide (MTT, Alfa Aesar) was dissolved in sterile PBS and the concentration was adjusted to 5 mg mL−1. The resulting yellow solution was filtered through a 0.2 μm filter and a 20 μL aliquot of the resulting MTT solution was added to each well. The plate was incubated at 37 °C and 5% CO2 for 4 h and the supernatant was removed from each well. DMSO (100 μL) was added to each well and the absorbance at 550 nm was recorded by using a plate reader. Blank readings were recorded on wells that contained only the medium. The assay was repeated in triplicate on different days, and the mean and standard deviation are reported.
Competing financial interests
A patent application covering GlcEnt–Amp/Amx has been filed.Acknowledgements
We gratefully acknowledge financial support from the Pacific Southwest Research Center of Excellence for Biodefense and Emerging Infectious Disease, the Searle Scholars Program (Kinship Foundation), the Department of Chemistry at MIT, and a Royal Thai Government Fellowship (P.C.). We thank Prof. Lynette Cegelski for providing E. coli UTI89 and Prof. Christopher T. Walsh for providing E. coli H9049.References
- Antimicrobial resistance: global report on surveillance 2014, World Heath Organization, Geneva, 2014 Search PubMed.
- Report to the president on combating antibiotic resistance, President's Council of Advisors on Science and Technology, 2014 Search PubMed.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089–1093 CrossRef CAS PubMed.
- D. J. Payne, M. N. Gwynn, D. J. Holmes and D. L. Pompliano, Nat. Rev. Drug Discovery, 2007, 6, 29–40 CrossRef CAS PubMed.
- A. E. Clatworthy, E. Pierson and D. T. Hung, Nat. Chem. Biol., 2007, 3, 541–548 CrossRef CAS PubMed.
- K. Lewis, Nat. Rev. Drug Discovery, 2013, 12, 371–387 CrossRef CAS PubMed.
- A. K. Barczak and D. T. Hung, Curr. Opin. Microbiol., 2009, 12, 490–496 CrossRef CAS PubMed.
- Antibiotic resistance threats in the United States, 2013, U.S. Department of Health and Human Services: Centers for Disease Control and Prevention (CDC), 2013 Search PubMed.
- N. Ruiz, D. Kahne and T. J. Silhavy, Nat. Rev. Microbiol., 2006, 4, 57–66 CrossRef PubMed.
- J. E. Cassat and E. P. Skaar, Cell Host Microbe, 2013, 13, 509–519 CAS.
- J. M. N. Roosenberg II, Y.-M. Lin, Y. Lu and M. J. Miller, Curr. Med. Chem., 2000, 7, 159–197 CrossRef.
- K. N. Raymond, E. A. Dertz and S. S. Kim, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3584–3588 CrossRef CAS PubMed.
- N. C. Andrews and P. J. Schmidt, Annu. Rev. Physiol., 2007, 69, 69–85 CrossRef CAS PubMed.
- M. A. Fischbach, H. Lin, D. R. Liu and C. T. Walsh, Nat. Chem. Biol., 2006, 2, 132–138 CrossRef CAS PubMed.
- R. C. Hider and X. Kong, Nat. Prod. Rep., 2010, 27, 637–657 RSC.
- K. Hantke, G. Nicholson, W. Rabsch and G. Winkelmann, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3677–3682 CrossRef CAS PubMed.
- A. J. Bäumler, R. M. Tsolis, A. W. M. van der Velden, I. Stojiljkovic, S. Anic and F. Heffron, Gene, 1996, 183, 207–213 CrossRef.
- A. J. Bäumler, T. L. Norris, T. Lasco, W. Voigt, R. Reissbrodt, W. Rabsch and F. Heffron, J. Bacteriol., 1998, 180, 1446–1453 Search PubMed.
- M. A. Fischbach, H. Lin, L. Zhou, Y. Yu, R. J. Abergel, D. R. Liu, K. N. Raymond, B. L. Wanner, R. K. Strong, C. T. Walsh, A. Aderem and K. D. Smith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16502–16507 CrossRef CAS PubMed.
