DOI:
10.1039/C5SC00814J
(Edge Article)
Chem. Sci., 2015,
6, 6407-6412
A highly convergent synthesis of the C1–C31 polyol domain of amphidinol 3 featuring a TST-RCM reaction: confirmation of the revised relative stereochemistry†
Received
5th March 2015
, Accepted 30th April 2015
First published on 6th August 2015
Abstract
The concise enantioselective synthesis of the revised C1–C31 fragment of the polyketide amphidinol 3 was accomplished in 16 steps and 12.8% overall yield. Salient features of the strategy include chemoselective Weinreb amide coupling and concomitant CBS reduction for the preparation of the C1–C15 tris-syn-1,5-diol motif and a temporary silicon-tethered ring-closing metathesis (TST-RCM) reaction in combination with a diastereoselective hydroboration for the construction of the C16–C31 polypropionate fragment. The union of the fragments was accomplished by a regioselective ring-opening of the terminal epoxide with a phenyl sulfone stabilized carbanion, which upon reduction and deprotection permits a comparison of the relative configuration with the natural product.
Introduction
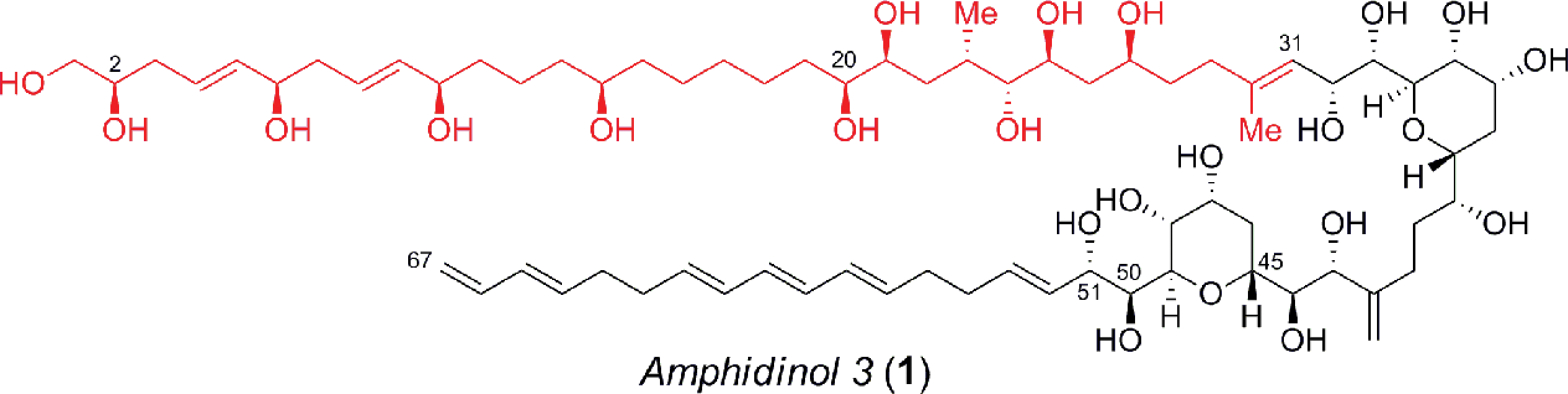
Amphidinols (AMs) and their congeners are structurally unique polyene–polyhydroxy secondary metabolites that belong to the linear polyether family isolated from the dinoflagellate Amphidinium species.1 In recent years there has been considerable interest in amphidinol 3 (1, Fig. 1), which was isolated in 1996 from A. klebsii in waters off the coast of Japan, due to its complex architecture and potent biological activity.1c For instance, the amphidinols exhibit antifungal, cytotoxic, hemolytic and anti-diatom activity, in which AM3 (1) exhibits the most potent antifungal activity (MEC = 4–9 μg per disk against Aspergillus niger), albeit with hemolytic action (EC50 = 0.009–0.4 μM against human erythrocyte cells). Interestingly, the mechanism of action for this agent has recently been attributed to its ability to form barrel-stave pores, similar to amphotericin B, which is induced by the stereospecific molecular recognition of membrane sterols.2,3 Specifically, the bis-tetrahydropyran core, which is highly conserved in this family, hydrogen bonds with the 3β-OH of ergosterol and cholesterol to permit the permeabilization of the membrane. The absolute and relative configuration of AM3 (1) was deduced using a combination of J-based configurational analysis (JBCA) for acyclic 1,2- and 1,3-dioxygenated systems, modified Mosher's method, NOE experiments and chiral HPLC analysis of degradation products.4 Nevertheless, the revision of the configuration at C2 and C51 has severely hampered progress towards the total synthesis of this agent.5 Hence, the unique molecular architecture and potent biological activity coupled with residual structural and mechanistic ambiguities have prompted several creative approaches6 to the C1–C31 polyol,7 C32–C51 bis-tetrahydropyran8 and the C52–C67 polyene,9 albeit many of which were accomplished prior to the stereochemical revisions outlined above. Herein, we now describe a novel and expeditious synthesis of the revised C1–C31 fragment of AM3 (1) using a highly convergent strategy, that confirms the relative configuration of this portion of the natural product.
