
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceIn vivo demonstration of an active tumor pretargeting approach with peptide nucleic acid bioconjugates as complementary system†
Anna
Leonidova‡
a,
Christian
Foerster‡§
b,
Kristof
Zarschler
b,
Maik
Schubert
b,
Hans-Jürgen
Pietzsch
b,
Jörg
Steinbach
b,
Ralf
Bergmann
b,
Nils
Metzler-Nolte
 c,
Holger
Stephan
*b and
Gilles
Gasser
*a
c,
Holger
Stephan
*b and
Gilles
Gasser
*a
aDepartment of Chemistry, University of Zurich, Winterthurerstrasse 190, CH-8057 Zurich, Switzerland. E-mail: gilles.gasser@chem.uzh.ch; Web: http://www.gassergroup.com Tel: +41 44 635 46 30
bHelmholtz-Zentrum Dresden – Rossendorf, Institute of Radiopharmaceutical Cancer Research, Bautzner Landstraße 400, D-01328 Dresden, Germany. E-mail: h.stephan@hzdr.de; Web: http://www.hzdr.de/NanoscalicSystems Tel: +49 351 260-3091
cLehrstuhl für Anorganische Chemie I – Bioanorganische Chemie, Fakultät für Chemie und Biochemie, Ruhr-Universität Bochum, Universitätsstrasse 150, D-44801 Bochum, Germany
First published on 17th June 2015
Abstract
A novel, promising strategy for cancer diagnosis and therapy is the use of a pretargeting approach. For this purpose, the non-natural DNA/RNA analogues Peptide Nucleic Acids (PNAs) are ideal candidates as in vivo recognition units due to their high metabolic stability and lack of unspecific accumulation. In the pretargeting approach, an unlabeled, highly specific antibody–PNA conjugate has sufficient time to target a tumor before administration of a small fast-clearing radiolabeled complementary PNA that hybridizes with the antibody–PNA conjugate at the tumor site. Herein, we report the first successful application of this multistep process using a PNA-modified epidermal growth factor receptor (EGFR) specific antibody (cetuximab) and a complementary 99mTc-labeled PNA. In vivo studies on tumor bearing mice demonstrated a rapid and efficient in vivo hybridization of the radiolabeled PNA with the antibody–PNA conjugate. Decisively, a high specific tumor accumulation was observed with a tumor-to-muscle ratio of >8, resulting in a clear visualization of the tumor by single photon emission computed tomography (SPECT).
Introduction
The excellent target specificity of monoclonal antibodies (mAbs) renders this class of biomacromolecules a beneficial platform to detect and treat tumor malignancies. In nuclear medicine, such tumor antigen-specific vehicles labeled with radionuclides would be applicable for non-invasive imaging of diseases and more importantly, for in vivo delivery of therapeutically relevant radioactivity doses to tumor sites. Unfortunately, the concept of utilizing radionuclide-carrying tumor-specific mAbs is afflicted with several drawbacks,1–3 mainly arising from the high molecular weight of mAbs (∼150 kDa). Due to size-related limitations in passing biological barriers,4 such as extravasation and the inability of glomerular filtration, mAbs exhibit a slow but gradual accumulation in tumor sites and long blood retention times of up to several days, respectively.5 The slow blood clearance rate of mAbs forces extensive waiting times before acquiring a diagnostic image with reasonable signal-to-background ratio as well as to label with appropriate isotopes.6,7 Detrimental radiation exposure for almost all tissues in the organism, especially during therapeutic applications, will be the result of their prolonged blood pool retention time.8 Despite innumerable research activities and efforts conducted so far, only two drugs, namely Bexxar® and Zevalin™, representing radiolabeled mAbs for treatment of Non-Hodgkin's lymphoma are currently approved by the FDA.9–11An attractive strategy to circumvent these limitations is the use of a pretargeting approach that involves an artificial in vivo recognition system composed of a nonradioactive antibody conjugate and a small radiolabeled “effector” molecule. As schematically represented in Scheme 1, in this multistep process, an unlabeled, highly tumor-specific antibody conjugate is first administrated into a patient. Upon injection, sufficient time is allowed for the antibody conjugate to reach the tumor and to be eliminated from the non targeted tissues. This is then followed by the administration of a small fast-clearing radiolabeled “effector” molecule that binds to the antibody conjugate at the tumor site.12,13 This approach allows for the rational use of long-circulating high-affinity mAbs for both non-invasive cancer radioimmunodetection and radioimmunotherapy.14,15
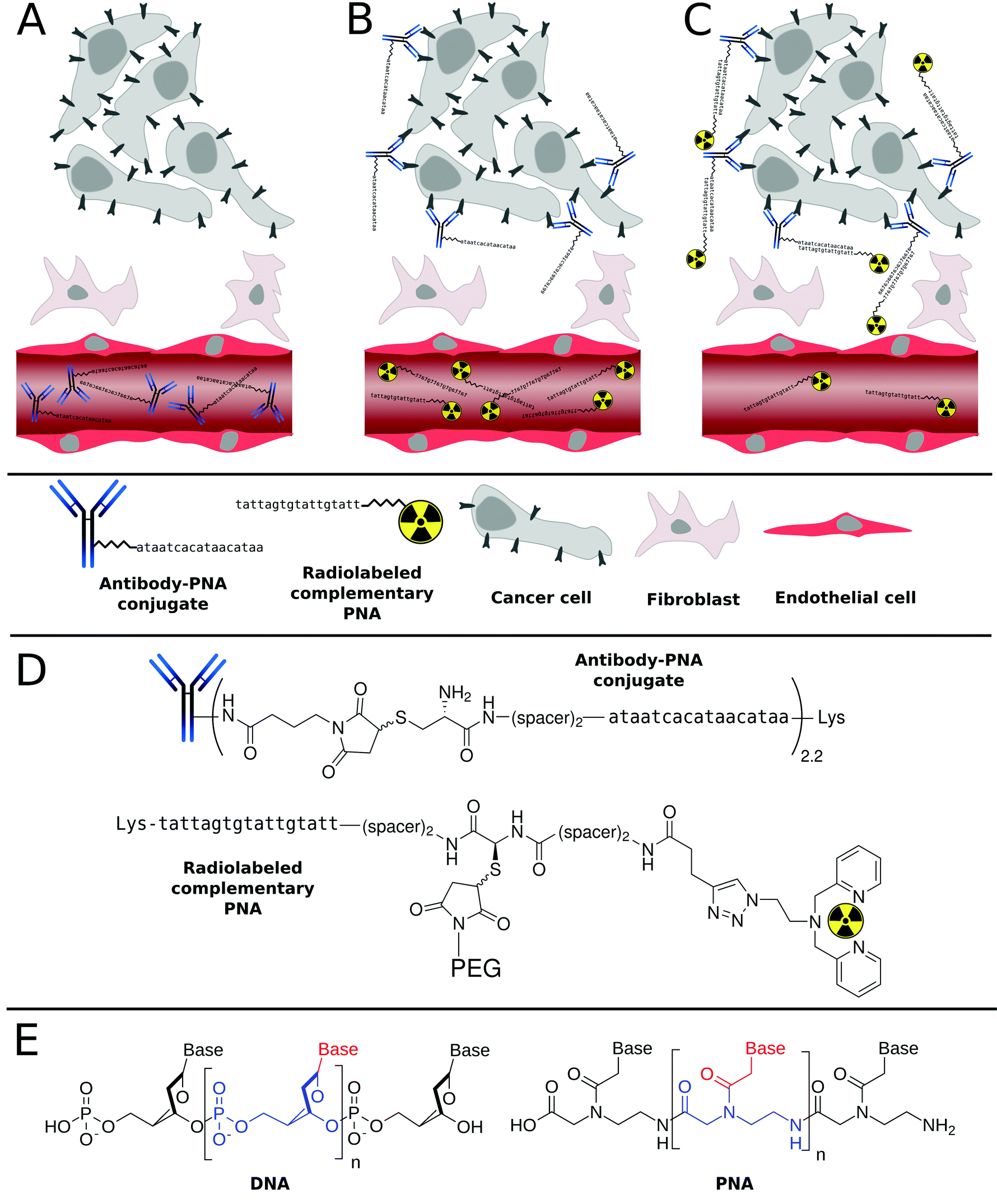 | ||
| Scheme 1 General principle of tumor pretargeting using Peptide Nucleic Acids (PNA). Firstly, unlabeled, highly specific antibody–PNA conjugates are administered intravenously into the patient (A). After accumulation of the antibody conjugates at the tumor site and clearance from non-target tissues, small fast-clearing radiolabeled complementary PNAs are injected (B), that hybridize with antibody–PNA conjugates (C). The radioactivity symbols inserted into the 2,2′-dipicolylamine (Dpa) chelator illustrate either 99mTc(CO)3 or 186Re(CO)3, which are used for diagnosis or therapy, respectively (D). While DNA has a deoxyribose sugar backbone, the PNA's backbone is composed of repeating N-(2-aminoethyl)-glycine units linked by amide bonds. Their various purine and pyrimidine bases are linked to the backbone by a methylene bridge and a carbonyl group (E). | ||
Several recognition systems have been investigated and to some extend clinically tested for different pretargeting approaches. Most prominent among them are streptavidin/biotin,16–19 bispecific antibody/hapten20–27 and synthetic complementary oligonucleotides/oligonucleotides such as morpholino and peptide nucleic acid derivatives.28–34 For more detailed information and secondary references, we highly recommend the review article of Goldenberg et al.35 Beside the “classical” recognition by supramolecular motifs, bio-orthogonal and ultra-fast click reactions have also been developed as complementary system in vivo.36–39
Among the range of synthetic oligonucleotides investigated for pretargeting, phosphorodiamidate morpholino oligomers (MORFs) and Peptide Nucleic Acids (PNAs, Scheme 1) have emerged as promising candidates. Both derivatives are non-charged mimics of the naturally occurring ribonucleic acids DNA and RNA. They exhibit a suitable degree of water solubility, are almost inert towards degradation in vivo,40,41 and insensitive towards chemical modifications even under harsh conditions. The superior intrinsic properties of PNAs over DNA/RNA have made them extremely interesting candidates for applications in (nuclear) medicine or biology. Radiolabeled PNA oligomers were indeed utilized as probes for molecular imaging of target specific mRNA sequences.42–48 However, the relatively low cellular uptake of PNAs has represented a serious drawback, which has undoubtedly delayed their use as antisense or antigen agents, although several techniques are now available to overcome this problem (e.g. use of cell-penetrating peptides, etc.). Nonetheless, the limited cellular uptake of PNAs creates a very interesting bio-orthogonal system. Indeed, administration of a radiolabeled PNA strand into a living organism rarely results into unspecific binding.34,48 In other words, the PNA strand is usually excreted in its intact form from the kidney/liver. This characteristic, in addition to the excellent physicochemical properties discussed above, have made PNAs a promising tool in the tumor pretargeting approach.
Pioneering work in this field of research was performed by Hnatowich and co-workers, who demonstrated a first proof-of-concept in 1997.32–34,49 In those studies, surrogates such as PNA-loaded polymeric32,34 and agarose-based avidin beads49 transplanted into mouse thighs were used. To the best of our knowledge, there is only a single report describing the utilization of PNA–streptavidin bioconjugates for (non-specific) tumor localization in a mouse model by passive diffusion.33 These PNA–protein conjugates were found to accumulate unspecifically in most tissues of the animals. Consequently, upon administration of the radiolabeled complementary PNA, radioactivity levels were significantly higher compared to control animals. However, tumor antigens have not been specifically targeted by anti-tumor antibody–PNA conjugates yet. Thus, a critical evaluation of a tumor pretargeting concept in vivo is still lacking.
In this work, we aim to demonstrate the suitability of PNA-based bioconjugates as versatile complementary system for the specific transportation and accumulation of radionuclides in tumors. More specifically, in this article, we first describe the preparation and characterization of several PNA bioconjugates that contained different building blocks such as a 2,2′-dipicolylamine (Dpa) to chelate the radioactive 99mTc as well as polyethylene glycol (PEG) units to tune the biodistribution of the PNA oligomers. In addition, radiolabeling of the Dpa-containing bioconjugates with [99mTc]Tc(H2O)3(CO)3+ as well as detailed radiopharmaceutical evaluation including biodistribution and metabolic profiling is presented.
Of note, to critically assess the PNA-based pretargeting system used in this work, the well-studied, FDA-approved therapeutic mAb cetuximab (C225; Erbitux®, ImClone LLC) was selected, since it is commercially available and shows chemical robustness as well as a high affinity to a clinically relevant tumor biomarker.50–54 The molecular target of cetuximab, namely the epidermal growth factor receptor (EGFR),55,56 is involved in regulating cell growth, differentiation and survival of cells.57,58 In a variety of human malignancies, EGFR is constitutively activated as a result of receptor overexpression, mutation or deregulation59–61 and has therefore been investigated as a major target for the treatment of uncontrolled tumor growth.62–64 All in all, this article demonstrates, for the first time, the successful tumor pretargeting approach using radiolabeled PNAs in combination with PNA–antibody bioconjugates in murine xenografts (human squamous carcinoma cell line A431). This report highlights the immense potential of this approach for both cancer radioimmunodetection as well as radioimmunotherapy.