- M. Raffatellu, M. D. George, Y. Akiyama, M. J. Hornsby, S.-P. Nuccio, T. A. Paixao, B. P. Butler, H. Chu, R. L. Santos, T. Berger, T. W. Mak, R. M. Tsolis, C. L. Bevins, J. V. Solnick, S. Dandekar and A. J. Bäumler, Cell Host Microbe, 2009, 5, 476–486 CAS.
- D. H. Goetz, M. A. Holmes, N. Borregaard, M. E. Bluhm, K. N. Raymond and R. K. Strong, Mol. Cell, 2002, 10, 1033–1043 CrossRef CAS PubMed.
- T. H. Flo, K. D. Smith, S. Sato, D. J. Rodriguez, M. A. Holmes, R. K. Strong, S. Akira and A. Aderem, Nature, 2004, 432, 917–921 CrossRef CAS PubMed.
- C. Ji, R. E. Juárez-Hernández and M. J. Miller, Future Med. Chem., 2012, 4, 297–313 CrossRef CAS PubMed.
- M. J. Miller, H. Zhu, Y. Xu, C. Wu, A. J. Walz, A. Vergne, J. M. Roosenberg, G. Moraski, A. A. Minnick, J. McKee-Dolence, J. Hu, K. Fennell, E. K. Dolence, L. Dong, S. Franzblau, F. Malouin and U. Möllmann, Biometals, 2009, 22, 61–75 CrossRef CAS PubMed.
- G. L. A. Mislin and I. J. Schalk, Metallomics, 2014, 6, 408–420 RSC.
- H. Budzikiewicz, Curr. Top. Med. Chem., 2001, 1, 73–82 CrossRef CAS PubMed.
- V. Braun, Drug Resist. Updates, 1999, 2, 363–369 CrossRef CAS PubMed.
- N. Ohi, B. Aoki, K. Moro, T. Kuroki, N. Sugimura, T. Noto, T. Nehashi, M. Matsumoto, H. Okazaki and I. Matsunaga, J. Antibiot., 1986, 39, 242–250 CrossRef CAS PubMed.
- P. Silley, J. W. Griffiths, D. Monsey and A. M. Harris, Antimicrob. Agents Chemother., 1990, 34, 1806–1808 CrossRef CAS PubMed.
- T. Hashizume, M. Sanada, S. Nakagawa and N. Tanaka, J. Antibiot., 1990, 43, 1617–1620 CrossRef CAS PubMed.
- H. Nikaido and E. Y. Rosenberg, J. Bacteriol., 1990, 172, 1361–1367 CAS.
- J. A. McKee, S. K. Sharma and M. J. Miller, Bioconjugate Chem., 1991, 2, 281–291 CrossRef CAS PubMed.
- N. A. C. Curtis, R. L. Eisenstadt, S. J. East, R. J. Cornford, L. A. Walker and A. J. White, Antimicrob. Agents Chemother., 1988, 32, 1879–1886 CrossRef CAS PubMed.
- M. S. Diarra, M. C. Lavoie, M. Jacques, I. Darwish, E. K. Dolence, J. A. Dolence, A. Ghosh, M. Ghosh, M. J. Miller and F. Malouin, Antimicrob. Agents Chemother., 1996, 40, 2610–2617 CAS.
- S. Wittmann, M. Schnabelrauch, I. Scherlitz-Hofmann, U. Möllmann, D. Ankel-Fuchs and L. Heinisch, Bioorg. Med. Chem., 2002, 10, 1659–1670 CrossRef CAS PubMed.
- U. Möllmann, L. Heinisch, A. Bauernfeind, T. Köhler and D. Ankel-Fuchs, Biometals, 2009, 22, 615–624 CrossRef PubMed.
- S. Yoganathan, C. S. Sit and J. C. Vederas, Org. Biomol. Chem., 2011, 9, 2133–2141 CAS.
- M. J. Miller, A. J. Walz, H. Zhu, C. Wu, G. Moraski, U. Möllmann, E. M. Tristani, A. L. Crumbliss, M. T. Ferdig, L. Checkley, R. L. Edwards and H. I. Boshoff, J. Am. Chem. Soc., 2011, 133, 2076–2079 CrossRef CAS PubMed.
