
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceConvergent chemo-enzymatic synthesis of mannosylated glycopeptides; targeting of putative vaccine candidates to antigen presenting cells†
Julie D.
McIntosh
bd,
Margaret A.
Brimble
*ad,
Anna E. S.
Brooks
bd,
P. Rod
Dunbar
*bd,
Renata
Kowalczyk
ad,
Yusuke
Tomabechi
cd and
Antony J.
Fairbanks
*cd
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds St, Auckland, New Zealand. E-mail: m.brimble@auckland.ac.nz
bSchool of Biological Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: r.dunbar@auckland.ac.nz
cDepartment of Chemistry, University of Canterbury, Private Bag 4800, Christchurch, 8140, New Zealand. E-mail: antony.fairbanks@canterbury.ac.nz
dMaurice Wilkins Centre for Molecular Biodiscovery, University of Auckland, Private Bag 92019, Auckland 1010, New Zealand
First published on 19th May 2015
Abstract
The combination of solid phase peptide synthesis and endo-β-N-acetylglucosaminidase (ENGase) catalysed glycosylation is a powerful convergent synthetic method allowing access to glycopeptides bearing full-length N-glycan structures. Mannose-terminated N-glycan oligosaccharides, produced by either total or semi-synthesis, were converted into oxazoline donor substrates. A peptide from the human cytomegalovirus (CMV) tegument protein pp65 that incorporates a well-characterised T cell epitope, containing N-acetylglucosamine at specific Asn residues, was accessed by solid phase peptide synthesis, and used as an acceptor substrate. High-yielding enzymatic glycosylation afforded glycopeptides bearing defined homogeneous high-mannose N-glycan structures. These high-mannose containing glycopeptides were tested for enhanced targeting to human antigen presenting cells (APCs), putatively mediated via the mannose receptor, and for processing by the APCs for presentation to human CD8+ T cells specific for a 9-mer epitope within the peptide. Binding assays showed increased binding of glycopeptides to APCs compared to the non-glycosylated control. Glycopeptides bearing high-mannose N-glycan structures at a single site outside the T cell epitope were processed and presented by the APCs to allow activation of a T cell clone. However, the addition of a second glycan within the T cell epitope resulted in ablation of T cell activation. We conclude that chemo-enzymatic synthesis of mannosylated glycopeptides enhances uptake by human APCs while preserving the immunogenicity of peptide epitopes within the glycopeptides, provided those epitopes are not themselves glycosylated.
Introduction
The mannose receptor (MR),1 a member of the calcium dependent lectin receptor family,2 recognises glycans attached to the surface of a wide variety of microbes and microorganisms, as well as extracellular glycoproteins. In addition to recognising mannose residues, the MR, which consists of multiple carbohydrate recognition domains (CRD's) in a single polypeptide chain, possesses a broader binding specificity than its name may initially suggest, and also binds oligosaccharides terminated in other monosaccharides, such as fucose and N-acetylglucosamine (GlcNAc), in a calcium dependent manner.3The roles that have been assigned to the MR are numerous, including internalisation and clearance of invading organisms by phagocytosis. Although the MR is expressed primarily by macrophages, the fact that it is expressed more widely on antigen presenting cells (APCs) led to the realisation4 that the MR is involved in antigen processing and presentation.5 This discovery in turn stimulated research into antigen mannosylation6 with the objective of enhancing antigen immunogenicity by targeting to APC's via the MR.7 The targeting of other species to dendritic cells via the MR has also been investigated for a variety of alternative purposes.8
Previous approaches at antigen mannosylation have typically involved conjugation techniques such as reductive amination of mannan-derived aldehydes,6e,f or the construction of neo-glycoconjugate structures in which the mannose residues were attached by linkers.6g,h In the former case concerns remain about both molecular heterogeneity and the lack of precise structural definition of such materials, whilst the latter may run the risk of the development of immune response to the ‘non-natural’ structural features of the linkers.
Chemical synthesis may address both of these concerns by providing defined homogenous materials in which the mannose-terminated oligosaccharide is attached to the peptide backbone by native linkages. Previously we accessed fluorescently-labelled glycopeptides containing single mannose residues through solid phase peptide synthesis (SPPS).9 A logical extension of this work is the construction of peptides that incorporate multivalent mannose residues via truncated or full-length branched N-glycan structures. However, the total chemical synthesis of homogeneous glycopeptides bearing complicated N-glycan structures which are linked to the peptide backbone through native Asn-linkages presents significant logistical challenges.
A solution to this problem is a combined chemo-enzymatic approach in which the peptide is accessed by synthesis, and the multi-antennary oligosaccharide is then attached enzymatically. In this respect we,10 together with Wang and co-workers,11 have been instrumental in developing the synthetic potential12 of the endo-β-N-acetylglucosaminidases (ENGases),13 a class of enzymes that specifically cleave the chitobiose core of N-linked glycans between the two innermost GlcNAc residues. Particularly noteworthy developments have been the development of N-glycan oxazolines as activated donors for these enzymes, first demonstrated by Shoda,14 and the production of a variety of ‘glycosynthase’15 mutants of enzymes such Endo M,16 Endo A,17 and Endo D18 which are able to process oxazolines as donors but which are incapable of performing product hydrolysis.19 Furthermore, the fact that the oxazoline substrates required for these bio-catalysed synthetic reactions may now be readily accessed from the corresponding reducing sugars in water,20 another seminal contribution by Shoda, has significantly increased the ease of access to donors, and so greatly broadened the scope of this potentially widely applicable methodology.
As part of an on-going program centred on the convergent synthesis of complex glycopeptides,21 we reasoned that the ENGase methodology is ideally suited to the production of glycopeptide vaccine candidates comprising high mannose N-glycans. To test the hypothesis that the conjugation of oligosaccharides bearing multiple mannose residues may enhance the binding of a peptide vaccine to APC's, glycans were incorporated into a 19-mer peptide from the cytomegalovirus (CMV) pp65 protein (pp65491–509; ILARNLVPMVATVQGQNLK) that incorporates a very well-characterised peptide epitope recognised by human cytotoxic T-lymphocytes (CTL) (pp65495–503; NLVPMVATV). The CMV pp65 protein22 is the most common tegument protein, and is capable of blocking innate and adaptive immune responses upon CMV invasion.23 Pp65 and pp65-derived peptide fragments stimulate the development of CTL in vivo and in vitro.24 The pp65495–503fragment is presented to T cells bound to HLA-A2, the most common MHC class I molecule (≈45% of the population), and T cell responses to this epitope are very well characterised,25 making it an ideal model antigen. The pp65491–509 peptide sequence chosen for the present study conveniently incorporates two Asn residues (Asn-495 and Asn-507) for the potential attachment of N-glycans, one of which sits (Asn-495) within the pp65495–503 T cell epitope. In order to facilitate binding analysis in cellular immunology screens, a fluorescence label was required, which in this instance was provided by the attachment of 5(6)-carboxyfluorescein (5(6)-CF)‡ in the final stages of SPPS. 5(6)-Carboxyfluorescein is a widely used bio-labelling reagent allowing for ready detection of peptides and proteins using both fluorescent microscopy and flow cytometry and can be readily incorporated into stepwise SPPS.9b,26 Glycopeptides 4–7 (Fig. 1) were therefore selected as synthetic targets, in addition to the non-glycosylated peptide 1, which was used as a control in the APC binding assay.