Results and discussion
Synthesis and characterization of PNA bioconjugates
All PNA oligomers and bioconjugates were synthesized manually on TentaGel S Fmoc-Lys(Boc)-RAM resin using commercially available Fmoc/Bhoc-protected PNA monomers and standard protocols previously reported by our groups.65 For sufficient stability of PNA–PNA hybrids, complementary PNA oligomers consisting of 17 bases were designed. Table 1 summarizes the PNA sequences used in this work. In order to radiolabel PNA with [99mTc]Tc(H2O)3(CO)3+, 2,2′-dipicolylamine (Dpa) was site-specifically introduced by copper-mediated 1,3-dipolar cycloaddition (“Click” Chemistry) as previously reported by our groups.66 In order to improve the pharmacokinetics and bioavailability of compounds and drug carriers, we envisaged PEGylating the PNA bioconjugates as described in the literature for different biomolecules.67–73 However, in the different studies having investigated the use of PEG-containing PNAs oligomers, the full impact of PEGylation on PNA's pharmacokinetics and biodistribution was not assessed.65,74–82| Entry | Abbreviation | Sequencea |
|---|---|---|
| a Spacer = –NH(CH2)2O(CH2)2OCH2CO–. | ||
| 1 | Dpa-PNA | H-Dpa-spacer-spacer-ttatgttatgtgattat-Lys-NH2 |
| 2 | Dpa-Cys-PNA | H-Dpa-spacer-spacer-Cys-spacer-spacer-ttatgttatgtgattat-Lys-NH2 |
| 3 | Cys-c-PNA | H-Cys-spacer-spacer-ataatcacataacataa-Lys-NH2 |
| 4 | Dpa-(Cys-PEG 2kDa )-PNA | H-Dpa-spacer-spacer-(Cys-PEG2kDa)-spacer-spacer-ttatgttatgtgattat-Lys-NH2 |
| 5 | Dpa-(Cys-PEG 10kDa )-PNA | H-Dpa-spacer-spacer-(Cys-PEG10kDa)-spacer-spacer-ttatgttatgtgattat-Lys-NH2 |
| 6 | (NOTA) 3 -C225-Cys-c-PNA | (NOTA)3-C225-mal-Cys-spacer-spacer-ataatcacataacataa-Lys-NH2 |
Of note, some of us recently demonstrated that PEGylation of 17-mer L-configured DNA-oligonucleotides – another promising complementary system – significantly altered radiopharmacokinetics. Indeed, a non-specific accumulations in kidneys was markedly reduced by 90%, while the blood circulation half-life was strongly increased by factor of 4.83 More specifically, in this work, two different PNA bioconjugates, namely Dpa-PNA and Dpa-Cys-PNA (Entries 1 and 2 in Table 1), were first synthesized as radionuclide carriers. Maleimido-PEG derivatives (2 and 10 kDa) were then inserted via Michael-type addition into the cysteine-containing PNA Dpa-Cys-PNA to give Dpa-(Cys-PEG2kDa)-PNA and Dpa-(Cys-PEG10kDa)-PNA, respectively (Entries 4 and 5 in Table 1). For this purpose, pretreatment of Dpa-Cys-PNA with the strong reducing agent tris(2-carboxyethyl)phosphine (TCEP) was found to be necessary to improve the yields of conjugation reaction by preventing PNA–PNA disulfide dimer formation.74,80 Prior to the addition of maleimido-PEG derivatives, excessive TCEP was removed by size exclusion chromatography to avoid reduction of C–C double bond of maleimide entity leading to sulfhydryl-unreactive succinimide derivatives. In addition, a cysteine-containing PNA oligomer, Cys-c-PNA (Entry 3 in Table 1), which is complementary to the other PNA sequences of this study, was prepared. The identity of all PNA analogues Dpa-PNA, Dpa-Cys-PNA, Cys-c-PNA, Dpa-(Cys-PEG2kDa)-PNA, and Dpa-(Cys-PEG10kDa)-PNA was confirmed by ESI-MS and MALDI-TOF MS. The high purity of the bioconjugates was verified by LC-MS (Fig. S2, S5, S8, S10 and S12†). Apart from the [M + nH]n+ peaks, additional [M + Cu + nH]n+, [M − picolyl + Cu + nH]n+ were also observed in both ESI and MALDI-TOF spectra for Dpa-containing products. This effect is due to the traces of copper ions still present after the introduction of Dpa to PNA sequence by “Click” Chemistry. Due to the polydispersity of PEG polymers combined with the multiple charged conjugates, ESI-MS generated spectra with multitudinous m/z peaks disabling conclusive results. The presence of Dpa-(Cys-PEG2kDa)-PNA and Dpa-(Cys-PEG10kDa)-PNA was, however, unambiguously confirmed by MALDI-TOF, where only single and double positive charged species were observed (Fig. S11, S13†).
Hybridization properties
In order to assess self-complementary interactions between the PNA strands, we performed UV-based melting curves for each single stranded PNA oligomers, namely Cys-c-PNA, Dpa-PNA, Dpa-(Cys-PEG2kDa)-PNA and Dpa-(Cys-PEG10kDa)-PNA. As displayed in Fig. S16†, no homo-hybridization was observed. In the case of hetero-hybridization, our data listed in Table S1† show that, even at room temperature, Cys-c-PNA forms perfect hybrids with all other PNA derivatives (complete match). The determined melting temperatures were almost independent of the degree of PEGylation.Bioconjugation between Cys-c-PNA and cetuximab
Cetuximab (C225; Erbitux®, ImClone LLC), a chimeric human-murine IgG1 monoclonal antibody, binds specifically to the extracellular domain of the epidermal growth factor receptor (EGFR) on both normal and tumor cells, and competitively inhibits the binding of epidermal growth factor (EGF) as well as other ligands. EGFR is often overexpressed in human malignancies and is associated with poor clinical prognosis.84,85 Cetuximab binding to EGFR blocks phosphorylation. This blockage results in inhibition of downstream cellular processes such as induction of apoptosis and cell growth. Due to its promising antitumor activity, cetuximab has been approved for the treatment of colorectal and head and neck squamous cell carcinoma as well as with external radiotherapy for the treatment of head and neck squamous cell carcinoma.86 Of note, we have recently shown that cetuximab labeled with the therapeutic β-emitter 90Y could improve permanent local tumor control after external radiotherapy.52 Since this well-studied anti-EGFR antibody possesses a high affinity to its molecular target, shows chemical robustness and is commercially available, we selected it as model for radiopharmaceutical evaluation of our PNA-based pretargeting system.Prior to attachment of Cys-c-PNA to cetuximab, the anti-EGFR antibody was modified with 1,4,7-triazacyclononane-1,4,7-triacetic acid (NOTA),87 a suitable [64Cu]Cu2+ chelator for PET-monitoring of aspired PNA–cetuximab bioconjugate.88–91 This allows us to quantify such important parameters as blood circulation half-life and tumor accumulation of PNA–cetuximab and therefore to optimize the administration regime of PNA–cetuximab conjugate and radiolabeled PNA. NOTA was successfully conjugated to cetuximab resulting in an average of three NOTA molecules per antibody to give (NOTA)3-C225.
Subsequent introduction of a maleimido group to (NOTA)3-C225 was successfully performed by reaction of (NOTA)3-C225 with 4-maleimido-butyric acid N-succinimidyl ester (GMBS) to obtain (NOTA)3-C225-mal.
Finally, Cys-c-PNA was linked to (NOTA)3-C225-mal under mild reaction conditions to give the bioconjugate (NOTA)3-C225–Cys-c-PNA (Entry 6 in Table 1). The average number of conjugated Cys-c-PNA to (NOTA)3-C225-mal was quantified by determination of the absorbance ratio 260 nm/280 nm in the UV spectrum.92–94 Based on this method, 2.2 ± 0.7 Cys-c-PNA moieties per antibody were found. MALDI-TOF MS analysis confirmed this result since the determined number of 2.4 bound PNA oligomers per antibody is in the same range as calculated by UV method (see Fig. S14†).
Radiochemistry
99mTc-radiolabeling of Dpa-PNA, Dpa-(Cys-PEG2kDa)-PNA and Dpa-(Cys-PEG10kDa)-PNA was performed via the precursor [99mTc(H2O)3(CO)3]+ generated by the IsoLink® kit “Carbonyl Labeling Agent”.95 Highly concentrated [99mTc(H2O)3(CO)3]+ precursor with radiochemical yields (rcy) of >95% was obtained by concentrating the solution at 100 °C for 30 min. Up to 580 MBq of 99mTc precursor was added to 10 nmol of each of the Dpa-bearing PNA conjugates to give the corresponding radiolabeled PNA conjugates with rcy >95% and high effective specific activities of up to 58 GBq μmol−1 (n = 19). Detailed studies to improve radiolabeling conditions have shown a strong dependency of rcy on the pH of the radiolabeling mixture. Radiolabeling at pH < 7 resulted in incomplete complexation of [99mTc(CO)3]+ with rcy of <85%, while the rcy went to up to >95% at optimized conditions (70 °C, 40 min, 10 nmol of PNA conjugate) when pH in the range from 7 to 8 was applied.After purification of 99mTc-labeled PNA derivatives by HPLC, partition experiments were performed in 1-octanol/buffer systems to assess the lipophilicity/hydrophobicity of the radiolabeled PNAs. Distribution ratio log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Do/w was determined at three different pH values (Table 2). Surprisingly, the logD values were almost independent on the degree of PEGylation within the tested pH range (7.2–7.6). Compared with previously published results based on a 12-mer PNA conjugate,48 it appears that the hydrophilicity is increased more by the lengthening of the PNA chain from 12-mer to 17-mer, rather than by the PEGylation.
Do/w was determined at three different pH values (Table 2). Surprisingly, the logD values were almost independent on the degree of PEGylation within the tested pH range (7.2–7.6). Compared with previously published results based on a 12-mer PNA conjugate,48 it appears that the hydrophilicity is increased more by the lengthening of the PNA chain from 12-mer to 17-mer, rather than by the PEGylation.
Do/w of radiolabeled PNA conjugates at different pH valuesa
| pH value | 12-mer [99mTc](Tc-Dpa)-PNA from ref. 48 | 17-mer [99mTc](Tc-Dpa)-PNA | 17-mer [99mTc](Tc-Dpa)-(Cys-PEG2kDa)-PNA | 17-mer [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA |
|---|---|---|---|---|
| a Shown are the averages of three independent experiments with the standard deviation in parentheses. | ||||
| 7.2 | −0.86 | −(2.35 ± 0.04) | −(2.28 ± 0.07) | −(2.40 ± 0.02) |
| 7.4 | −0.85 | −(2.22 ± 0.03) | −(2.20 ± 0.02) | −(2.22 ± 0.04) |
| 7.6 | −0.84 | −(2.35 ± 0.07) | −(2.42 ± 0.06) | −(2.35 ± 0.02) |
In order to examine if the modification of cetuximab with Cys-c-PNA resulted in loss of affinity to the EGFR, the antibody derivatives (NOTA)3-C225 and (NOTA)3-C225-Cys-c-PNA were radiolabeled with [64Cu]CuCl2. Of note, the pH of the [64Cu]CuCl2 labeling solution had to be adjusted to around 6 prior addition to the solutions containing the cetuximab conjugates to avoid antibody denaturation. In addition, due to the sensitivity of the antibody, mild reaction conditions (30 °C without shaking) were used. (NOTA)3-C225 and (NOTA)3-C225-Cys-c-PNA were labeled with effective specific activities of up to 16.7 GBq μmol−1 and radiochemical yields >99%.