- C. Ji, P. A. Miller and M. J. Miller, J. Am. Chem. Soc., 2012, 134, 9898–9901 CrossRef CAS PubMed.
- A. P. Tomaras, J. L. Crandon, C. J. McPherson, M. A. Banevicius, S. M. Finegan, R. L. Irvine, M. F. Brown, J. P. O'Donnell and D. P. Nicolau, Antimicrob. Agents Chemother., 2013, 57, 4197–4207 CrossRef CAS PubMed.
- T. A. Wencewicz, T. E. Long, U. Möllmann and M. J. Miller, Bioconjugate Chem., 2013, 24, 473–486 CrossRef CAS PubMed.
- T. A. Wencewicz and M. J. Miller, J. Med. Chem., 2013, 56, 4044–4052 CrossRef CAS PubMed.
- J. Starr, M. F. Brown, L. Aschenbrenner, N. Caspers, Y. Che, B. S. Gerstenberger, M. Huband, J. D. Knafels, M. M. Lemmon, C. Li, S. P. McCurdy, E. McElroy, M. R. Rauckhorst, A. P. Tomaras, J. A. Young, R. P. Zaniewski, V. Shanmugasundaram and S. Han, J. Med. Chem., 2014, 57, 3845–3855 CrossRef CAS PubMed.
- T. Zheng and E. M. Nolan, J. Am. Chem. Soc., 2014, 136, 9677–9691 CrossRef CAS PubMed.
- T. Zheng, J. L. Bullock and E. M. Nolan, J. Am. Chem. Soc., 2012, 143, 18388–18400 CrossRef PubMed.
- J. K. Nicholson, E. Holmes, J. Kinross, R. Burcelin, G. Gibson, W. Jia and S. Pettersson, Science, 2012, 336, 1262–1267 CrossRef CAS PubMed.
- J. K. Nicholson and I. D. Wilson, Nat. Rev. Drug Discovery, 2003, 2, 668–676 CrossRef CAS PubMed.
- S. I. Müller, M. Valdebenito and K. Hantke, Biometals, 2009, 22, 691–695 CrossRef PubMed.
- X. Yu, Y. Dai, T. Yang, M. R. Gagné and H. Gong, Tetrahedron, 2011, 67, 144–151 CrossRef CAS.
- R. Lagos, M. Baeza, G. Corsini, C. Hetz, E. Strahsburger, J. A. Castillo, C. Vergara and O. Monasterio, Mol. Microbiol., 2001, 42, 229–243 CrossRef CAS PubMed.
- M. A. Fischbach, H. Lin, D. R. Liu and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 571–576 CrossRef CAS PubMed.
- E. M. Nolan, M. A. Fischbach, A. Koglin and C. T. Walsh, J. Am. Chem. Soc., 2007, 129, 14336–14347 CrossRef CAS PubMed.
- M. Luo, H. Lin, M. A. Fischbach, D. R. Liu, C. T. Walsh and J. T. Groves, ACS Chem. Biol., 2006, 1, 29–32 CrossRef CAS PubMed.
- W. R. Harris, C. J. Carrano, S. R. Cooper, S. R. Sofen, A. E. Avdeef, J. V. McArdle and K. N. Raymond, J. Am. Chem. Soc., 1979, 101, 6097–6104 CrossRef CAS.
- H. L. T. Mobley, D. M. Green, A. L. Trifillis, D. E. Johnson, G. R. Chippendale, C. V. Lockatell, B. D. Jones and J. W. Warren, Infect. Immun., 1990, 58, 1281–1289 CAS.
- M. A. Mulvey, J. D. Schilling and S. J. Hultgren, Infect. Immun., 2001, 69, 4572–4579 CrossRef CAS PubMed.
- R. A. Welch, V. Burland, G. Plunkett III, P. Redford, P. Roesch, D. Rasko, E. L. Buckles, S.-R. Liou, A. Boutin, J. Hackett, D. Stroud, G. F. Mayhew, D. J. Rose, S. Zhou, D. C. Schwartz, N. T. Perna, H. L. T. Mobley, M. S. Donnenberg and F. R. Blattner, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 17020–17024 CrossRef CAS PubMed.