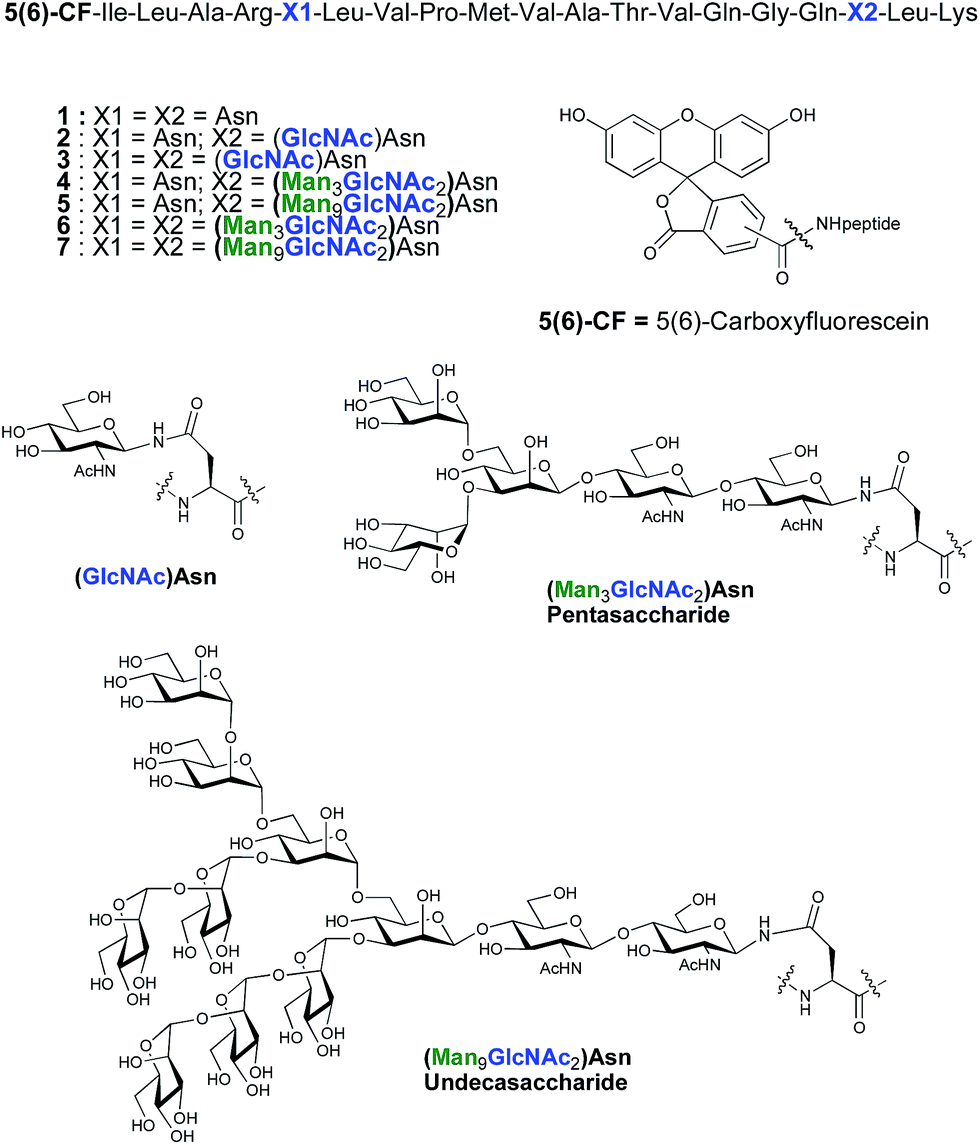 | ||
| Fig. 1 Structures of CMV-glycopeptides prepared by enzymatic addition of mannose-containing oligosaccharides. | ||
Results and discussion
Peptide synthesis
ENGase catalysed glycosylation requires a protein or peptide to contain a GlcNAc at Asn residues that are ultimately destined to bear full length N-glycans. Glycopeptides 2 and 3 (Fig. 1), which contained GlcNAc at Asn-5, and GlcNAc at both Asn-5 and Asn-17 respectively, were targeted for peptide synthesis. It was envisaged that both could be readily accessed by SPPS in which the protected glycosyl amino acid building block Fmoc-[GlcNAc(OAc)3]Asn-OH 827 was incorporated as required. Firstly, the non-glycosylated control peptide 1 was synthesised using aminomethyl polystyrene resin,28 to which Fmoc-Lys(Boc)-(4-(hydroxymethyl)phenoxypropionic acid, HMPP) was attached using N,N′-diisopropylcarbodiimide (DIC) as coupling agent (Scheme 1). Microwave-assisted SPPS using the appropriate Fmoc protected amino acid building blocks and 2-(6-chloro-1H-benzotriazole-1-yl)-1,1,3,3-tetramethylammonium hexafluorophosphate (HCTU) as coupling agent was followed by HCTU-mediated coupling with 5(6)-carboxyfluorescein (5(6)-CF). Peptide 9 was cleaved from the resin using trifluoroacetic acid (TFA). Subsequent reduction of the methionine sulfoxide was performed using dimethylsulfide (DMS) and tetrabutylammonium iodide in TFA.29 Finally purification by reverse phase (RP) HPLC yielded pure 1. Similarly microwave-assisted SPPS using building block 8 with (1-[bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium 3-oxid hexafluorophosphate (HATU) activation, followed by incorporation of the 5(6)-CF label and cleavage from the resin yielded glycopeptides 10 and 11. Removal of the acetate protecting groups on the attached GlcNAcs by treatment with sodium methoxide in methanol, subsequent reduction of the methionine sulfoxide, and purification by RP-HPLC yielded targets 2 and 3, comprising one and two GlcNAc residues respectively.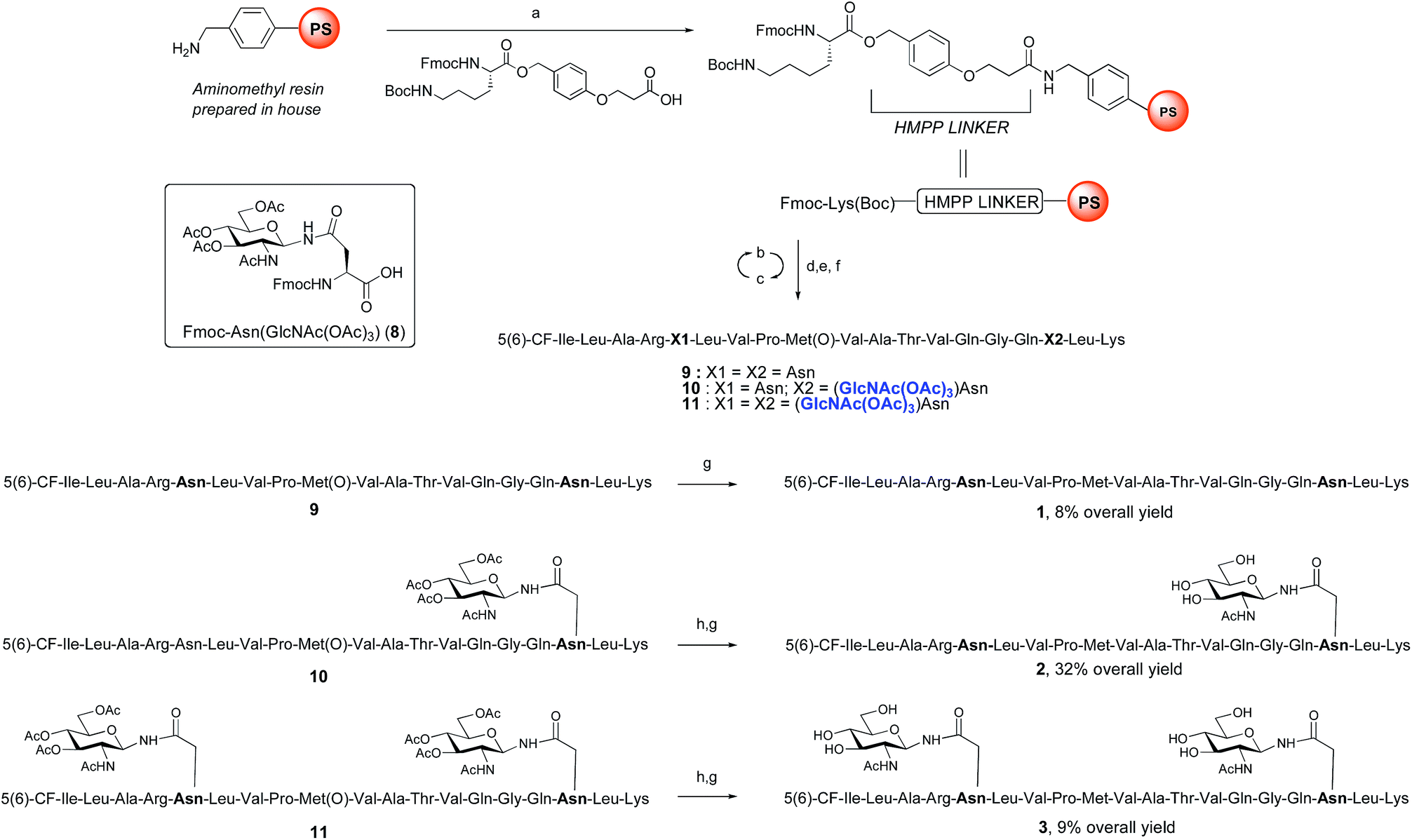 | ||
| Scheme 1 Synthesis of CMV control peptide 1 and glycopeptides 2 and 3 containing a GlcNAc residue either at Asn-17, or at both Asn-5 and Asn-17. Reagents and conditions: (a) DIC, CH2Cl2, rt, 2 h; (b) 20% piperidine, DMF, 35 W, 75 °C, 1 × 0.5 min + 1 × 3 min; (c) Fmoc-AA-OH, HCTU, iPr2EtN, DMF, 20 W, 75 °C, 5 min; or Fmoc-Arg(Pbf)-OH, HCTU, iPr2EtN, DMF, rt, 25 min + 25 W, 75 °C, 5 min; or 8, HATU, 2,4,6-trimethylpyridine, DMF, 20 W, 75 °C, 15 min; (d) 5(6)-carboxyfluorescein, HCTU, iPr2EtN, DMF, 20 W, 75 °C, 20 min; (e) 20% piperidine, DMF, 35 W, 75 °C, 4 × 3 min; (f) TFA, iPr3SiH, DODT, H2O, rt, 2 h; (g) DMS, TBAI, TFA, 0 °C, 1 h (for 1) or 1.5 h (for 2) or 9.5 h (for 3); (h) 1 M NaOMe in MeOH, rt, pH 12.2, 1.5 h (for 2) or pH 10.4, 3 h (for 3). | ||
Enzymatic glycosylation