Affinity of [64Cu]Cu-(NOTA)3-C225-Cys-c-PNA and [64Cu]Cu-(NOTA)3-C225 to the EGFR
In order to evaluate the influence of PNA-conjugation on the antibody's binding specificity and affinity to human EGFR, the dissociation characteristics of [64Cu]Cu-(NOTA)3-C225-Cys-c-PNA and [64Cu]Cu-(NOTA)3-C225 were determined comparatively using two dimensional cell cultures of epidermoid carcinoma (A431) and squamous carcinoma (FaDu) cells. These tumor cell lines present different expression levels of the receptor on their cell surface.96Fig. 1 shows the saturation binding curves of radiolabeled cetuximab conjugates for both cell lines. For [64Cu]Cu-(NOTA)3-C225-Cys-c-PNA and [64Cu]Cu-(NOTA)3-C225, Scatchard analysis was applied to determine dissociation constants (Table 3). A Kd of 5.4 ± 0.9 nM and Bmax of 13.3 pmol per mg protein for A431 and a Kd of 1.1 ± 0.2 nM and Bmax of 1.9 pmol per mg protein for FaDu cells were obtained for [64Cu]Cu-(NOTA)3-C225-Cys-c-PNA. [64Cu]Cu-(NOTA)3-C225 showed slightly different dissociation constants with a Kd of 7.7 ± 0.8 nM and Bmax of 15.9 pmol per mg protein for A431 and a Kd of 2.0 ± 0.3 nM and Bmax of 2.9 pmol per mg protein for FaDu cells. Modification of the monoclonal antibody with PNAs does not affect its binding behavior to EGFR expressing tumor cells. This is of particular importance since the immunoreactivity as well as the high affinity of cetuximab to EGFR has to be conserved after chemical conjugation.56
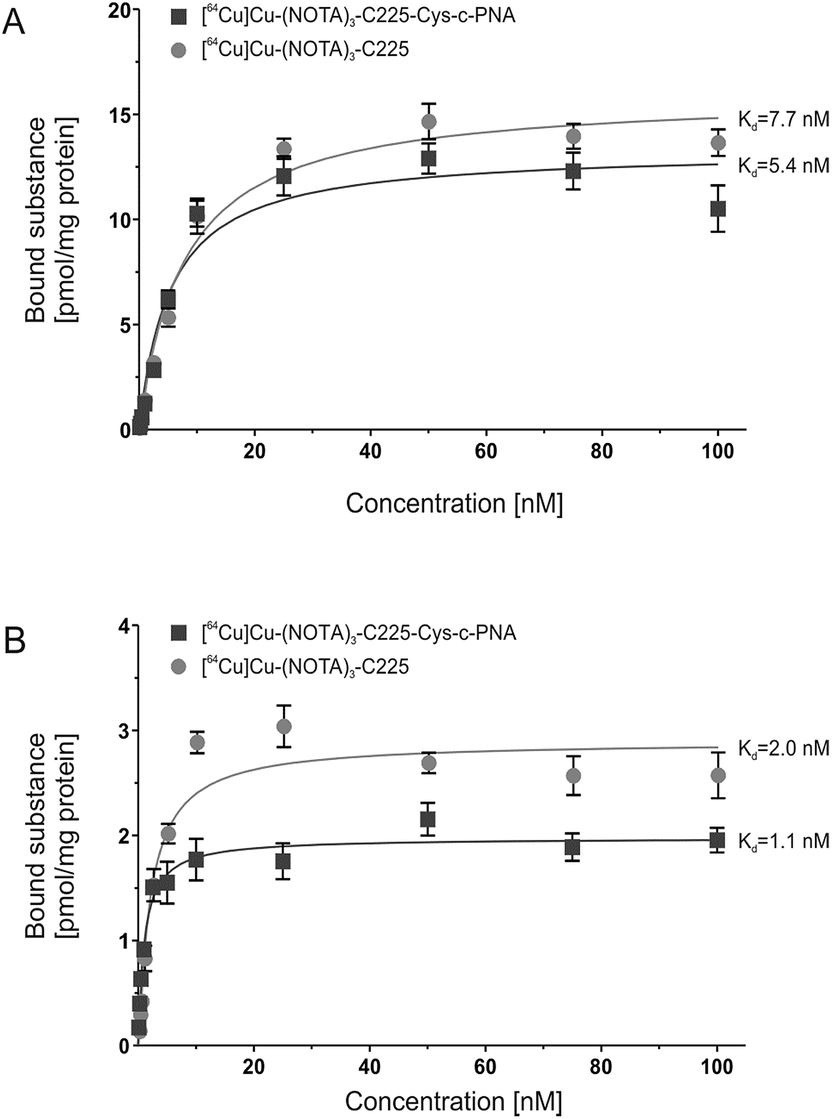 | ||
| Fig. 1 In vitro binding studies of radiolabeled cetuximab conjugates. Saturation curves of [64Cu]Cu-(NOTA)3-C225-Cys-c-PNA and [64Cu]Cu-(NOTA)3-C225 upon incubation with human EGFR-positive tumor cell lines A431 (A) and FaDu (B). | ||
| Cell line | [ 64 Cu]Cu-(NOTA) 3 -C225-Cys-c-PNA | [ 64 Cu]Cu-(NOTA) 3 -C225 | |
|---|---|---|---|
| A431 | K d | 5.4 ± 0.9 nM | 7.7 ± 0.8 nM |
| B max | 13.3 ± 0.6 pmol mg−1 | 15.9 ± 0.4 pmol mg−1 | |
| FaDu | K d | 1.1 ± 0.2 nM | 2.0 ± 0.3 nM |
| B max | 1.9 ± 0.1 pmol mg−1 | 2.9 ± 0.1 pmol mg−1 |
The variation in the affinity of the antibody conjugates between A431 and FaDu cells can be explained by the different cellular context. Such an effect has been previously reported for EGF.97 Björkelund and co-workers indeed observed an important influence of the investigated cell lines on the binding characteristics and on the multiple ligand–receptor interactions. The authors explained these phenomena by the occurrence of varying ratios of EGFR homodimers and heterodimers composed of EGFR and the human epidermal growth factor receptor 2 (HER2) due to different expression levels of these receptors. The different ratios of EGFR and HER2 may also account for the herein described variation in the affinity of the cetuximab conjugates between A431 and FaDu cells. The former cell line overexpresses EGFR with 1–3 × 106 receptors per cell98 and has a lower HER2 expression,96,99,100 whereas the latter cell line possesses a large HER2 population and presents less EGFR on the cell surface (7 × 105 receptors per cell).96,99,101
Biodistribution studies
The data previously presented by Hnatowich and co-workers32–34,49 and by our laboratories48 demonstrated favorable properties of radiolabeled PNAs towards in vivo tumor pretargeting applications. These include very fast distribution, low non-specific accumulation in non-targeted tissue as well as renal elimination as preferred elimination pathway from organism. However, these experiments also showed that specific accumulation of radiolabeled PNA derivatives in pretargeted tumor tissue might not be sufficiently high for therapeutic approaches.33,34,49 The reason behind it is certainly the rapid elimination of the radiolabeled PNA from blood. The major aim of attaching large PEG moieties onto our 17-mer PNA oligomers was therefore to increase blood retention and subsequently enhance blood availability. This should result into increased hybridization incidences in pretargeted tumor tissue.As an initial step of this evaluation process, we determined the impact of the degree of PEGylation on the radiopharmacological behavior by conducting biodistribution studies and dynamic SPECT scans in healthy male Wistar rats. To ensure comparability and compatibility with further animal studies as well as published results, the data presented are the means ± standard deviation of standard uptake values (SUV), defined as the tracer concentration at a certain time point normalized to injected dose per unit body weight. Detailed biodistribution data presented as SUV and %ID are summarized in Tables S2 and S3.† At 5 min post injection (Table 4), the 99mTc-labeled 17-mer PNA conjugates clearly showed an elevated level of activity concentration in the blood pool with increasing degree of PEGylation. Compared to the non-PEGylated 17-mer Dpa-PNA, the attachment of PEG led to about 10% and 45% higher activity concentration in the blood pool for 2 kDa PEG and 10 kDa PEG, respectively.
| 17-mer [99mTc](Tc-Dpa)-PNA | 17-mer [99mTc](Tc-Dpa)-(Cys-PEG2kDa)-PNA | 17-mer [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA | ||||
|---|---|---|---|---|---|---|
| 5 min p.i. (n = 12) | 60 min p.i. (n = 11) | 5 min p.i. (n = 8) | 60 min p.i. (n = 8) | 5 min p.i. (n = 8) | 60 min p.i. (n = 8) | |
| Blood | 1.36 ± 0.21 | 0.24 ± 0.10 | 1.51 ± 0.13 | 0.24 ± 0.06 | 2.01 ± 0.38 | 0.34 ± 0.11 |
| Kidneys | 11.7 ± 1.60 | 13.1 ± 1.99 | 12.0 ± 1.42 | 9.29 ± 1.10 | 11.3 ± 1.86 | 9.75 ± 2.64 |
| Adrenals | 0.56 ± 0.12 | 0.14 ± 0.04 | 0.80 ± 0.25 | 0.32 ± 0.21 | 0.82 ± 0.15 | 0.25 ± 0.08 |
| Liver | 0.91 ± 0.23 | 0.85 ± 0.26 | 1.12 ± 0.42 | 0.98 ± 0.42 | 1.15 ± 0.21 | 0.73 ± 0.30 |
| Spleen | 0.59 ± 0.31 | 0.22 ± 0.04 | 0.92 ± 0.29 | 0.71 ± 0.34 | 0.60 ± 0.13 | 0.54 ± 0.46 |
| Pancreas | 0.51 ± 0.30 | 0.14 ± 0.16 | 0.35 ± 0.04 | 0.26 ± 0.25 | 0.51 ± 0.06 | 0.61 ± 1.24 |
| Thymus | 0.46 ± 0.09 | 0.10 ± 0.02 | 0.46 ± 0.08 | 0.10 ± 0.01 | 0.45 ± 0.06 | 0.12 ± 0.04 |
| Muscles | 0.38 ± 0.16 | 0.05 ± 0.01 | 0.35 ± 0.03 | 0.09 ± 0.04 | 0.30 ± 0.08 | 0.13 ± 0.09 |
| Lung | 1.06 ± 0.18 | 0.22 ± 0.06 | 2.30 ± 0.69 | 2.16 ± 1.36 | 1.41 ± 0.24 | 0.42 ± 0.16 |
| Heart | 0.58 ± 0.07 | 0.10 ± 0.04 | 0.64 ± 0.06 | 0.13 ± 0.04 | 0.81 ± 0.21 | 0.17 ± 0.06 |
| Femur | 0.56 ± 0.03 | 0.16 ± 0.02 | 0.45 ± 0.01 | 0.14 ± 0.03 | 0.51 ± 0.07 | 0.16 ± 0.04 |
| Testicles | 0.25 ± 0.13 | 0.09 ± 0.02 | 0.34 ± 0.14 | 0.13 ± 0.03 | 0.34 ± 0.08 | 0.16 ± 0.03 |
| Hadrian glands | 0.57 ± 0.08 | 0.12 ± 0.03 | 0.58 ± 0.18 | 0.18 ± 0.15 | 0.59 ± 0.09 | 0.17 ± 0.04 |
| Brain | 0.04 ± 0.01 | 0.01 ± 0.00 | 0.04 ± 0.00 | 0.02 ± 0.02 | 0.07 ± 0.02 | 0.01 ± 0.01 |
| Hair & Skin | 0.79 ± 0.07 | 0.24 ± 0.11 | 0.93 ± 0.08 | 0.24 ± 0.03 | 0.86 ± 0.19 | 0.28 ± 0.07 |
In agreement with the concept of tumor pretargeting,102 all radiolabeled PNA conjugates were distributed by the blood stream very rapidly and were almost completely eliminated from the blood pool 60 min after administration. This minimizes unpredictable whole-body radiation exposure. As expected for compounds with molecular weights significantly lower than 30 kDa and of highly hydrophilic nature (see logDo/w values from Table 2), the activity was almost exclusively eliminated via the renal pathway.
Compared with previously published results on a 12-mer PNA conjugate [99mTc](Tc-Dpa-PNA),48 the expansion to a 17-mer conjugate enhanced blood availability 5 min p.i. by about 12% (Table 5).
| 12-mer [99mTc]-(Tc-Dpa)-PNA from ref. 48 | 17-mer [99mTc]-(Tc-Dpa)-PNA | 17-mer [99mTc](Tc-Dpa)-(Cys-PEG2kDa)-PNA | 17-mer [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA | |
|---|---|---|---|---|
| Blood 5 min p.i. (60 min p.i.) | 1.21 ± 0.05 (0.26 ± 0.10) | 1.36 ± 0.21 (0.24 ± 0.10) | 1.51 ± 0.13 (0.24 ± 0.06) | 2.01 ± 0.38 (0.34 ± 0.11) |
| Kidneys 5 min p.i. (60 min p.i.) | 7.12 ± 0.43 (5.45 ± 0.45) | 11.7 ± 1.60 (13.11 ± 1.99) | 12.0 ± 1.42 (9.29 ± 1.10) | 11.3 ± 1.86 (9.75 ± 2.64) |
| Liver 5 min p.i. (60 min p.i.) | 0.99 ± 0.03 (0.67 ± 0.10) | 0.91 ± 0.23 (0.85 ± 0.26) | 1.12 ± 0.42 (0.98 ± 0.42) | 1.15 ± 0.21 (0.73 ± 0.30) |
In combination with the attachment of a PEG moiety, we were able to further elevate blood availability to 25% and 66% for 2 kDa PEG and 10 kDa PEG, respectively. The increase in the length of the PNA sequence from 12-mer to 17-mer also led to higher kidney uptake from (7.12 ± 0.43) SUV to (11.71 ± 1.60) SUV and (5.45 ± 0.45) SUV to (13.11 ± 1.99) SUV 5 min and 60 min post injection, respectively. Similar relationship between the length of oligonucleotide sequence and kidney retention has been reported for morpholino-type oligonucleotides92 and has also been observed, in a much greater extent, for L-configured DNA-oligonucleotides in our laboratories.83
Based on compiled data (see Fig. 2), among the 17-mer PNA conjugates used in this study, [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA exhibits the highest activity concentration in the blood pool and the lowest activity concentration in liver and kidney tissue (60 min p.i.) combined with a trend of wash-out from those organs. This promising 17-mer PNA conjugate was therefore further evaluated by dynamic SPECT scans.