- J. P. Henderson, J. R. Crowley, J. S. Pinkner, J. N. Walker, P. Tsukayama, W. E. Stamm, T. M. Hooton and S. J. Hultgren, PLoS Pathog., 2009, 5, e1000305 Search PubMed.
- P. Kuhnert, J. Nicolet and J. Frey, Appl. Environ. Microbiol., 1995, 61, 4135–4139 CAS.
- P. Daegelen, F. W. Studier, R. E. Lenski, S. Cure and J. F. Kim, J. Mol. Biol., 2009, 394, 634–643 CrossRef CAS PubMed.
- G. Reid and A. W. Bruce, World J. Urol., 2006, 24, 28–32 CrossRef PubMed.
- M. A. J. Beerepoot, G. ter Riet, S. Nys, W. M. van der Wal, C. A. J. M. de Borgie, T. M. de Reijke, J. M. Prins, J. Koeijers, A. Verbon, E. Stobberingh and S. E. Geerlings, Arch. Intern. Med., 2012, 172, 704–712 CrossRef CAS PubMed.
- E. D. Weinberg, Perspect. Biol. Med., 1997, 40, 578–583 CrossRef CAS PubMed.
- J. R. Bailey, C. S. J. Probert and T. A. Cogan, PLoS One, 2011, 6, e26507 CAS.
- M. G. Blango, E. M. Ott, A. Erman, P. Veranic and M. A. Mulvey, PLoS One, 2014, 9, e93327 Search PubMed.
- I. Sekirov, S. L. Russell, L. C. M. Antunes and B. B. Finlay, Physiol. Rev., 2010, 90, 859–904 CrossRef CAS PubMed.
- F. Rafii, J. B. Sutherland and C. E. Cerniglia, Ther. Clin. Risk Manage., 2008, 4, 1343–1357 CAS.
- L. J. Searle, G. Méric, I. Porcelli, S. K. Sheppard and S. Lucchini, PLoS One, 2015, 10, e0117906 Search PubMed.
- A. L. Lloyd, D. A. Rasko and H. L. T. Mobley, J. Bacteriol., 2007, 189, 3532–3546 CrossRef CAS PubMed.
- S. L. Chen, M. Wu, J. P. Henderson, T. M. Hooton, M. E. Hibbing, S. J. Hultgren and J. I. Gordon, Sci. Transl. Med., 2013, 5, 184ra60 Search PubMed.
- S. Subashchandrabose and H. L. T. Mobley, Metallomics, 2015 10.1039/c4mt00329b.
- W. Rabsch, W. Voigt, R. Reissbrodt, R. M. Tsolis and A. J. Bäumler, J. Bacteriol., 1999, 181, 3610–3612 CAS.
- E. C. Garcia, A. R. Brumbaugh and H. L. T. Mobley, Infect. Immun., 2011, 79, 1225–1235 CrossRef CAS PubMed.
- T. J. Wiles, R. R. Kulesus and M. A. Mulvey, Exp. Mol. Pathol., 2008, 85, 11–19 CrossRef CAS PubMed.
- R. J. A. Ramirez, L. Karamanukyan, S. Ortiz and C. G. Gutierrez, Tetrahedron Lett., 1997, 38, 749–752 CrossRef CAS.
- R. C. Scarrow, D. J. Ecker, C. Ng, S. Liu and K. N. Raymond, Inorg. Chem., 1991, 30, 900–906 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Tables of bacterial strains employed in this study, iron content of the antimicrobial activity medium, characterization of GlcEnt–Amp/Amx 7–10, GlcEnt-PEG3-N312–13, and BLAST search for iroN sequence. Figures of HPLC traces of MceC- and IroB-catalyzed glucosylation of Ent-PEG3-N311, optical absorption spectra of GlcEnt–Amp/Amx 7–10, additional antimicrobial activity assays, time-kill kinetics, competition assays for FepA and IroN recognition, mixed-species antimicrobial activity assays, Lcn2 effect on antibacterial activity of GlcEnt–Amp/Amx 7–10, and cytotoxicity assays against T84 cells. See DOI: 10.1039/c5sc00962f |
| This journal is © The Royal Society of Chemistry 2015 |