The synthesis of glycopeptides 4 and 6 bearing Man3-terminated pentasaccharide N-glycans (Scheme 2) was achieved using the tetrasaccharide oxazoline donor 12, which was synthesized as previously described.10c The mutant ENGase Endo A E173H, which had previously been demonstrated17a,21a to transfer oxazoline 12 to a variety of acceptors bearing GlcNAc residues, was used for conjugation to both peptides 2 and 3. In both cases the E173H mutant was effective in producing the desired glycopeptide products 4 and 6 in good yield. Subsequent purification by RP-HPLC allowed separation of the two regioisomeric materials that arose from the use of the 5(6)-carboxyfluorescein label.‡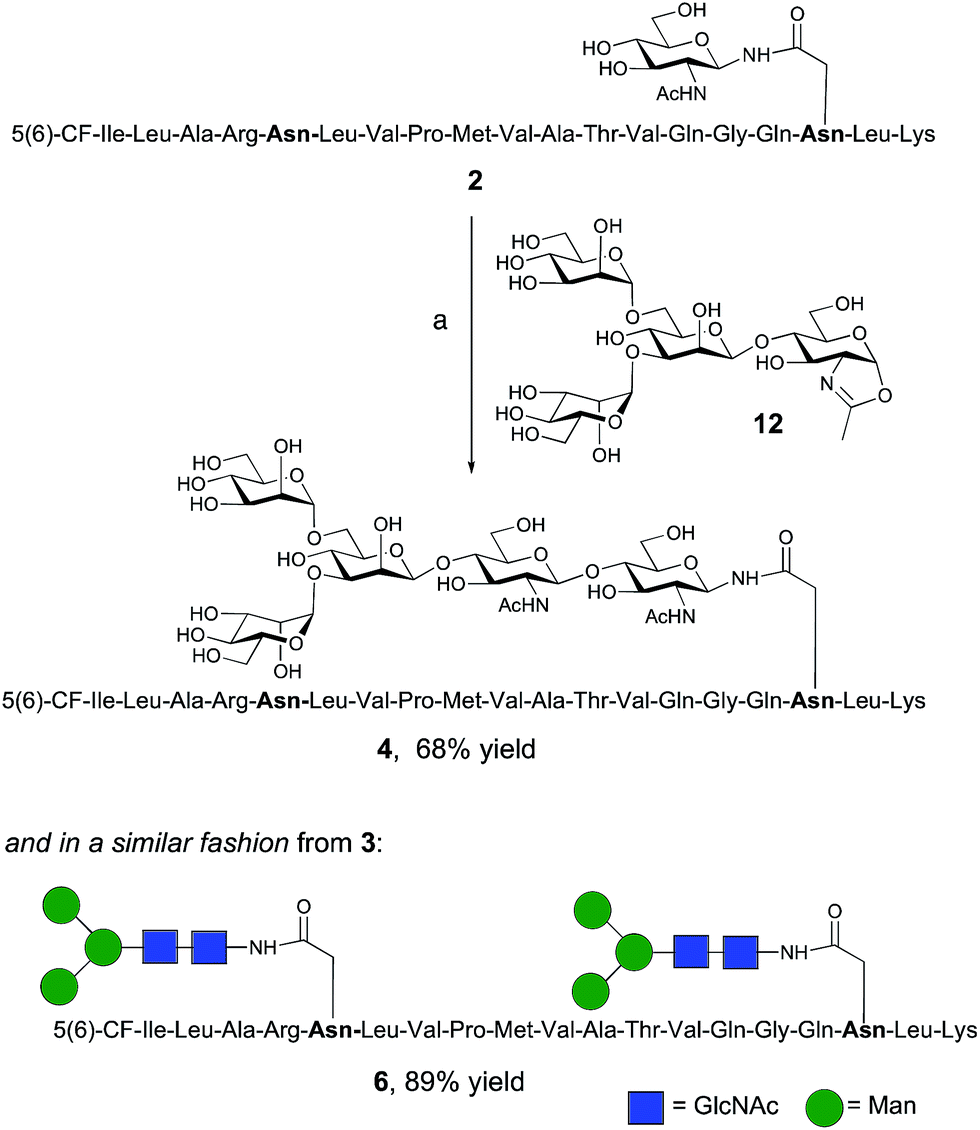 | ||
| Scheme 2 Reagents and conditions: (a) oxazoline 12, Endo A E173H, sodium phosphate buffer (100 mM, pH 6.5), 23 °C, 7 h. | ||
The synthesis of glycopeptides bearing full-length high mannose N-glycans bearing nine mannose residues required the use of the decasaccharide oxazoline donor 13 (Scheme 3). Although highly complex oligosaccharide oxazolines, such as 13, may be accessed by total chemical synthesis, a considerably more efficient procedure is to access them by semi-synthesis following isolation of a high mannose containing oligosaccharide/glycoconjugate from natural sources. In the case of oxazoline 13, isolation from soybean flour is a convenient if somewhat protracted method, and the reported procedure11g allowed access to multi-milligram quantities of oxazoline 13. Oxazoline 13 was then assayed as a substrate for the Endo A E173H mutant enzyme, which had previously been applied for the synthesis of a variety of truncated high mannose structures in addition to those reported above.17a,21a
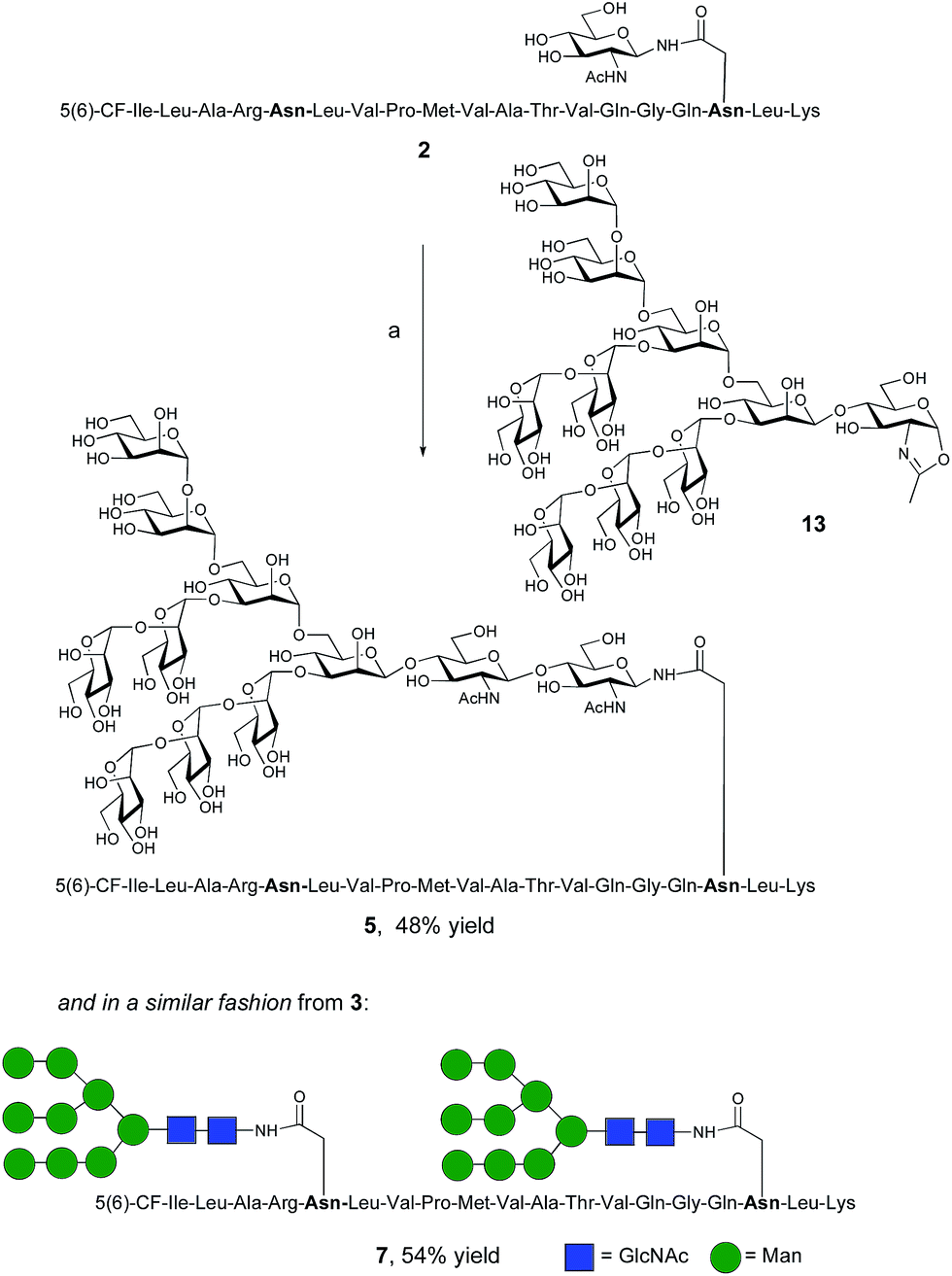 | ||
| Scheme 3 Reagents and conditions: (a) oxazoline 13, Endo M N175Q, sodium phosphate buffer (100 mM, pH 6.5), 23 °C. | ||
Surprisingly it was found that the E173H mutant of Endo A was incapable of catalyzing the transfer of oxazoline 13 to either peptide 2 or 3; indeed oxazoline 13 was recovered intact from these reactions indicating that it was not a hydrolytic substrate for the E173H mutant. This was a somewhat perplexing result because Endo A is known to cleave full-length high mannose oligosaccharides efficiently; indeed WT Endo A is itself incapable of performing synthetic reactions using oxazoline 13 as the donor because product hydrolysis is considerably faster than synthesis. Clearly the E173H mutation had subtly affected the substrate tolerance of the enzyme so that it was no longer capable of processing full-length N-glycan structures. In light of this negative result, attention turned to the commercially available N175Q mutant of Endo M.16a This enzyme has previously been shown to accept donor 13 as a substrate, and indeed in the present study was effective in producing the desired glycopeptides 5 and 7, from peptides 2 and 3, respectively in good yield. Purification by RP-HPLC again enabled separation of the two regioisomeric fluorescently-labeled materials.‡