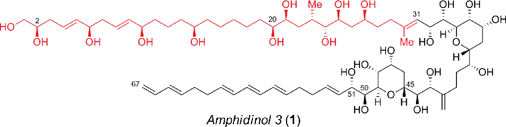 |
| | Fig. 1 Structure of the polyene–polyhydroxy secondary metabolite, amphidinol 3 (1). | |
Retrosynthetic analysis
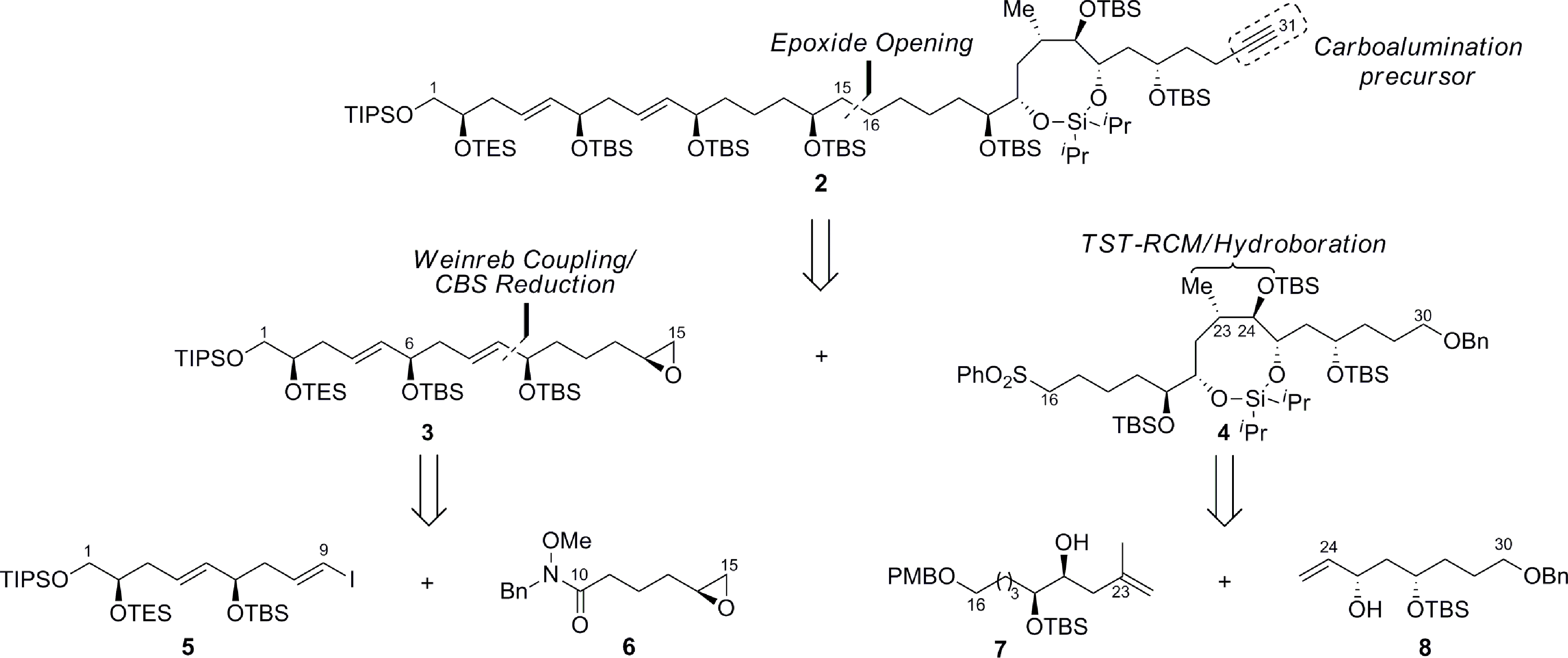
We envisioned the C1–C31 fragment, which is challenging due to the complications posed by the installation of remote stereochemistry in the acyclic linear carbon backbone, would be derived using the strategy outlined in Scheme 1. For instance, this motif has three syn-1,5-diols, two of which are separated by E-configured double bonds, coupled to a highly functionalized polyacetate/polypropionate type domain that is terminated with a trisubstituted E-olefin.