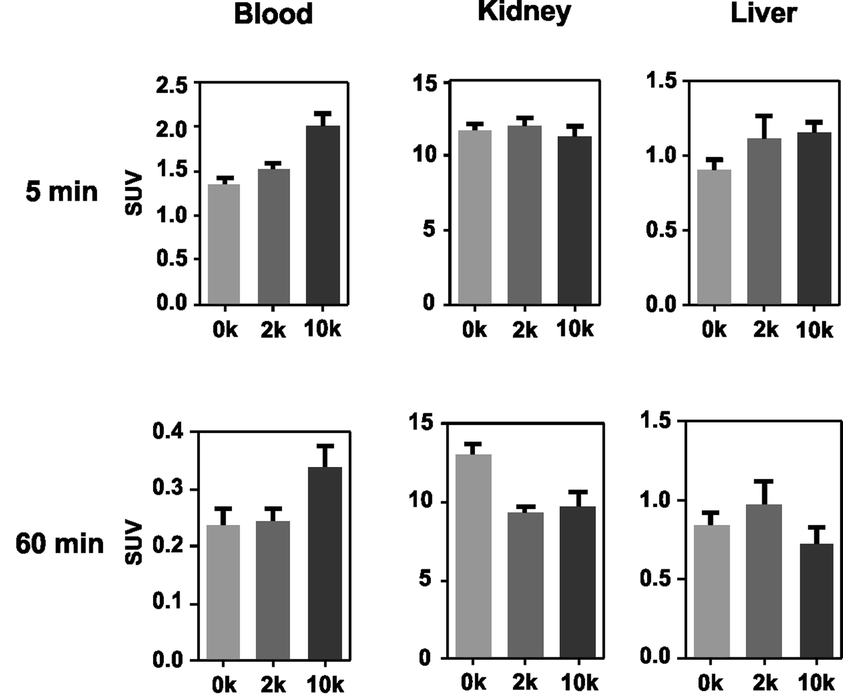 | ||
| Fig. 2 Comparison of activity concentration (SUV) in the blood, kidneys and liver of rats after single intravenous injection following sacrifice at 5 and 60 min p.i. | ||
SPECT image 5 min post injection (Fig. 3) substantiated an almost homogeneous blood distribution with enhanced activity concentration in heart, left and right carotids, both kidneys with hotspots at the renal pelvis, and bladder (urine). As demonstrated with the SPECT images at 60 min post injection, the majority of activity has been eliminated from blood pool via kidneys into the bladder. The calculated activity-under-curve (AUC) projection clearly shows removal of activity by renal pathway.
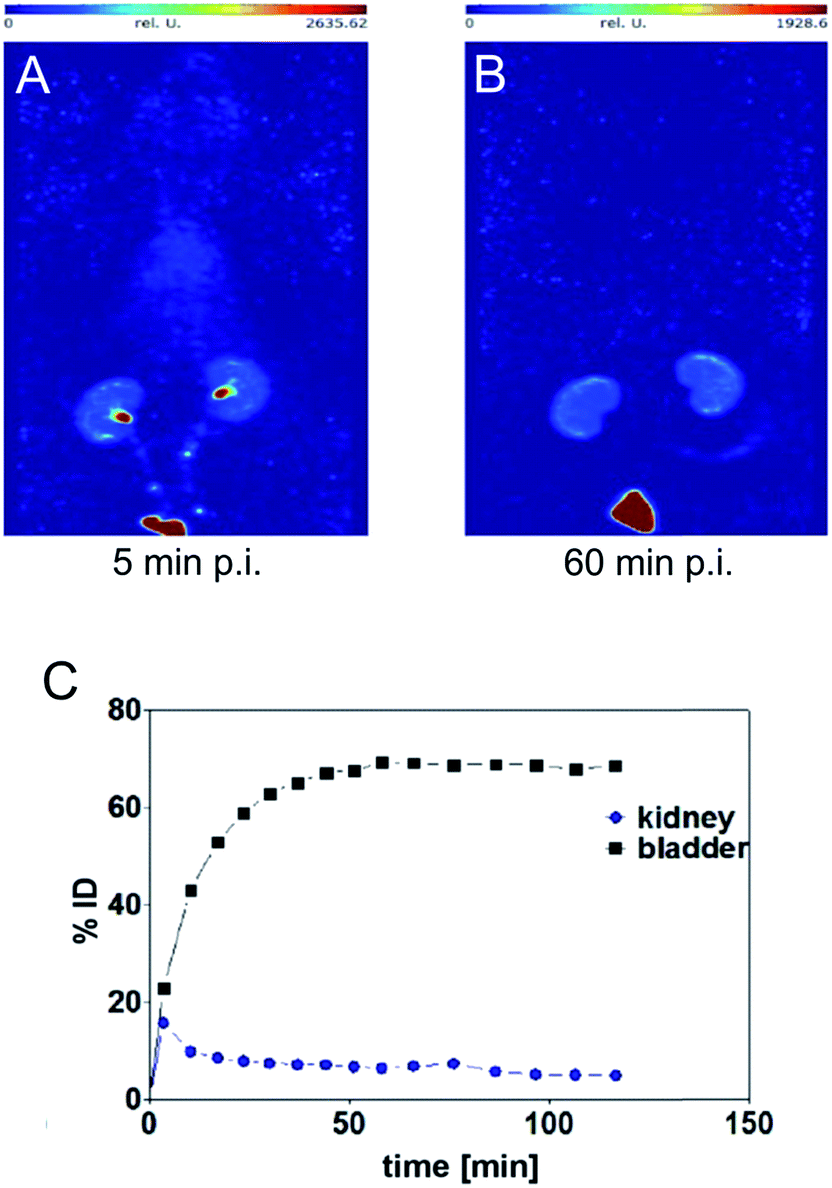 | ||
| Fig. 3 Maximum intensity projections generated from dynamic SPECT (A and B) and calculated activity-under-curve for bladder and kidney tissue of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA (C) after single intravenous administration in single healthy male Wistar rat (SPECT/CT images and tumor mouse are presented in Fig. S18†). | ||
Radio-HPLC analysis of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA samples from rat arterial blood, kidney extracts and urine showed no metabolic degradation over a time period of 120 min post injection (Fig. S17†).
Evaluation of the pretargeting approach
These promising radiopharmaceutical results strongly encouraged us to investigate the tumor pretargeting approach using Dpa-(Cys-PEG10kDa)-PNA. Tumor xenografts (epidermoid carcinoma) were obtained after subcutaneous injection of A431 cells into the right thigh of female NMRI nu/nu mice. Imaging and biodistribution studies were performed when the tumor were in the range of 8 to 13 mm.The control experiment in this xenograft model without pretreatment of (NOTA)3-C225-Cys-c-PNA before injection of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA showed no radioactivity localization in tumor site (Fig. 4A). The remaining radioactivity after 1 h p.i. is mainly located in the kidneys as expected on the basis of biodistribution experiments with rats described above.
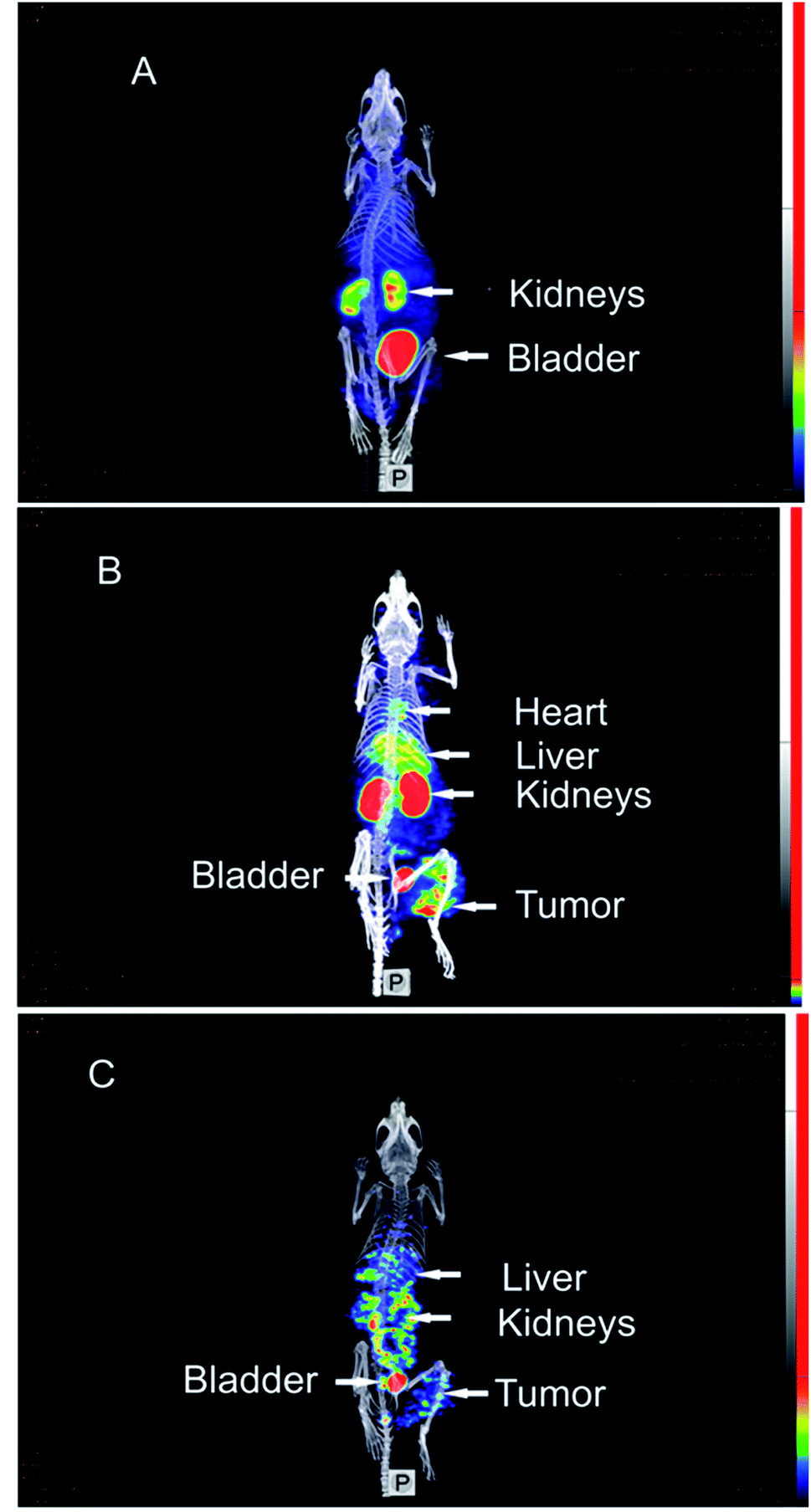 | ||
| Fig. 4 SPECT/CT maximum intensity projection images of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA in murine A431 tumor xenograft (NMRI nu/nu mice; tumor located at right thigh). (A) 1 h post injection of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA without preinjection of (NOTA)3-C225-Cys-c-PNA. (B) 1 h post injection of radiotracer; (NOTA)3-C225-Cys-c-PNA was administered 24 h before injection of radiotracer. (C) 20 h post injection of radiotracer; (NOTA)3-C225-Cys-c-PNA was administered 24 h before injection of radiotracer. | ||
When mice were pretreated with (NOTA)3-C225-Cys-c-PNA 24 h prior to administration of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA, SPECT images clearly demonstrated an accumulation of radioactivity at tumor site referring to efficient and rapid in vivo hybridization at pretargeted tumor tissue. The tumor is clearly visible after 60 min (Fig. 4B) and the radioactivity can be detected for at least 1 day (Fig. 4C). The enhanced radioactivity level in the kidneys, liver and blood compared to the control experiment is due to the circulating antibody conjugate. The activity wash-out from blood and tissues is faster relative to the tumor. Altogether, the tumor pretargeting using PNA allows for fast tumor localization and can be considered for improved in vivo targeting.