Ability of glycosylated peptides to target antigen to MR-expressing APC
Peptides were assessed for their ability to target monocyte derived dendritic cells (hereafter referred to as antigen presenting cells, APC) from 3 different human donors; monocytes isolated from blood were cultured for 6 days and then used on day 6 for binding assays. These in vitro-derived cells express high levels of MR (CD206; Fig. 2A) as well as moderate levels of DC-SIGN (CD209; data not shown) that can also bind high order mannose glycans. APC treated with peptide 1 exhibited non-specific binding at 37 °C across all donors tested (Fig. 2B–D); the nature of this interaction was not further analysed in this study. However, there was enhanced targeting of glycopeptides 4–7 to the APC across all donors (Fig. 2B–D). That this enhanced targeting required the presence of MR expression was demonstrated by analysis of MR-deficient cells (lymphocytes) present in the same samples, which indicated there was no consistent enhanced targeting of glycopeptides 4–7 as compared to peptide 1 (see ESI, Fig. S9†). The observed magnitude of targeting to APC was found to be dependent on both the number and structure of the attached N-glycans. Fig. 2B and C show that glycopeptides 6 and 7, each bearing two N-glycans, exhibited slightly stronger binding to the APC as compared to their mono-glycosylated counterparts 4 and 5. Fig. 2D shows that on average peptides 5 and 7, which both contain Man9GlcNAc2 structures, showed stronger binding to the APC as compared to 4 and 6, which bear truncated Man3GlcNAc2 oligosaccharides. A caveat to these observations is that although the binding affinities do appear to correlate with N-glycan structure, further analysis is required to determine whether the peptides are actually binding to MR (CD206), DC-SIGN (CD209), or indeed both.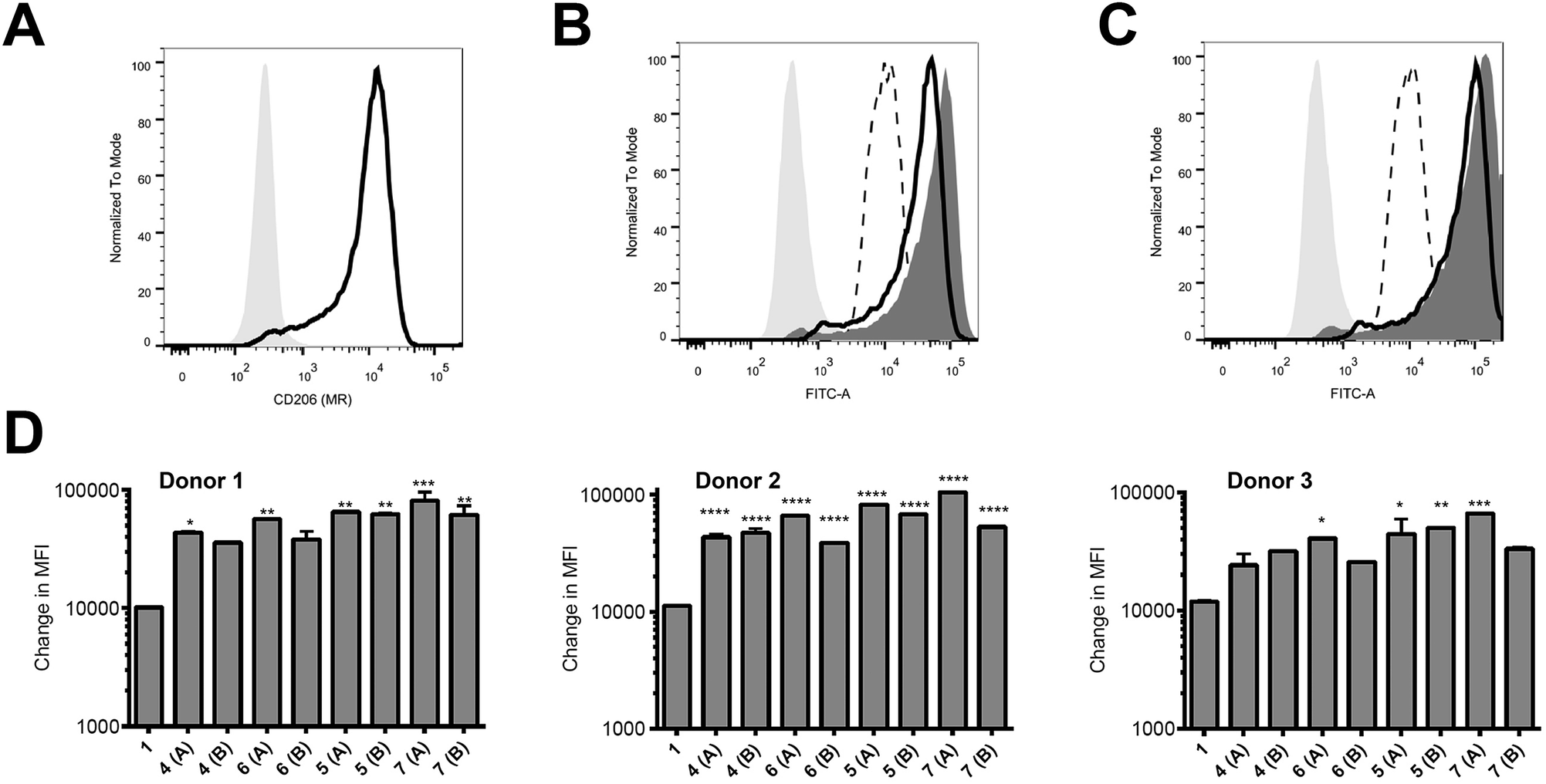 | ||
| Fig. 2 Increased targeting of glycosylated peptides to MR-positive APC. (A) Expression levels of CD206 (MR) on the APC were determined using an anti-CD206 antibody. (B and C) APC were treated with 10 μM of peptides 1 and 4–7 at 37 °C for 60 minutes and analysed by flow cytometry. Results in (B) show the binding for a representative donor of 1 (dotted line) compared to 4 (black line) and 6 (dark grey histogram) relative to the unstained sample (light grey shaded histogram). Results in (C) show the binding of 1 (dotted line) compared to 5 (black line) and 7 (dark grey histogram) relative to the unstained sample (light grey shaded histogram). In (D), results are given as the change in measured fluorescence intensity (MFI, calculated as the MFI of sample minus the MFI of the untreated control); each sample was tested in duplicate. Monocytes cultured from three individual donors are shown. Statistical significance for each sample (compared to 1) was calculated using a one way Anova followed by Dunnett's multiple comparison test; *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Ability of glycosylated peptides to induce activation of a NLV-specific T cell clone