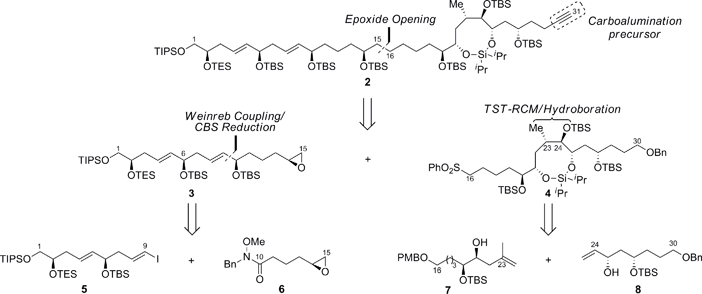 |
| | Scheme 1 Retrosynthetic analysis of the C1–C31 polyol fragment of amphidinol 3. TIPS = triisopropylsilyl, TBS = tert-butyldimethylsilyl, TES = triethylsilyl, Bn = benzyl, PMB = p-methoxybenzyl. | |
Hence, the ability to develop a highly convergent route to 2 would provide an opportunity to facilitate a Negishi carboalumination/Cram addition10 to enable the union with the C32–C67 segment and elaboration to the natural product. The retrosynthetic analysis of 2 affords two fragments, 3 and 4, of similar size and complexity, which we assumed could be coupled via the ring-opening of the terminal epoxide 3 with the lithiated sulfone derived from 4. The masked syn-1,5-tetraol 3 would in turn be prepared by the alkylation of the Weinreb amide 6 with an organometallic reagent derived from the vinyl iodide 5 followed by an enantioselective reduction of the resulting ketone. The preparation of the cyclic silaketal 4, which constitutes the aforementioned polyacetate/polypropionate type domain, relies on a Z-selective TST-RCM reaction for coupling 7 and 8 with concomitant diastereoselective hydroboration to facilitate the construction of the C23–C24 stereocenters using medium-ring stereocontrol.11,12
Results and discussion
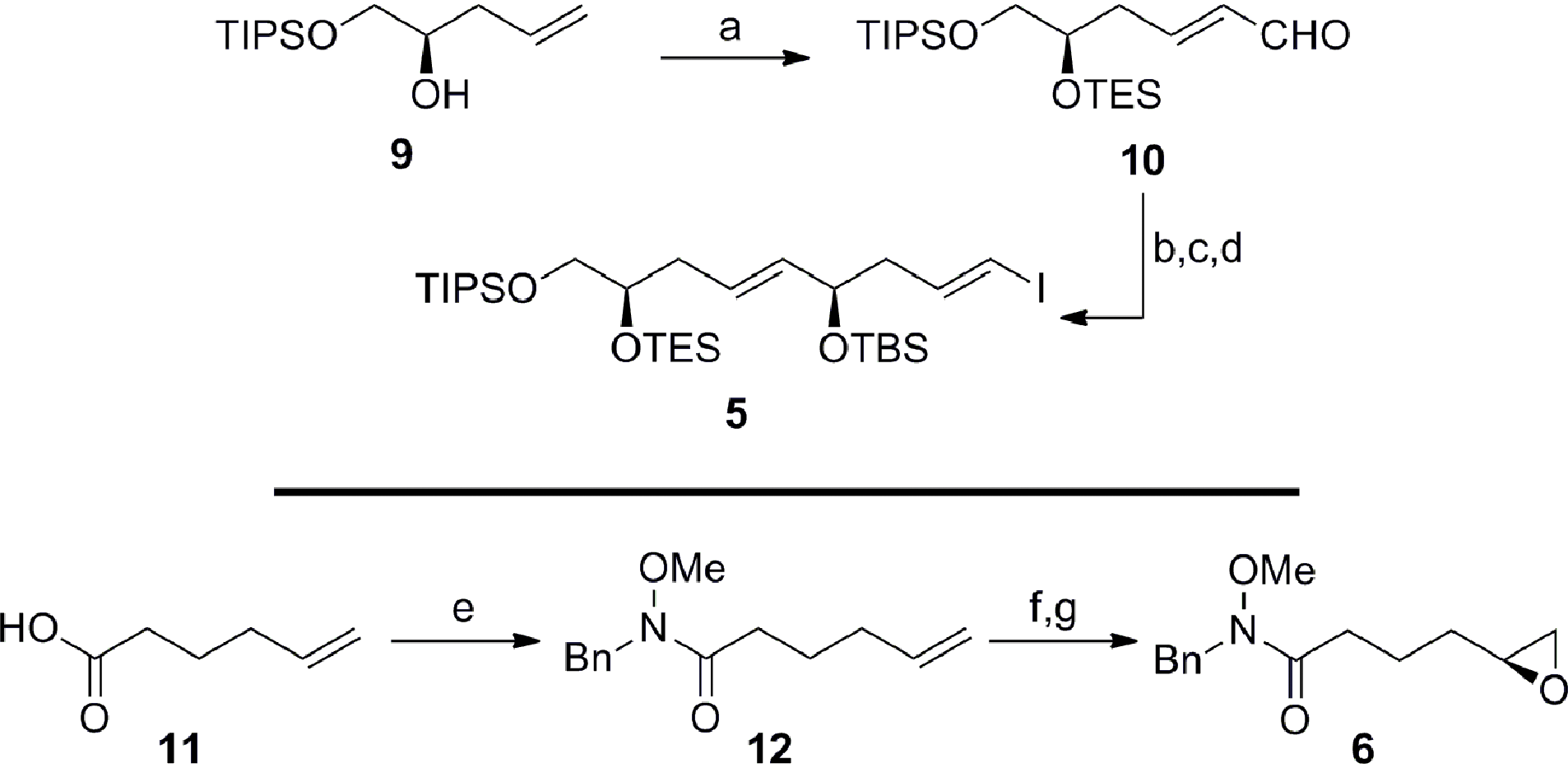
Guided by this strategy, we began our synthesis of the C1–C15 fragment 3 with the preparation of Weinreb coupling partners 5 and 6 (Scheme 2). Cross metathesis of the homoallylic alcohol 913 with excess acrolein using Hoveyda–Grubbs’ second-generation catalyst,14 followed by in situ protection of the secondary alcohol furnished enal 10 in 93% yield (E/Z ≥ 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by NMR). Treatment of the α,β-unsaturated aldehyde 10 with the chiral tin boronate derived from the combination of the allenyl stannane with (lIpc)2BH in diethyl ether at −78 °C, afforded the requisite vinyl stannane in 89% yield with excellent stereocontrol (ds ≥ 19:1 by NMR).15 Protection of the resulting secondary alcohol as the tert-butyldimethylsilyl ether and halogen–metal exchange of the vinyl stannane gave iodide 5 in 94% (over 2 steps), thereby