For more detailed evaluation of our pretargeting approach, we also conducted biodistribution studies in eight murine xenografts, applying the same experimental conditions as described for SPECT imaging (single intravenous injection of (NOTA)3-C225-Cys-c-PNA 24 h prior to administration of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA). 24 h post injection of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA, an enhanced radioactivity concentration of SUV 0.63 ± 0.27 was determined in tumor tissue (Fig. 5). Compared with non-targeted muscle tissue, a high contrast of tumor-to-muscle ratio of 8.29 ± 1.28 was achieved. The elevated levels of activity concentration in blood, liver, heart, and lung may be explained by incomplete blood elimination of (NOTA)3-C225-Cys-c-PNA. It is very likely that circulating (NOTA)3-C225-Cys-c-PNA formed hybrids with [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA in blood pool leading to enhanced retention time of activity in blood and aforementioned organs. This is in agreements with findings for PNA–streptavidin conjugates circulating in the blood to be able to efficiently bound radiolabeled complementary PNA.33
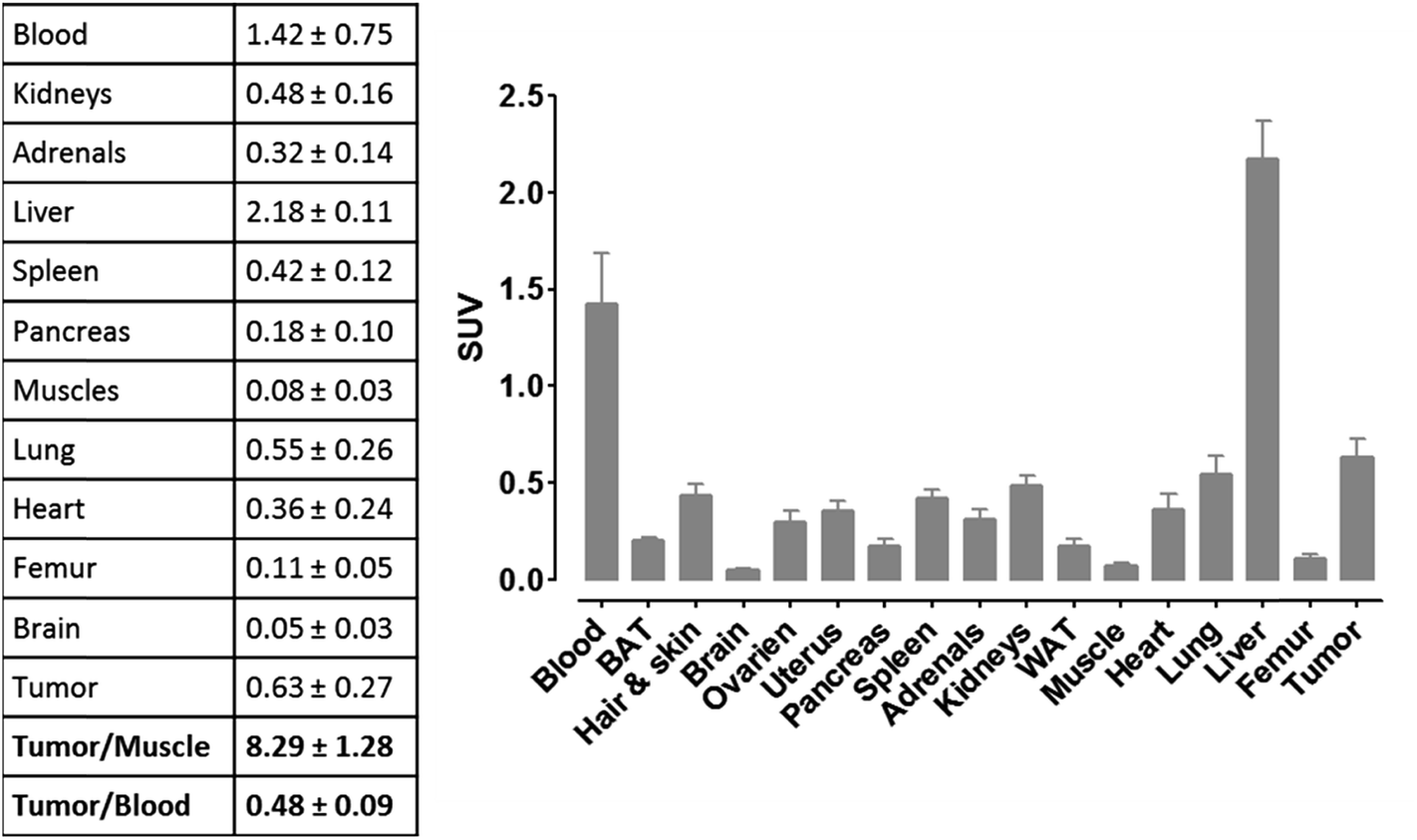 | ||
| Fig. 5 Biodistribution of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA (24 h post injection of radiotracer) in murine xenograft (NMRI nu/nu mice; A431 tumor cells transplanted into right thigh) 24 h after single administration of (NOTA)3-C225-Cys-c-PNA. Values in the table are averages in SUV including standard deviation (8 mice). | ||
Although a distinct accumulation of activity in the tumor site was observed 60 min after the administration of radiolabeled complementary PNAs to pretargeted mice, the resulting tumor-to-blood ratios might not be ideal. We determined that the waiting period of 24 h between both administrations represented the optimal balance in terms of rate of blood clearance and rate of internalization of PNA–cetuximab conjugates. On one hand, extended intervals such as 72 h would enhance blood clearance of the antibody–PNA conjugates as well as decrease activity concentration in blood, liver, heart, and lung. On the other hand, PNA–cetuximab conjugates bound to EGF-receptors are undergoing internalization via receptor-mediated endocytosis, which gradually diminishes the amount of hybridization events with radiolabeled complementary PNAs. For this reason, we believe that extended waiting will not significantly enhance tumor-to-background ratios.
Conclusions
The search for novel radiotheragnostic modalities for the detection and treatment of cancer is currently attracting a lot of attention worldwide. Among the different options investigated, the use of a pretargeting approach with the non-natural DNA/RNA analogues Peptide Nucleic Acids (PNAs) as recognition units is extremely attractive.33 By temporally separated administration of the antibody–PNA conjugate and its radiolabeled complementary PNA counterpart, the important limitations of conventional directly radiolabeled antibodies are overcome. Among these limitations, slow blood clearance has been identified as a major hurdle for diagnostic tumor discrimination, requiring extensive waiting times, which can last up to a week after administration before image acquisition. Furthermore, mAb accumulation in non-target tissues results in radiation damage to non-tumor cells and subsequently in severe toxicity.8 The herein exemplified pretargeting approach using PNAs facilitates the rational use of mAb conjugates for diagnostic and therapeutic purposes since it allows for sufficient time for the antibody–PNA conjugate to find the target tissue and for the rapid clearance of the radioactive PNA construct from circulation and normal tissues.By eliminating the hitherto existing limitations of PNAs such as insufficient water solubility as well as unfavorable biodistribution,41 we successfully optimized this complementary system for future pretargeting approaches. Nonetheless, the herein obtained results for our PNA-based approach cannot be juxtaposed with reported studies applying phosphorodiamidate morpholino oligomers (MORFs) due to major divergences with respect to animal models, tumor entities as well as tumor cell-related molecular targets and corresponding antibodies. Neglecting these facts, similar tumor uptake and non-target ratios were achieved.28,31 All in all, as demonstrated in this article, PNAs are a favorable alternative to MORFs for this field of research, especially when considering their relatively facile synthesis.
More specifically, in this article, we initially described the first detailed radiopharmaceutical evaluation of PNA bioconjugates for tumor pretargeting. We could then demonstrate that the PEGylation of PNA oligomers resulted into optimized pharmacokinetic properties. Compared with their non-PEGylated analogue, PEGylated PNAs showed lower kidney and liver accumulation, better renal excretion and a more beneficial residence time in blood. We also present a versatile conjugation protocol to modify the EGFR specific therapeutic antibody cetuximab. Coupling of a cysteine-functionalized PNA oligomer to the mAb equipped with maleimido functional groups was achieved at ambient temperature. As expected, under these conditions, the modification of cetuximab with the PNA conjugate did not affect its binding properties towards EGFR-positive tumor cells showing hence that this modified antibody could be used in our study. Very importantly, in vivo studies in tumor bearing mice demonstrated the high potential of the described pretargeting approach. Rapid and efficient in vivo hybridization of a fast-clearing radiolabeled complementary PNA with a cetuximab-PNA conjugate led to high specific tumor accumulation. The studies performed have shown that the 17-mer PNAs investigated are promising candidates for further preclinical studies. All in all, this study opens up new avenues not only in the field of radioimaging but also in the field of cancer radioimmunotherapy. We are currently analyzing if such an approach could be used to treat cancer by using therapeutic radionuclides such as 90Y, 177Lu, 186Re or 188Re.
Experimental section
Material and methods
Chemicals and solvents were of reagent grade or better and purchased from commercial suppliers and were used without further purification unless otherwise specified. Alpha-methoxy-omega-ethyl-maleimide poly(ethylene glycol) (MeO-PEG-mal; MeO-PEG2kDa-maleimide with PDI = 1.03; MeO-PEG10kDa-maleimide with PDI = 1.08) and N-alpha-(9-fluorenylmethyloxycarbonyl)-S-trityl-L-cysteine (Fmoc-L-Cys(Trt)-OH) were purchased from Iris Biotech. PNA monomers were supplied by Link technologies. TentaGel S RAM Lys(Boc) Fmoc resin was purchased from Rapp Polymere. 2,2′-dipicolylamine (Dpa-N3) was prepared following a previously reported procedure.48 All other chemicals were of reagent-grade and sourced from Sigma-Aldrich.ESI-MS spectra were recorded on a Bruker Esquire 6000. The matrix-assisted laser desorption/ionization time of flight mass spectrometry (MALDI-TOF) mass spectra were measured on a Bruker Daltonics Autoflex. The experiments were performed in reflector (RP) or linear (LP) mode with positive polarity using α-cyano-4-hydroxy-cinnamic acid on a Prespotted AnchorChip (PAC HCCA) or sinapinic acid (SA) as the matrix. LC-MS spectra were measured on an Acquity™ from Waters system equipped with a PDA detector and an auto sampler using an Agilent Zorbax 300SB-C18 analytical column (3.5 μm particle size, 300 Å pore size, 150 × 4.6 mm). This LC was coupled to an Esquire HCT from Bruker (Bremen, Germany) for the MS measurements. The LC run (flow rate: 0.3 mL min−1) was performed with a linear gradient of A (distilled water containing 0.1% v/v formic acid) and B (acetonitrile (Sigma-Aldrich HPLC-grade), containing 0.1% v/v formic acid); t = 0 min, 5% B; t = 3 min, 5% B; t = 17 min, 100% B; t = 20 min, 100% B; t = 25 min, 5% B. HPLC purification was performed on a Varian ProStar system equipped with a UV/Vis spectrometer and an Agilent Zorbax 300SB-C18 prep column (5 μm particle size, 300 Å pore size, 150 × 21.1 mm. Flow rate: 20 mL min−1). The runs were performed with a linear gradient of A (distilled water containing 0.1% v/v TFA) and B (acetonitrile (Sigma-Aldrich HPLC-grade), containing 0.1% v/v TFA). Preparative run: t = 0 min, 5% B; t = 17 min, 42% B; t = 25 min, 100% B; t = 30 min, 100% B; t = 32 min, 5% B (for Dpa-PNA, Dpa-Cys-PNA and Cys-c-PNA). Preparative runs: t = 0 min, 10% B; t = 24 min, 60% B; t = 25 min, 100% B; t = 30 min, 100% B; t = 32 min, 10% B (for Dpa-(Cys-PEGx)-PNA). The size exclusion purification was performed on an AKTAprime Plus system using HiTrap Desalting 5 × 5 mL GE Healthcare (10 mM HCl in distilled water, flow rate 3 mL min−1). Radio-HPLC of the 99mTc labeled Dpa-PNA derivatives were performed on a Perkin-Elmer system with quaternary pump (series 200 LC pump) equipped with a radio-detector (RAMONA from raytest), a UV/Vis-detector (LC 290 from Perkin-Elmer) and an Eurosphere 100 column (5 μm particle size, 200 mm × 4.5 mm, flow rate: 1 mL min−1). The runs were performed with a linear gradient of A (distilled water containing 0.1% v/v TFA) and B (acetonitrile Fisher HPLC-grade, containing 0.1% v/v TFA): t = 0 min, 0% B; t = 20 min, 100% B. Supernatants from samples of arterial blood plasma were analyzed on a Hewlett Packard system (series 1100) equipped with a radio-detector (RAMONA from raytest) and a Zorbax C18 300SB column (4 μm particle size, 9.4 × 250 mm, flow rate: 2 mL min−1, column temperature 30 °C). The runs were performed with a linear gradient of A (50 mM aqueous triethylamine-acetic acid buffer pH = 6.45) and B (acetonitrile Fisher HPLC-grade): t = 0 min, 5% B, t = 15 min, 50% B, t = 16 min 95% B, t = 20 min, 95% B. Radio-TLC of the 64Cu-labeled antibody conjugates [64Cu]Cu-(NOTA)3-C225 and [64Cu]Cu-(NOTA)3-C225-Cys-c-PNA were performed at ITLC-SA plates and 0.9% sodium chloride solution as mobile phase. UV/Vis measurements and hybridization studies were performed on a Specord 210 from Analytik Jena AG. To determine the concentrations of PNA-derivatives measurements carried out at 260 nm by 90 °C with following extinction coefficients: Cys-c-PNAε = 197 μL × nmol−1 × cm−1, Dpa-PNA, Dpa-(Cys-PEG2kDa)-PNA, and Dpa-(Cys-PEG10kDa)-PNAε = 182 μL × nmol−1 × cm−1. Cetuximab derivatives were measured at 280 nm by room temperature. The extinction coefficient was determined via UV/Vis calibration curve and linear regression analysis: C225ε = 217 ± 14 μL nmol−1 cm−1.