The ability of the mannosylated peptides to be processed and induce T cell activation was determined. The T cell clone used in this study recognises the epitope pp65495–503 (![[N with combining low line]](https://www.rsc.org/images/entities/char_004e_0332.gif)
![[L with combining low line]](https://www.rsc.org/images/entities/char_004c_0332.gif)
![[V with combining low line]](https://www.rsc.org/images/entities/char_0056_0332.gif) PMVATV) and is hereafter referred to as the NLV-specific clone. HLA-A2+ APC were incubated with the peptides overnight to allow processing and presentation of the peptide epitope. The pp65495–503 peptide itself (“NLV” in Fig. 3) binds HLA-A2 molecules on the APCs and activates the NLV-specific T cell clone, as measured by secretion of the cytokine TNFα (Fig. 3). The non-glycosylated longer peptide pp65491–509 (1) is also processed by the APCs to present pp65495–503 bound to HLA-A2 to activate the NLV-specific T cell clone (Fig. 3). Mono-glycosylated peptides 4 and 5, each bearing a glycan at Asn-17 – outside the pp65495–503 T cell epitope – were both processed and presented at the surface of the APC by MHC class I, resulting in activation of the NLV-specific T cell clone (Fig. 3). Interestingly, APC pulsed with glycopeptides 6 and 7, which each bear an additional N-glycan located at Asn-5 – the first amino acid in the T cell epitope – induced no activation of the NLV-specific clone cells in any of the individuals tested. Hence glycosylation at Asn-5, within the epitope recognised by the T cell clone, inhibits processing of the peptide by APCs, and therefore ablates T cell activation.
PMVATV) and is hereafter referred to as the NLV-specific clone. HLA-A2+ APC were incubated with the peptides overnight to allow processing and presentation of the peptide epitope. The pp65495–503 peptide itself (“NLV” in Fig. 3) binds HLA-A2 molecules on the APCs and activates the NLV-specific T cell clone, as measured by secretion of the cytokine TNFα (Fig. 3). The non-glycosylated longer peptide pp65491–509 (1) is also processed by the APCs to present pp65495–503 bound to HLA-A2 to activate the NLV-specific T cell clone (Fig. 3). Mono-glycosylated peptides 4 and 5, each bearing a glycan at Asn-17 – outside the pp65495–503 T cell epitope – were both processed and presented at the surface of the APC by MHC class I, resulting in activation of the NLV-specific T cell clone (Fig. 3). Interestingly, APC pulsed with glycopeptides 6 and 7, which each bear an additional N-glycan located at Asn-5 – the first amino acid in the T cell epitope – induced no activation of the NLV-specific clone cells in any of the individuals tested. Hence glycosylation at Asn-5, within the epitope recognised by the T cell clone, inhibits processing of the peptide by APCs, and therefore ablates T cell activation.