General chemistry
:2.5:5 v/v/v [3 × 1.5 mL (90 min each)]. The resulting solutions were first evaporated to dryness before being precipitated with ice-cold ether. The solids were centrifuged, washed with ice-cold ether and finally air-dried. The obtained crude oligomers were lyophilized in acetonitrile–water, purified and analyzed with RP-HPLC, and finally characterized with ESI and/or MALDI-TOF mass spectrometry.
Cys-c-PNA (H-Cys-spacer-spacer-ataatcacataacataa-Lys-NH2). To obtain Cys-c-PNA, Fmoc-Cys(Trt)-OH was added to the growing PNA chain on the beads by using the same protocol as for PNA monomers (described above). The resulting Cys-c-PNA was cleaved off the resin following the general protocol shown above and purified by preparative HPLC to yield white powder. Characterization: ESI-MS m/z 852.2 [M + 6H]6+, 730.8 [M + 7H]7+, 639.4 [M + 8H]8+, 568.8 [M + 9H]9+; MALDI-TOF (PAC HCCA, RP) m/z 5108.2 [M + H]1+, 5130.2 [M + Na]1+.
Dpa-PNA (H-Dpa-spacer-spacer-ttatgttatgtgattat-Lys-NH2). To append the Dpa ligand to PNA, 4-pentynoic acid was added to the N-terminus of the PNA sequence according to the procedure previously reported by Gasser et al.48 Dpa (5 equiv.) and CuI (2 equiv.) dissolved in a mixture DIPEA–DMF 1
:6 v/v (4.207 mL) were introduced into the syringe and the mixture was shaken overnight. The resin was thoroughly washed by DMF, DCM, ACN, EDTA 0.1 M, shaken in EDTA 0.1 M for 2 h (3×) and washed by ACN, DCM and DMF. The product was then cleaved off the resin (see the General procedure above) and isolated by preparative HPLC as white powder. Characterization: ESI-MS m/z 1085.2 [M + 5H]5+, 906.6 [M + 6H]6+, 775.5 [M + 7H]7+, 678.7 [M + 8H]8+, 603.4 [M + 9H]9+; 1097.9 [M + Cu+5H]5+, 915.3 [M + Cu+6H]6+, 785.1 [M + Cu+7H]7+, 686.6 [M + Cu+8H]8+, 610.5 [M + Cu + 9H]9+; 1066.8 [M − py(CH)2 + 5H]5+, 889.0 [M − py(CH)2 + 6H]6+, 762.3 [M + –py(CH)2 + 7H]7+, 667.1 [M + –py(CH)2 + 8H]8+, 593.1 [M + –py(CH)2 + 9H]9+. MALDI-TOF (SA, LP) m/z 5422.2 [M + H]1+, 5444.2 [M + Na]1+, 5485.9 [M + Cu + H]1+, 5393.8 [M − py(CH2) + Cu + H]1+, 5330.2 [M − py(CH2) + H]1+.
Dpa-Cys-PNA (H-Dpa-spacer-spacer-Cys-spacer-spacer-ttatgttatgtgattat-Lys-NH2). To allow PEG conjugation via Michael addition, cysteine residue was added to the PNA sequence as described above for Cys-PNA. Dpa ligand was introduced as specified above for DPA-PNA. The product was cleaved off the resin (see the General procedure) and purified by preparative HPLC to obtain white powder. Characterization: ESI-MS m/z 831.4 [M + 7H]7+, 727.6 [M + 8H]8+, 647.1 [M + 9H]9+. MALDI-TOF (SA, LP) m/z 5815.9 [M + H]1+, 5837.8 [M + Na]1+, 5879.4 [M + Cu + H]1+, 5787.2 [M − py(CH2) + Cu + H]1+, 5723.7 [M − py(CH2) + H]1+, 5745.7 [M − py(CH2) + Na]1+.
Dpa-(Cys-PEG x )-PNA . Dpa-Cys-PNA and TCEP (10 equiv.) were dissolved in distilled water (30 mL) and shaken overnight. The reaction mixture was then lyophilized, redissolved in 10 mM HCl and separated by size exclusion. The collected Dpa-Cys-PNA fractions were combined and split into two flasks (∼20 mL each). MeO-PEG2kDa-maleimide (5 equiv.) or MeO-PEG10kDa-Maleimide (5 equiv.) was added and the mixture was then shaken overnight. The reaction mixtures were lyophilized, purified and analyzed with RP-HPLC and characterized by ESI and MALDI-TOF mass spectrometry.
588.1 [M + H]1+, Dpa-PEG242-PNAm/z 16632.2 [M + H]1+, Dpa-PEG243-PNAm/z 16676.2 [M + H]1+, Dpa-PEG244-PNAm/z 16720.3 [M + H]1+, Dpa-PEG245-PNAm/z 16764.3 [M + H]1+, Dpa-PEG246-PNAm/z 16808.4 [M + H]1+, Dpa-PEG247-PNAm/z 16852.4 [M + H]1+, Dpa-PEG248-PNAm/z 16896.5 [M + H]1+, Dpa-PEG249-PNAm/z 16940.1 [M + H]1+, Dpa-PEG250-PNAm/z 16984.6 [M + H]1+, Dpa-PEG251-PNAm/z 17028.6 [M + H]1+, Dpa-PEG252-PNAm/z 17072.7 [M + H]1+, Dpa-PEG253-PNAm/z 17116.7 [M + H]1+, Dpa-PEG254-PNAm/z 17160.8 [M + H]1+, Dpa-PEG255-PNAm/z 17204.8 [M + H]1+, Dpa-PEG256-PNAm/z 17248.9 [M + H]1+, Dpa-PEG257-PNAm/z 17292.9 [M + H]1+, Dpa-PEG258-PNAm/z 17336.9 [M + H]1+, Dpa-PEG259-PNAm/z 17381.0 [M + H]1+, Dpa-PEG260-PNAm/z 17425.1 [M + H]1+. Corresponding [M + 2H]2+ states, such as m/z 8382.7 (Dpa-PEG245-PNA) were also observed.
Polydispersity index (PDI) calculation for PEGylated PNAs. PDI index was calculated using the following formula:
| PDI = Mw/Mn |
and Mn is number average molecular weight defined as
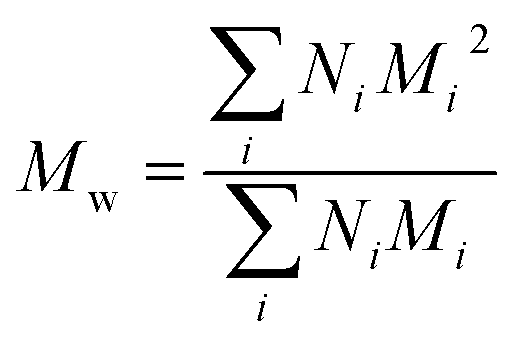
where Mi is mass of a polymer of a certain length and Ni is the amount of this polymer present.
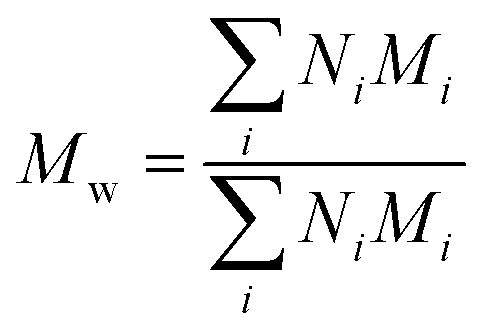
M n and Mw were estimated from MALDI spectra. As PEG polymers used for synthesis had PDI <1.1 and PNA was monodisperse, PDI of PEGylated PNAs was expected to be <1.1. Therefore, no mass discrimination effect should have interfered, so Ni was assumed to be proportional to peak intensity in MALDI spectra. Calculated PDIs corresponded to those of PEG starting material.
Bioconjugation chemistry
:1 with a resulting pH value of 6.9. The reaction mixture was left for 22 h at room temperature and the vial was swirled occasionally.
The reaction mixture was worked up by centrifugal filtration for 6 times (Jumbosep™ centrifugal devices; 30 kDa cut-off; 50 mM sodium bicarbonate saline buffer; pH 6.4; 2500 min−1, 60 min, 10 °C). Finally, the product solution was further concentrated to circa 1.0 mL by Macrosep™ Advance centrifugal device (30 kDa cut-off; 2500 min−1, 90 min, 10 °C). The recovery of the antibody was almost quantitatively (98% by UV/Vis measurement). This value was assumed as yield. MALDI-TOF (SA, LP): Gaussian distribution of peaks was observed; most intense peaks: m/z 154106 [M + H]1+, m/z 77184 [M + 2H]2+.
477 [M + H]1+, m/z 77984 [M + 2H]2+.
:1. The reaction mixture was left for 4 days at room temperature and the vial was swirled occasionally. The high viscosity of DMSO containing solution led to extraordinary slow centrifugal filtration (Macrosep™ Advance device, 30 kDa cut-off). The addition of 12 mL phosphate buffer was required to dilute the reaction mixture, which also led to precipitation of non-reacted Cys-c-PNA. Subsequently, the solution was transferred into several Protein low-bind tubes from Eppendorf, cooled to 10 °C for 2 h, and centrifuged for 60 min (2500 min−1, 10 °C). The resulting clear supernatant was transferred carefully into a Jumbosep™ device. After adding 40 mL of phosphate buffer the diluted reaction mixture was purified by centrifugal filtration. The purification using Jumbosep™ devices was performed 6 times. Finally, the product solution was further concentrated to circa 1.0 mL by Macrosep™ Advance centrifugal device (30 kDa cut-off; 2500 min−1, 90 min, 10 °C). MALDI-TOF (SA, LP): Gaussian distribution of peaks was observed; most intense peaks: m/z 168614.1 [M + H]1+, m/z 86022 [M + 2H]2+.
The number of bound Cys-c-PNA to cetuximab was also spectrophotometrically determined by measuring the absorbance at different wavelengths.92–94 The maximum absorbance of cetuximab was found to be at 280 nm and of Cys-c-PNA at 260 nm. Assuming that the conjugation of Cys-c-PNA to cetuximab will not influence the extinction coefficients of the individual compounds Lambert–Beer law were formulated at 260 nm and 280 nm. The extinction coefficients of cetuximab and Cys-c-PNA were determined via UV/Vis calibration curves and linear regression analysis: εcetuximab 280 nm = 217 ± 14 μL nmol−1 cm−1, εcetuximab 260 nm = 97 ± 6 μL nmol−1 cm−1, εCys-c-PNA 280 nm = 114 ± 4 μL nmol−1 cm−1 and εCys-c-PNA 260 nm = 192 ± 6 μL nmol−1 cm−1. Based on the 260 nm/280 nm absorbance ratio, the conjugation degree can be calculated with following equation:
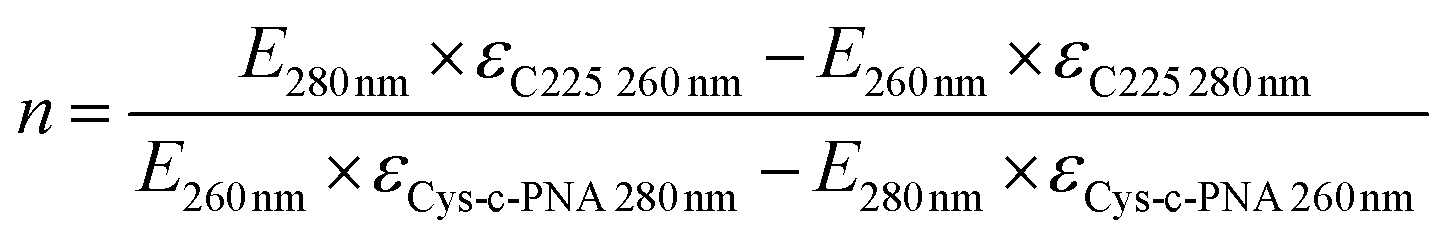
Radiochemistry
For each radiolabeling experiment 10 nmol of particular Dpa-PNA derivative from stock solution diluted in 400 μL of phosphate buffer (pH 5.4) were used. The labeling tube containing the PNA solution was gently flushed with argon for 5 min. Subsequently, approximately 400 μL [99mTc]Tc(CO)3(H2O)3+ kit solution (250–580 MBq) were added. The pH value of the radiolabeling mixture was tested by a triple zone pH-paper (Tritest pH 1–11). Optimal pH for radiolabeling ranges from 7 to 8. Occasionally, the pH value had to be adjusted by addition of further phosphate buffer (pH 5.4 or 8.2). The mixture was heated to 70 °C for 40 min and cooled to room temperature. The radiochemical yield (rcy) was determined by radio-HPLC. For HPLC injection purposes, 10 μL of labeling mixture were added to 90 μL of HPLC solvent A. For all radiolabeling experiments rcy of >95% determined from reaction mixtures (n = 19) were obtained. Decay corrected effective specific activities of up to 58 GBq μmol−1 were achieved. For in vivo studies (biodistribution and SPECT imaging) the radiolabeling mixtures were concentrated and re-buffered (sterile PBS) by centrifugal filtration (13200 min−1; 20 min; 25 °C; recovery of activity 70–80%). Centrifugal filtration was applied for purification purposes, if insufficient rcy (<95%) occurred. A typical volume of radiolabeled Dpa-PNA derivatives was 150–250 μL sterile phosphate buffered saline (PBS). Characterization: Radio-HPLC tR: 99mTcO4− 3.0 min; [99mTc(H2O)3(CO)3]+ 5.0–6.0 min; Dpa-PNA 10.5 min; Dpa-(Cys-PEG2kDa)-PNA 12.0 min; Dpa-(Cys-PEG10kDa)-PNA 12.9 min.