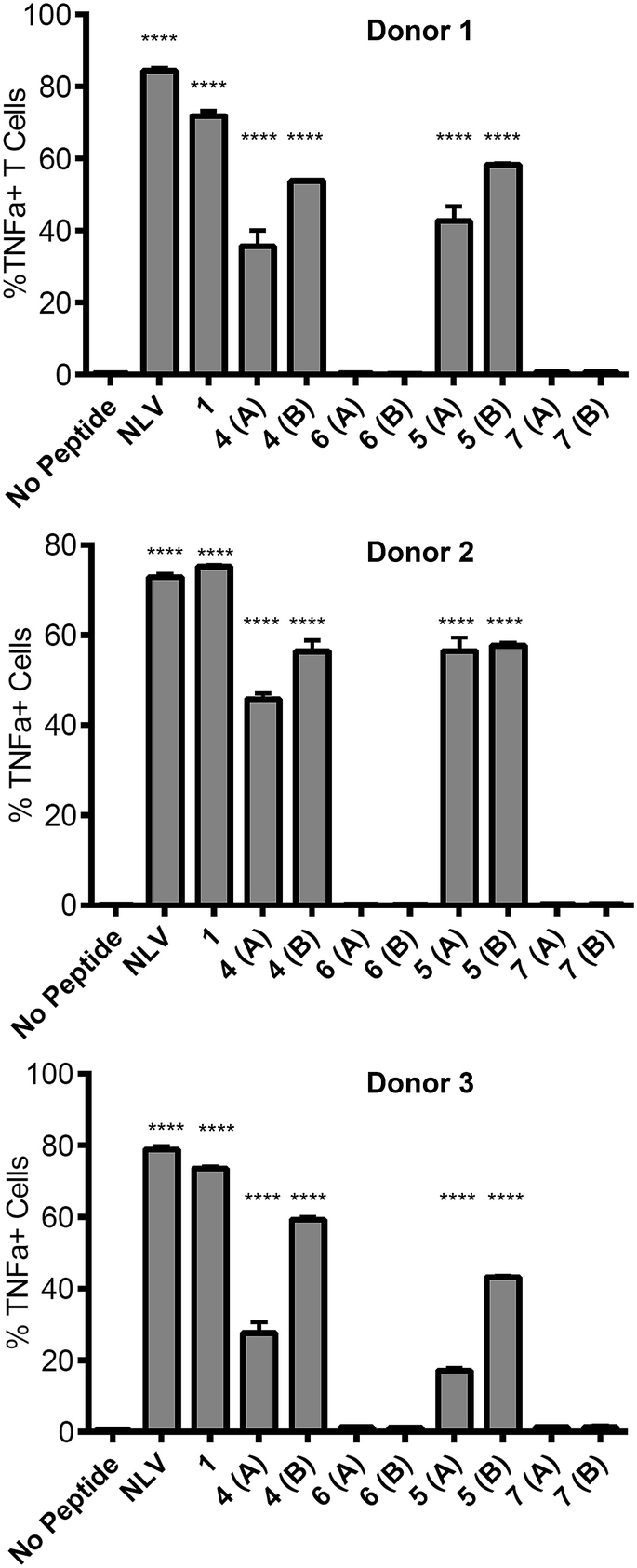 | ||
| Fig. 3 Ability of peptides to induce T cell activation. Activation of the NLV-specific clone was measured by intracellular staining for the cytokine TNFα and analysed by flow cytometry. Results are given as the percent TNFα positive cells of the NLV-specific clone cells. NLV refers to the peptide NLVPMVATV, which served as a positive control for the assay. APC were prepared from 3 individual donors as shown; each compound was tested in duplicate. Statistical significance for each sample (compared to 1) was calculated using a one way Anova followed by Dunnett's multiple comparison test; *p < 0.05, **p < 0.01, ***p < 0.001. | ||
This result provides key information for the design of glycopeptide vaccine candidates using this type of modification; care should be taken to ensure that the addition of any targeting N-glycans should be distant from any epitopes of interest to guarantee that correct processing of the epitope is possible.30 However, importantly the ability of the mono-glycosylated peptides 4 and 5 to induce T cell activation indicates that the addition of the mannose-terminated oligosaccharides did not restrict antigen processing of unglycosylated peptide epitopes within the construct, suggesting this strategy may indeed be highly useful to target antigen to MR-expressing cells for presentation to T cells.
Conclusions
We have demonstrated the effectiveness of a convergent chemo-enzymatic strategy to prepare pp65-based glycopeptides bearing truncated and full-length high mannose N-glycans. The combination of Fmoc-SPPS, the use of oxazoline donors of synthetic or semi-synthetic origin, and ENGase-catalysed glycosylation afforded glycopeptides in good yield. Experiments to test the levels of glycopeptide binding to APCs indicated both that the addition of high mannose N-glycans resulted in improved targeting of the peptide cargo to MR-expressing cells, and that the targeted peptides were able to be processed and presented by the APCs to human T cells; the latter with the proviso that the site of glycosylation was not within the epitope sequence. Thus the controlled addition of high mannose N-glycans to peptides at judiciously selected sites has the potential to afford superior constructs for vaccine development.Acknowledgements
We gratefully acknowledge financial support from the Royal Society of New Zealand Marsden Fund (UOC0910), the University of Auckland (Biopharma Thematic Research Initiative), and the Freemasons Roskill Foundation (Postdoctoral Fellowship to RK).Notes and references
- (a) L. Martinez-Pomares, J. Leukocyte Biol., 2012, 96, 1177–1186 CrossRef PubMed; (b) L. East and C. M. Isacke, Biochim. Biophys. Acta, 2002, 1572, 364–386 CrossRef CAS.
- W. I. Weis, M. E. Taylor and K. Drickamer, Immunol. Rev., 1998, 163, 19–34 CrossRef CAS PubMed.
- M. E. Taylor, K. Bezouska and K. Drickamer, J. Biol. Chem., 1992, 267, 1719–1726 CAS.
- (a) A. J. Engering, M. Cella, D. Fluitsma, M. Brockhaus, E. C. Hoefsmit, A. Lanzavecchia and J. Pieters, Eur. J. Immunol., 1997, 27, 2417–2425 CrossRef CAS PubMed; (b) F. Sallusto, M. Cella, C. Danieli and A. Lanzavecchia, J. Exp. Med., 1995, 182, 389–400 CrossRef CAS; (c) T. I. Prigozy, P. A. Sieling, D. Clemens, P. L. Stewart, S. M. Behar, S. A. Porcelli, M. B. Brenner, R. L. Modlin and M. Kronenberg, Immunity, 1997, 6, 187–197 CrossRef CAS.
- U. Gazi and L. Martinez-Pomares, Immunobiology, 2009, 214, 554–561 CrossRef CAS PubMed.