Hybridization studies
Biological studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) Do/w at 25 ± 1 °C.
Information on the lipophilicity of 99mTc-labeled Dpa-PNA, Dpa-(Cys-PEG2kDa)-PNA and Dpa-(Cys-PEG10kDa)-PNA was obtained by distribution experiments in a water/1-octanol system. All radiolabeled PNA derivatives were isolated by HPLC in radiochemical purity of >98%. Aliquots of 250 kBq were added to phosphate buffered saline with pH values of 7.2, 7.4 and 7.6 reaching a total volume of 500 μL in 2 mL microcentrifuge tubes. To this solution, 500 μL of 1-octanol were added and the two phases were agitated in a thermomixer for 30 min at (25 ± 1) °C. After centrifugation of samples, aqueous and organic phases were separated and aliquots of both phases were measured using an automated gamma counter (PerkinElmer Life and Analytical Sciences). Each value was recorded as triplet.
Do/w at 25 ± 1 °C.
Information on the lipophilicity of 99mTc-labeled Dpa-PNA, Dpa-(Cys-PEG2kDa)-PNA and Dpa-(Cys-PEG10kDa)-PNA was obtained by distribution experiments in a water/1-octanol system. All radiolabeled PNA derivatives were isolated by HPLC in radiochemical purity of >98%. Aliquots of 250 kBq were added to phosphate buffered saline with pH values of 7.2, 7.4 and 7.6 reaching a total volume of 500 μL in 2 mL microcentrifuge tubes. To this solution, 500 μL of 1-octanol were added and the two phases were agitated in a thermomixer for 30 min at (25 ± 1) °C. After centrifugation of samples, aqueous and organic phases were separated and aliquots of both phases were measured using an automated gamma counter (PerkinElmer Life and Analytical Sciences). Each value was recorded as triplet.
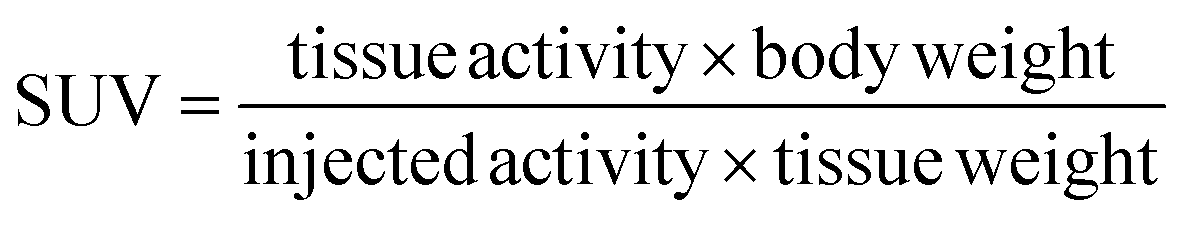 | (1) |
The activity amounts in the urine were calculated as difference between the injected dose and the recovery from all individual organs, tissues, blood and carcass.
Acknowledgements
We thank Karin Landrock for excellent technical assistance. This work was financially supported by the Swiss National Science Foundation (Professorships no. PP00P2_133568133568 and PP00P2PP00P2_157545157545 as well as Research Grants no. 200021_129910 and no. 200020_146776 to G.G), the University of Zurich (G.G), the Stiftung für Wissenschaftliche Forschung of the University of Zurich (G.G.), the Helmholtz Virtual Institute NanoTracking (Agreement no. VH-VI-421), the Research Department Interfacial Systems Chemistry at Ruhr University Bochum (N.M.-N.) and the COST Action CM1105 (N.M.-N and G.G.). This study is part of a research initiative Technologie und Medizin – Multimodale Bildgebung zur Aufklärung des in-vivo-Verhaltens von polymeren Biomaterialien of the Helmholtz-Portfoliothema.References
- J. Barbet, M. Bardies, M. Bourgeois, J. F. Chatal, M. Cherel, F. Davodeau, A. Faivre-Chauvet, J. F. Gestin and F. Kraeber-Bodere, Methods Mol. Biol., 2012, 907, 681–697 CAS
.
- I. Navarro-Teulon, C. Lozza, A. Pelegrin, E. Vives and J. P. Pouget, Immunotherapy, 2013, 5, 467–487 CrossRef CAS PubMed
- D. M. Goldenberg and R. M. Sharkey, Expert Opin. Biol. Ther., 2012, 12, 1173–1190 CrossRef CAS PubMed
- R. K. Jain, J. Natl. Cancer Inst., 1989, 81, 570–576 CrossRef CAS PubMed
- M. A. Tabrizi, C. M. Tseng and L. K. Roskos, Drug Discovery Today, 2006, 11, 81–88 CrossRef CAS PubMed
- S. M. Knowles and A. M. Wu, J. Clin. Oncol., 2012, 30, 3884–3892 CrossRef PubMed
- I. Vaneycken, M. D'Huyvetter, S. Hernot, J. De Vos, C. Xavier, N. Devoogdt, V. Caveliers and T. Lahoutte, Curr. Opin. Biotechnol., 2011, 22, 877–881 CrossRef PubMed
- P. Carter, Nat. Rev. Cancer, 2001, 1, 118–129 CrossRef CAS PubMed
- R. M. Sharkey and D. M. Goldenberg, Immunotherapy, 2011, 3, 349–370 CrossRef PubMed
- S. J. Goldsmith, Semin. Nucl. Med., 2010, 40, 122–135 CrossRef PubMed
- F. Kyle and R. Pettengell, Targeted Oncol., 2007, 2, 173–179 CrossRef
- E. Frampas, C. Rousseau, C. Bodet-Milin, J. Barbet, J. F. Chatal and F. Kraeber-Bodere, Front. Oncol., 2013, 3, 159 CAS
- R. M. Sharkey, H. Karacay, T. M. Cardillo, C. H. Chang, W. J. McBride, E. A. Rossi, I. D. Horak and D. M. Goldenberg, Clin. Cancer Res., 2005, 11, 7109s–7121s CrossRef CAS PubMed
- D. M. Goldenberg, R. M. Sharkey, G. Paganelli, J. Barbet and J. F. Chatal, J. Clin. Oncol., 2006, 24, 823–834 CrossRef CAS PubMed
- R. M. Sharkey, C. H. Chang, E. A. Rossi, W. J. McBride and D. M. Goldenberg, Tumor Biol., 2012, 33, 591–600 CrossRef CAS PubMed
- D. J. Hnatowich, F. Virzi and M. Rusckowski, J. Nucl. Med., 1987, 28, 1294–1302 CAS
- S. J. Knox, M. L. Goris, M. Tempero, P. L. Weiden, L. Gentner, H. Breitz, G. P. Adams, D. Axworthy, S. Gaffigan, K. Bryan, D. R. Fisher, D. Colcher, I. D. Horak and L. M. Weiner, Clin. Cancer Res., 2000, 6, 406–414 CAS
- S. Shen, A. Forero, A. F. LoBuglio, H. Breitz, M. B. Khazaeli, D. R. Fisher, W. Wang and R. F. Meredith, J. Nucl. Med., 2005, 46, 642–651 CAS
- C. Grana, M. Chinol, C. Robertson, C. Mazzetta, M. Bartolomei, C. De Cicco, M. Fiorenza, M. Gatti, P. Caliceti and G. Paganelli, Br. J. Cancer, 2002, 86, 207–212 CrossRef CAS PubMed
- E. Gautherot, J. Bouhou, J. M. Le Doussal, C. Manetti, M. Martin, E. Rouvier and J. Barbet, Cancer, 1997, 80, 2618–2623 CrossRef CAS PubMed
- J. M. Le Doussal, A. Chetanneau, A. Gruaz-Guyon, M. Martin, E. Gautherot, P. A. Lehur, J. F. Chatal, M. Delaage and J. Barbet, J. Nucl. Med., 1993, 34, 1662–1671 CAS
- E. Janevik-Ivanovska, E. Gautherot, M. Hillairet de Boisferon, M. Cohen, G. Milhaud, A. Tartar, W. Rostene, J. Barbet and A. Gruaz-Guyon, Bioconjugate Chem., 1997, 8, 526–533 CrossRef CAS PubMed
- R. M. Sharkey, W. J. McBride, H. Karacay, K. Chang, G. L. Griffiths, H. J. Hansen and D. M. Goldenberg, Cancer Res., 2003, 63, 354–363 CAS
- R. M. Sharkey, H. Karacay, W. J. McBride, E. A. Rossi, C. H. Chang and D. M. Goldenberg, Clin. Cancer Res., 2007, 13, 5577s–5585s CrossRef CAS PubMed
- F. Kraeber-Bodere, S. Bardet, C. A. Hoefnagel, M. R. Vieira, J. P. Vuillez, A. Murat, T. C. Ferreira, M. Bardies, L. Ferrer, I. Resche, E. Gautherot, E. Rouvier, J. Barbet and J. F. Chatal, Clin. Cancer Res., 1999, 5, 3190s–3198s CAS
- F. Kraeber-Bodere, C. Rousseau, C. Bodet-Milin, L. Ferrer, A. Faivre-Chauvet, L. Campion, J. P. Vuillez, A. Devillers, C. H. Chang, D. M. Goldenberg, J. F. Chatal and J. Barbet, J. Nucl. Med., 2006, 47, 247–255 CAS
- R. Schoffelen, O. C. Boerman, D. M. Goldenberg, R. M. Sharkey, C. M. van Herpen, G. M. Franssen, W. J. McBride, C. H. Chang, E. A. Rossi, W. T. van der Graaf and W. J. Oyen, Br. J. Cancer, 2013, 109, 934–942 CrossRef CAS PubMed
- G. Liu, K. Mang'era, N. Liu, S. Gupta, M. Rusckowski and D. J. Hnatowich, J. Nucl. Med., 2002, 43, 384–391 CAS
- G. Liu, S. Dou, Y. Liu, Y. Wang, M. Rusckowski and D. J. Hnatowich, Bioconjugate Chem., 2011, 22, 2539–2545 CrossRef CAS PubMed
- G. Liu, S. Dou, S. Baker, A. Akalin, D. Cheng, L. Chen, M. Rusckowski and D. J. Hnatowich, Cancer Biol. Ther., 2010, 10, 767–774 CrossRef CAS PubMed
- G. Liu, C. Liu, S. Zhang, J. He, N. Liu, S. Gupta, M. Rusckowski and D. J. Hnatowich, Nucl. Med. Commun., 2003, 24, 697–705 CrossRef CAS PubMed
- G. Mardirossian, K. Lei, M. Rusckowski, F. Chang, T. Qu, M. Egholm and D. J. Hnatowich, J. Nucl. Med., 1997, 38, 907–913 CAS
- M. Rusckowski, T. Qu, F. Chang and D. J. Hnatowich, Cancer, 1997, 80, 2699–2705 CrossRef CAS PubMed
- Y. Wang, F. Chang, Y. Zhang, N. Liu, G. Liu, S. Gupta, M. Rusckowski and D. J. Hnatowich, Bioconjugate Chem., 2001, 12, 807–816 CrossRef CAS PubMed
- D. M. Goldenberg, C. H. Chang, E. A. Rossi, W. J. McBride and R. M. Sharkey, Theranostics, 2012, 2, 523–540 CrossRef CAS PubMed
- R. Rossin, S. M. van Duijnhoven, T. Lappchen, S. M. van den Bosch and M. S. Robillard, Mol. Pharmaceutics, 2014, 11, 3090–3096 CrossRef CAS PubMed
- T. Reiner and B. M. Zeglis, J. Labelled Compd. Radiopharm., 2014, 57, 285–290 CrossRef CAS PubMed
- T. Reiner, J. S. Lewis and B. M. Zeglis, J. Visualized Exp., 2015, e52335 Search PubMed
- F. C. van de Watering, M. Rijpkema, M. Robillard, W. J. Oyen and O. C. Boerman, Front. Med., 2014, 1, 44 Search PubMed
- J. Summerton and D. Weller, Antisense Nucleic Acid Drug Dev., 1997, 7, 187–195 CrossRef CAS PubMed
- K. O. Mang'era, G. Liu, W. Yi, Y. Zhang, N. Liu, S. Gupta, M. Rusckowski and D. J. Hnatowich, Eur. J. Nucl. Med. Mol. Imaging, 2001, 28, 1682–1689 CrossRef PubMed
- M. R. Lewis and F. Jia, J. Cell. Biochem., 2003, 90, 464–472 CrossRef CAS PubMed
- S. Scarfi, M. Giovine, R. Pintus, E. Millo, E. Clavarino, M. Pozzolini, L. Sturla, R. P. Stock, U. Benatti and G. Damonte, Biotechnol. Appl. Biochem., 2003, 38, 61–69 CrossRef CAS PubMed