- See for example: (a) L.-Z. He, A. Crocker, J. Lee, J. Mendoza-Ramirez, X.-T. Wang, L. A. Vitale, T. O'Neill, C. Petromilli, H.-F. Zhang, J. Lopez, D. Rohrer, T. Keler and R. Clynes, J. Immunol., 2007, 178, 6259–6267 CrossRef CAS; (b) M. Luong, J. S. Lam, J. Chen and S. M. Levitz, Vaccine, 2007, 25, 4340–4344 CrossRef CAS PubMed; (c) D. Betting, X. Mu, Ka. Kafi, D. McDonnel, F. Rosas, D. Gold and J. Timmerman, Vaccine, 2009, 27, 250–259 CrossRef CAS PubMed; (d) S. K. Singh, I. Streng-Ouwehand, M. Litjens, H. Kalay, S. Burgdorf, E. Saeland, C. Kurts, W. W. Unger and Y. van Kooyk, Eur. J. Immunol., 2011, 41, 916–925 CrossRef CAS PubMed; (e) V. Apostolopoulos, N. Barnes, G. A. Pietersz and I. F. C. McKenzie, Vaccine, 2000, 18, 3174–3184 CrossRef CAS; (f) V. Apostolopoulos, G. A. Pietersz, B. E. Loveland, M. S. Sandrin and I. F. C. McKenzie, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 10128–10132 CrossRef CAS; (g) K.-C. Sheng, M. Kalkanidis, D. Pouniotis, S. Esparon, C. Tang, V. Apostolopoulos and G. Pietersz, Eur. J. Immunol., 2008, 38, 424–436 CrossRef CAS PubMed; (h) M. C. Agnes, A. Tan, R. Jordens, A. Geluk, B. O. Roep, T. Ottenhoff, J. W. Drijfhout and F. Koning, Int. Immunol., 1998, 10, 1299–1304 CrossRef CAS.
- T. Keler, V. Ramakrishna and M. W. Fanger, Expert Opin. Biol. Ther., 2004, 4, 1953–1962 CrossRef CAS PubMed.
- (a) U. Hillaert, M. Verdoes, B. I. Florea, A. Saragliadis, K. L. L. Habets, J. Kuiper, S. Van Calenbergh, F. Ossendorp, G. A. Van Der Marel, C. Driessen and H. S. Overkleeft, Angew. Chem., Int. Ed., 2009, 48, 1629–1632 CrossRef CAS PubMed; (b) J.-S. Thomann, B. Heurtault, S. Weidner, M. Brayé, J. Beyrath, S. Fournel, F. Schuber and B. Frisch, Biomaterials, 2011, 32, 4574–4583 CrossRef CAS PubMed; (c) E.-A. Raiber, C. Tulone, Y. Zhang, L. Martinez-Pomares, E. Steed, A. Sponaas, J. Langhorne, M. Noursadeghi, B. Chain and A. Tabor, ACS Chem. Biol., 2010, 5, 461–476 CrossRef CAS PubMed; (d) H.-K. Kim, H. Wei, A. Kulkarni, R. M. Pogranichniy and D. H. Thompson, Biomacromolecules, 2012, 13, 636–644 CrossRef CAS PubMed; (e) B. Carrillo-Conde, E.-H. Song, A. Chavez-Santoscoy, Y. Phanse, A. E. Ramer-Tait, N. L. B. Pohl, M. J. Wannemuehler, B. H. Bellaire and B. Narasimhan, Mol. Pharmaceutics, 2011, 8, 1877–1886 CrossRef CAS PubMed.
- (a) M. A. Brimble, R. Kowalczyk, P. W. R. Harris, P. R. Dunbar and V. J. Muir, Org. Biomol. Chem., 2008, 6, 112–121 RSC; (b) R. Kowalczyk, P. W. R. Harris, R. P. Dunbar and M. A. Brimble, Synthesis, 2009, 2210–2222 CAS.
- (a) T. W. D. F. Rising, T. D. W. Claridge, N. Davies, D. P. Gamblin, J. W. B. Moir and A. J. Fairbanks, Carbohydr. Res., 2006, 341, 1574–1596 CrossRef CAS PubMed; (b) T. W. D. F. Rising, T. D. W. Claridge, J. W. B. Moir and A. J. Fairbanks, ChemBioChem, 2006, 7, 1177–1180 CrossRef CAS PubMed; (c) T. W. D. F. Rising, C. D. Heidecke, J. W. B. Moir, Z. Ling and A. J. Fairbanks, Chem.–Eur. J., 2008, 14, 6444–6464 CrossRef CAS PubMed; (d) T. B. Parsons, J. W. B. Moir and A. J. Fairbanks, Org. Biomol. Chem., 2009, 7, 3128–3140 RSC; (e) C. D. Heidecke, T. B. Parsons and A. J. Fairbanks, Carbohydr. Res., 2009, 344, 2433–2438 CrossRef CAS PubMed; (f) Y. Tomabechi, M. A. Squire and A. J. Fairbanks, Org. Biomol. Chem., 2014, 12, 942–955 RSC.
- (a) B. Li, Y. Zeng, S. Hauser, H. J. Song and L.-X. Wang, J. Am. Chem. Soc., 2005, 127, 9692–9693 CrossRef CAS PubMed; (b) H. Li, B. Li, H. Song, L. Breydo, I. V. Baskakov and L.-X. Wang, J. Org. Chem., 2005, 70, 9990–9996 CrossRef CAS PubMed; (c) L.-X. Wang, H. Song, S. Liu, H. Lu, S. Jiang, J. Ni and H. Li, ChemBioChem, 2005, 6, 1068–1074 CrossRef CAS PubMed; (d) H. Li, S. Singh, Y. Zeng, H. Song and L.-X. Wang, Bioorg. Med. Chem. Lett., 2005, 15, 895–898 CrossRef CAS PubMed; (e) Y. Zeng, J. S. Wang, B. Li, S. Hauser, H. Li and L.-X. Wang, Chem.–Eur. J., 2006, 12, 3355–3364 CrossRef CAS PubMed; (f) B. Li, H. Song, S. Hauser and L.-X. Wang, Org. Lett., 2006, 8, 3081–3084 CrossRef CAS PubMed; (g) M. Umekawa, W. Huang, B. Li, K. Fujita, H. Ashida, L.-X. Wang and K. Yamamoto, J. Biol. Chem., 2008, 283, 4469–4479 CrossRef CAS PubMed; (h) L.-X. Wang, Carbohydr. Res., 2008, 343, 1509–1522 CrossRef CAS PubMed; (i) Y. Wei, C. Li, W. Huang, B. Li, S. Strome and L.-X. Wang, Biochemistry, 2008, 47, 10294–10304 CrossRef CAS PubMed; (j) H. Ochiai, W. Huang and L.-X. Wang, J. Am. Chem. Soc., 2008, 130, 13790–13803 CrossRef CAS PubMed; (k) H. Ochiai, W. Huang and L.-X. Wang, Carbohydr. Res., 2009, 344, 592–598 CrossRef CAS PubMed.
- For some reviews see: (a) A. J. Fairbanks, Pure Appl. Chem., 2013, 85, 1847–1863 CrossRef CAS; (b) A. J. Fairbanks, C. R. Chim., 2011, 14, 44–58 CrossRef CAS PubMed; (c) W. Huang, H. Ochiai, X. Zhang and L.-X. Wang, Carbohydr. Res., 2008, 343, 2903–2913 CrossRef CAS PubMed.