- J. Segura, C. Fillat, D. Andreu, J. Llop, O. Millan, B. G. de la Torre, Z. Nikolovski, V. Gomez, N. Andreu, A. Pinyot, R. Castelo, J. D. Gispert and J. A. Pascual, Ther. Drug Monit., 2007, 29, 612–618 CrossRef CAS PubMed
- Y. Shen, R. Shrestha, A. Ibricevic, S. P. Gunsten, M. J. Welch, K. L. Wooley, S. L. Brody, J. S. Taylor and Y. Liu, Interface Focus, 2013, 3, 20120059 CrossRef PubMed
-
C. Y. Shiue and S. Eck, in Handbook of Radiopharmaceuticals, ed. M. J. Welch and C. S. Redvanly, Wiley, New-York, USA, 2003, pp. 467–479 Search PubMed
- X. Tian, M. R. Aruva, H. R. Wolfe, W. Qin, E. R. Sauter, M. L. Thakur, S. A. Waldman and E. Wickstrom, Nucleosides, Nucleotides Nucleic Acids, 2005, 24, 1085–1091 CAS
- G. Gasser, K. Jäger, M. Zenker, R. Bergmann, J. Steinbach, H. Stephan and N. Metzler-Nolte, J. Inorg. Biochem., 2010, 104, 1133–1140 CrossRef CAS PubMed
- F. Chang, T. Qu, M. Rusckowski and D. J. Hnatowich, Appl. Radiat. Isot., 1999, 50, 723–732 CrossRef CAS PubMed
- I. Eke, M. Ingargiola, C. Förster, L. A. Kunz-Schughart, M. Baumann, R. Runge, R. Freudenberg, J. Kotzerke, J. M. Heldt, H. J. Pietzsch, J. Steinbach and N. Cordes, Int. J. Radiat. Biol., 2014, 90, 678–686 CrossRef CAS PubMed
- M. Ingargiola, R. Runge, J. M. Heldt, R. Freudenberg, J. Steinbach, N. Cordes, M. Baumann, J. Kotzerke, G. Brockhoff and L. A. Kunz-Schughart, Int. J. Cancer, 2014, 135, 968–980 CrossRef CAS PubMed
- L. Koi, R. Bergmann, K. Brüchner, J. Pietzsch, H. J. Pietzsch, M. Krause, J. Steinbach, D. Zips and M. Baumann, Radiother. Oncol., 2014, 110, 362–369 CrossRef CAS PubMed
- J. Saker, M. Kriegs, M. Zenker, J. M. Heldt, I. Eke, H. J. Pietzsch, R. Grenman, N. Cordes, C. Petersen, M. Baumann, J. Steinbach, E. Dikomey and U. Kasten-Pisula, J. Nucl. Med., 2013, 54, 416–423 CrossRef CAS PubMed
- M. Saki, M. Toulany, W. Sihver, M. Zenker, J. M. Heldt, B. Mosch, H. J. Pietzsch, M. Baumann, J. Steinbach and H. P. Rodemann, Strahlenther. Onkol., 2012, 188, 823–832 CrossRef CAS PubMed
- J. Harding and B. Burtness, Drugs Today, 2005, 41, 107–127 CrossRef CAS PubMed
- W. Sihver, J. Pietzsch, M. Krause, M. Baumann, J. Steinbach and H. J. Pietzsch, Pharmaceuticals (Basel), 2014, 7, 311–338 CrossRef CAS PubMed
- S. R. Hubbard and W. T. Miller, Curr. Opin. Cell Biol., 2007, 19, 117–123 CrossRef CAS PubMed
- J. Schlessinger, Cell, 2000, 103, 211–225 CrossRef CAS PubMed
- A. Gschwind, O. M. Fischer and A. Ullrich, Nat. Rev. Cancer, 2004, 4, 361–370 CrossRef CAS PubMed
- T. Holbro, G. Civenni and N. E. Hynes, Exp. Cell Res., 2003, 284, 99–110 CrossRef CAS PubMed
- N. E. Hynes and G. MacDonald, Curr. Opin. Cell Biol., 2009, 21, 177–184 CrossRef CAS PubMed
- G. Lurje and H. J. Lenz, Oncology, 2009, 77, 400–410 CrossRef CAS PubMed
- N. Tebbutt, M. W. Pedersen and T. G. Johns, Nat. Rev. Cancer, 2013, 13, 663–673 CrossRef CAS PubMed
- C. Yewale, D. Baradia, I. Vhora, S. Patil and A. Misra, Biomaterials, 2013, 34, 8690–8707 CrossRef CAS PubMed
-
G. Gasser, in Peptide Nucleic Acids: Methods and Protocols, ed. P. E. Nielsen and D. H. Appella, Humana Press, 2014, vol. 1050, pp. 55–72 Search PubMed
- G. Gasser, N. Hüsken, S. D. Köster and N. Metzler-Nolte, Chem. Commun., 2008, 3675–3677 RSC
-
T. Bruckdorfer, in European Biopharmaceutical Review, Spring, 2008, pp. 96, 98, 100, 102, 104 Search PubMed
- K. P. García, K. Zarschler, L. Barbaro, J. A. Barreto, W. O'Malley, L. Spiccia, H. Stephan and B. Graham, Small, 2014, 10, 2516–2529 CrossRef PubMed
- M. Hamidi, P. Rafiei and A. Azadi, Expert Opin. Drug Discovery, 2008, 3, 1293–1307 CrossRef CAS PubMed
-
S. Jevsevar and R. Kontermann, Half-life extension through PEGylation Therapeutic Proteins, Strategies to Modulate Their Plasma Half-life, 2012 Search PubMed
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288–6308 CrossRef CAS PubMed
- F. Veronese and G. Pasut, Drug Discovery Today, 2005, 10, 1451–1458 CrossRef CAS PubMed
- J. M. Harris and R. B. Chess, Nat. Rev. Drug Discovery, 2003, 2, 214–221 CrossRef CAS PubMed
- P. Anstaett, Y. Zheng, T. Thai, A. M. Funston, U. Bach and G. Gasser, Angew. Chem., Int. Ed., 2013, 52, 4217–4220 CrossRef CAS PubMed
- R. Bahal, N. A. McNeer, D. H. Ly, W. M. Saltzman and P. M. Glazer, Artificial DNA: PNA & XNA, 2013, 4, 49–57 Search PubMed
- G. M. Bonora, S. Drioli, M. Ballico, A. Faccini, R. Corradini, S. Cogoi and L. Xodo, Nucleosides, Nucleotides Nucleic Acids, 2007, 26, 661–664 CAS
- A. Cattani-Scholz, D. Pedone, F. Blobner, G. Abstreiter, J. Schwartz, M. Tornow and L. Andruzzi, Biomacromolecules, 2009, 10, 489–496 CrossRef CAS PubMed
- M. Dettin, D. Silvestri, R. Danesin, E. Cretaio, G. Picariello, E. Casarin, A. Sonato, F. Romanato and M. Morpurgo, Molecules, 2012, 17, 11026–11045 CrossRef CAS PubMed
- J. M. Goldman, L. A. Zhang, A. Manna, B. A. Armitage, D. H. Ly and J. W. Schneider, Biomacromolecules, 2013, 14, 2253–2261 CrossRef CAS PubMed
- L. M. Kundu, H. Tsukada, Y. Matsuoka, N. Kanayama, T. Takarada and M. Maeda, Anal. Chem., 2012, 84, 5204–5209 CrossRef CAS PubMed
- B. Sahu, I. Sacui, S. Rapireddy, K. J. Zanotti, R. Bahal, B. A. Armitage and D. H. Ly, J. Org. Chem., 2011, 76, 5614–5627 CrossRef CAS PubMed
- Z. Zhang, Y. Liu, C. Jarreau, M. J. Welch and J.-S. A. Taylor, Biomater. Sci., 2013, 1, 1055–1064 RSC
-
C. Foerster, M. Schubert, R. Bergmann, S. Vonhoff, S. Klussmann, M. Walther, J. Pietzsch, H.-J. Pietzsch and J. Steinbach, in Technetium and Other Radiometals in Chemistry and Medicine, ed. U. Mazzi, W. C. Eckelman and W. A. Volkert, SGEditoriali, Padova, Italy, 2010, pp. 357–362 Search PubMed
- Y. Humblet, Expert Opin. Pharmacother., 2004, 5, 1621–1633 CrossRef CAS PubMed
- B. Vincenzi, A. Zoccoli, F. Pantano, O. Venditti and S. Galluzzo, Curr. Cancer Drug Targets, 2010, 10, 80–95 CrossRef CAS PubMed
- T. M. Brand, M. Iida and D. L. Wheeler, Cancer Biol. Ther., 2011, 11, 777–792 CrossRef CAS PubMed
- K. Zarschler, K. Prapainop, E. Mahon, L. Rocks, M. Bramini, P. M. Kelly, H. Stephan and K. A. Dawson, Nanoscale, 2014, 6, 6046–6056 RSC
- C. S. Cutler, H. M. Hennkens, N. Sisay, S. Huclier-Markai and S. S. Jurisson, Chem. Rev., 2013, 113, 858–883 CrossRef CAS PubMed
- E. W. Price and C. Orvig, Chem. Soc. Rev., 2014, 43, 260–290 RSC
- C. F. Ramogida and C. Orvig, Chem. Commun., 2013, 49, 4720–4739 RSC
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS PubMed
- G. Liu, S. Zhang, J. He, N. Liu, S. Gupta, M. Rusckowski and D. J. Hnatowich, Q. J. Nucl. Med., 2002, 46, 233–243 CAS
- J. He, G. Liu, S. Dou, S. Gupta, M. Rusckowski and D. Hnatowich, Bioconjugate Chem., 2007, 18, 983–988 CrossRef CAS PubMed
- W. H. Kuijpers, E. S. Bos, F. M. Kaspersen, G. H. Veeneman and C. A. van Boeckel, Bioconjugate Chem., 1993, 4, 94–102 CrossRef CAS PubMed
- R. Alberto, R. Schibli, A. Egli, A. P. Schubiger, U. Abram and T. A. Kaden, J. Am. Chem. Soc., 1998, 120, 7987–7988 CrossRef CAS
- J. Y. Song, S. W. Lee, J. P. Hong, S. E. Chang, H. Choe and J. Choi, Cancer Lett., 2009, 283, 135–142 CrossRef CAS PubMed
- H. Björkelund, L. Gedda and K. Andersson, PLoS One, 2011, 6, e16536 Search PubMed
- Z. Novy, P. Barta, J. Mandikova, M. Laznicek and F. Trejtnar, Nucl. Med. Biol., 2012, 39, 893–896 CrossRef CAS PubMed
- M. Azemar, M. Schmidt, F. Arlt, P. Kennel, B. Brandt, A. Papadimitriou, B. Groner and W. Wels, Int. J. Cancer, 2000, 86, 269–275 CrossRef CAS
- M. M. Moasser, A. Basso, S. D. Averbuch and N. Rosen, Cancer Res., 2001, 61, 7184–7188 CAS
- I. King and A. C. Sartorelli, Cancer Res., 1989, 49, 5677–5681 CAS
- D. T. Reardan, C. F. Meares, D. A. Goodwin, M. McTigue, G. S. David, M. R. Stone, J. P. Leung, R. M. Bartholomew and J. M. Frincke, Nature, 1985, 316, 265–268 CrossRef CAS PubMed
- K. Zarschler, S. Witecy, F. Kapplusch, C. Foerster and H. Stephan, Microb. Cell Fact., 2013, 12, 97 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Characterization of the PNAs used this study (Fig. S1–14), radio HPLC of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA (Fig. S15), melting temperatures (TM) for three complementary 17-mer PNA systems (Table S1), melting curves of different PNAs (Fig. S16), radio HPLC of the [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA (original), in rat arterial blood plasma (Fig. S17), biodistribution of radiolabeled PNAs in Wistar rats (%ID mean ± SD) (Table S2), biodistribution of radiolabeled PNAs in Wistar rats (SUV mean ± SD) (Table S3), SPECT/CT comparison of the biodistribution of [99mTc](Tc-Dpa)-(Cys-PEG10kDa)-PNA in rat and mouse (Fig. S18). See DOI: 10.1039/c5sc00951k |
| ‡ These authors have contributed equally to the work. |
| § Current address: Department of Oncology, University of Alberta, 11560 University Avenue, Edmonton, Alberta, T6G1Z2, Canada. |
| This journal is © The Royal Society of Chemistry 2015 |