- For some early demonstrations of the synthetic potential of enzymes such as Endo A and Endo M see: (a) J.-Q. Fan, K. Takegawa, S. Iwahara, A. Kondo, I. Kato, C. Abeygunawardana and Y. C. Lee, J. Biol. Chem., 1995, 270, 17723–17729 CrossRef CAS PubMed; (b) J.-Q. Fan, M. S. Quesenberry, K. Takegawa, S. Iwahara, A. Kondo, I. Kato and Y. C. Lee, J. Biol. Chem., 1995, 270, 17730–17735 CrossRef CAS PubMed; (c) K. Yamamoto, K. Fujimori, K. Haneda, M. Mizuno, T. Inazu and H. Kumagai, Carbohydr. Res., 1997, 305, 415–422 CrossRef CAS; (d) K. Haneda, T. Inazu, M. Mizuno, R. Iguchi, K. Yamamoto, H. Kumagai, S. Aimoto, H. Suzuki and T. Noda, Bioorg. Med. Chem. Lett., 1998, 8, 1303–1306 CrossRef CAS; (e) T. Yamanoi, M. Tsutsumida, Y. Oda, E. Akaike, K. Osumi, K. Yamamoto and K. Fujita, Carbohydr. Res., 2004, 339, 1403–1406 CrossRef CAS PubMed.
- M. Fujita, S.-I. Shoda, K. Haneda, T. Inazu, K. Takegawa and K. Yamamoto, Biochim. Biophys. Acta, 2001, 1528, 9–14 CrossRef CAS.
- (a) L. F. Mackenzie, Q. R. Wang, R. A. J. Warren and S. G. Withers, J. Am. Chem. Soc., 1998, 120, 5583–5584 CrossRef CAS; (b) C. Malet and A. Planas, FEBS Lett., 1998, 440, 208–212 CrossRef CAS.
- (a) M. Umekawa, C. Li, T. Higashiyama, W. Huang, H. Ashida, K. Yamamoto and L.-X. Wang, J. Biol. Chem., 2010, 285, 511–521 CrossRef CAS PubMed; (b) M. Umekawa, W. Huang, B. Li, K. Fujita, H. Ashida, L.-X. Wang and K. Yamamoto, J. Biol. Chem., 2008, 283, 4469–4479 CrossRef CAS PubMed.
- (a) C. D. Heidecke, Z. Ling, N. C. Bruce, J. W. B. Moir, T. B. Parsons and A. J. Fairbanks, ChemBioChem, 2008, 9, 2045–2051 CrossRef CAS PubMed; (b) W. Huang, C. Li, B. Li, M. Umekawa, K. Yamamoto, X. Zhang and L.-X. Wang, J. Am. Chem. Soc., 2009, 131, 2214–2223 CrossRef CAS PubMed.
- S.-Q. Fan, W. Huang and L.-X. Wang, J. Biol. Chem., 2012, 287, 11272–11281 CrossRef CAS PubMed.
- W. Huang, J. Giddens, S.-Q. Fan, C. Toonstra and L.-X. Wang, J. Am. Chem. Soc., 2012, 134, 12308–12318 CrossRef CAS PubMed.
- M. Noguchi, T. Tanaka, H. Gyakushi, A. Kobayashi and S.-I. Shoda, J. Org. Chem., 2009, 74, 2210–2212 CrossRef CAS PubMed.
- (a) Y. Tomabechi, G. Krippner, P. M. Rendle, M. A. Squire and A. J. Fairbanks, Chem.–Eur. J., 2013, 19, 15084–15088 CrossRef CAS PubMed; (b) R. Kowalczyk, M. A. Brimble, Y. Tomabechi, A. J. Fairbanks, M. Fletcher and D. L. Hay, Org. Biomol. Chem., 2014, 12, 8142–8151 RSC.
- (a) A. A. Scalzo, A. J. Corbett, W. D. Rawlinson, G. M. Scott and M. A. Degli-Esposti, Immunol. Cell Biol., 2007, 85, 46–54 CrossRef CAS PubMed; (b) J. P. Tomtishen III, Virol. J., 2012, 9, 22–29 CrossRef PubMed.
- R. F. Kalejta, Microbiol. Mol. Biol. Rev., 2008, 72, 249–265 CrossRef CAS PubMed.
- E. McLaughlin-Taylor, H. Pande, S. J. Forman, B. Tanamachi, C. R. Li, J. A. Zaia, P. D. Greenberg and S. R. Riddell, J. Med. Virol., 1994, 43, 103–110 CrossRef CAS PubMed.
- (a) C. La Rosa, R. Krishnan, S. Markel, J. P. Schneck, R. Houghten, C. Pinilla and D. J. Diamond, Blood, 2001, 97, 1776–1786 CrossRef CAS PubMed; (b) V. Appay, P. R. Dunbar, M. Callan, P. Klenerman, G. M. A. Gillespie, L. Papagno, G. S. Ogg, A. King, F. Lechner, C. A. Spina, S. Little, D. V. Havlir, D. D. Richman, N. Gruener, G. Pape, A. Waters, P. Easterbrook, M. Salio, V. Cerundolo, A. J. McMichael and S. L. Rowland-Jones, Nat. Med., 2002, 8, 379–385 CrossRef CAS PubMed.
- (a) M. A. Fara, J. J. Diaz-Mochon and M. Bradley, Tetrahedron Lett., 2006, 47, 1011–1014 CrossRef CAS PubMed; (b) R. Fischer, O. Mader, G. Jung and R. Brock, Bioconjugate Chem., 2003, 14, 653–660 CrossRef CAS PubMed.
- (a) T. Inazu and K. Kobayashi, Synlett, 1993, 869–871 CrossRef CAS; (b) T. Tanaka, H. Nagai, M. Noguchi, A. Kobayashi and S.-I. Shoda, Chem. Commun., 2009, 3378–3379 RSC; (c) P. Kirsch, N. Kusunose, J. Aikawa, T. Kigawa, S. Yokoyama and T. Ogawa, Bioorg. Med. Chem., 1995, 3, 1631–1636 CrossRef CAS.
- (a) A. R. Mitchell, S. B. H. Kent, M. Engelhard and R. B. Merrifield, J. Org. Chem., 1978, 43, 2845–2852 CrossRef CAS; (b) P. W. R. Harris, S. H. Yang and M. A. Brimble, Tetrahedron Lett., 2011, 52, 6024–6026 CrossRef CAS PubMed.
- E. Nicolas, M. Vilaseca and E. Giralt, Tetrahedron, 1995, 51, 5701–5710 CrossRef CAS.
- A reduction in epitope presentation due to glycosylation has previously been observed in O-linked glycopeptide anti-cancer vaccine candidates. See for example: (a) B. L. Wilkinson, S. Day, L. R. Malins, V. Apostolopoulos and R. J. Payne, Angew. Chem., Int. Ed., 2011, 50, 1635–1639 CrossRef CAS PubMed; (b) B. L. Wilkinson, S. Day, R. Chapman, S. Perrier, V. Apostolopoulos and R. J. Payne, Chem.–Eur. J., 2012, 18, 16540–16548 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details for the synthesis of all peptides and glycopeptides and spectra. See DOI: 10.1039/c5sc00952a |
| ‡ The 5(6)-isomeric mixture of carboxyfluorescein is more affordable than the corresponding single isomer 5-CF or isomer 6-CF, and can be separated using reverse phase high-performance liquid chromatography (RP-HPLC) once the labeled peptide is constructed. In this work the separated regioisomers were arbitrarily labeled as either (A) or (B). See ESI.† |
| This journal is © The Royal Society of Chemistry 2015 |