completing the pronucleophile component. The preparation of the enantiomerically enriched Weinreb amide 6 originated with the conversion of 5-hexenoic acid 11 to the Weinreb amide 12 using carbonyldiimidazole and N-benzyl-O-methylhydroxylamine.16 Epoxidation of the terminal olefin in 12 with in situ generated DMDO provided the racemic epoxide,17 which was subjected to Jacobsen's hydrolytic kinetic resolution to furnish the enantiomerically enriched epoxide 6 (≥99% ee by HPLC).18
:1 by NMR). Treatment of the α,β-unsaturated aldehyde 10 with the chiral tin boronate derived from the combination of the allenyl stannane with (lIpc)2BH in diethyl ether at −78 °C, afforded the requisite vinyl stannane in 89% yield with excellent stereocontrol (ds ≥ 19:1 by NMR).15 Protection of the resulting secondary alcohol as the tert-butyldimethylsilyl ether and halogen–metal exchange of the vinyl stannane gave iodide 5 in 94% (over 2 steps), thereby completing the pronucleophile component. The preparation of the enantiomerically enriched Weinreb amide 6 originated with the conversion of 5-hexenoic acid 11 to the Weinreb amide 12 using carbonyldiimidazole and N-benzyl-O-methylhydroxylamine.16 Epoxidation of the terminal olefin in 12 with in situ generated DMDO provided the racemic epoxide,17 which was subjected to Jacobsen's hydrolytic kinetic resolution to furnish the enantiomerically enriched epoxide 6 (≥99% ee by HPLC).18
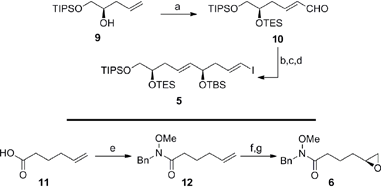 |
| | Scheme 2 Preparation of the C1–C9 iodide 5 and the C10–C15 epoxide 6. Conditions: (a) Acrolein, HG-II, CH2Cl2, 40 °C, then TESOTf, Et3N, CH2Cl2, −78 °C, 93%, E/Z ≥ 19:1; (b) AllenylSnBu3, (lIpc)2BH, Et2O, −40 °C to −20 °C, then 10, Et2O, −78 °C, 89%, ds ≥ 19:1; (c) TBSOTf, Et3N, CH2Cl2, 0 °C, 95%; (d) I2, Et2O, 0 °C, 99%; (e) CDI, then BnNH(OMe), CH2Cl2, 0 °C to RT, 92%; (f) Acetone, Oxone®, NaHCO3, EtOAc/H2O (1:1), RT, 98%; (g) (S,S)-Co-OAc, H2O, THF, RT, 60% (based on 50% conv.), ≥99% ee; HG-II = Hoveyda–Grubbs’ second-generation catalyst, Tf = trifluoromethanesulfonyl, Ipc = isopinocampheyl, CDI = 1,1′-carbonyldiimidazole, Oxone® = potassium peroxymonosulfate, THF = tetrahydrofuran. | |
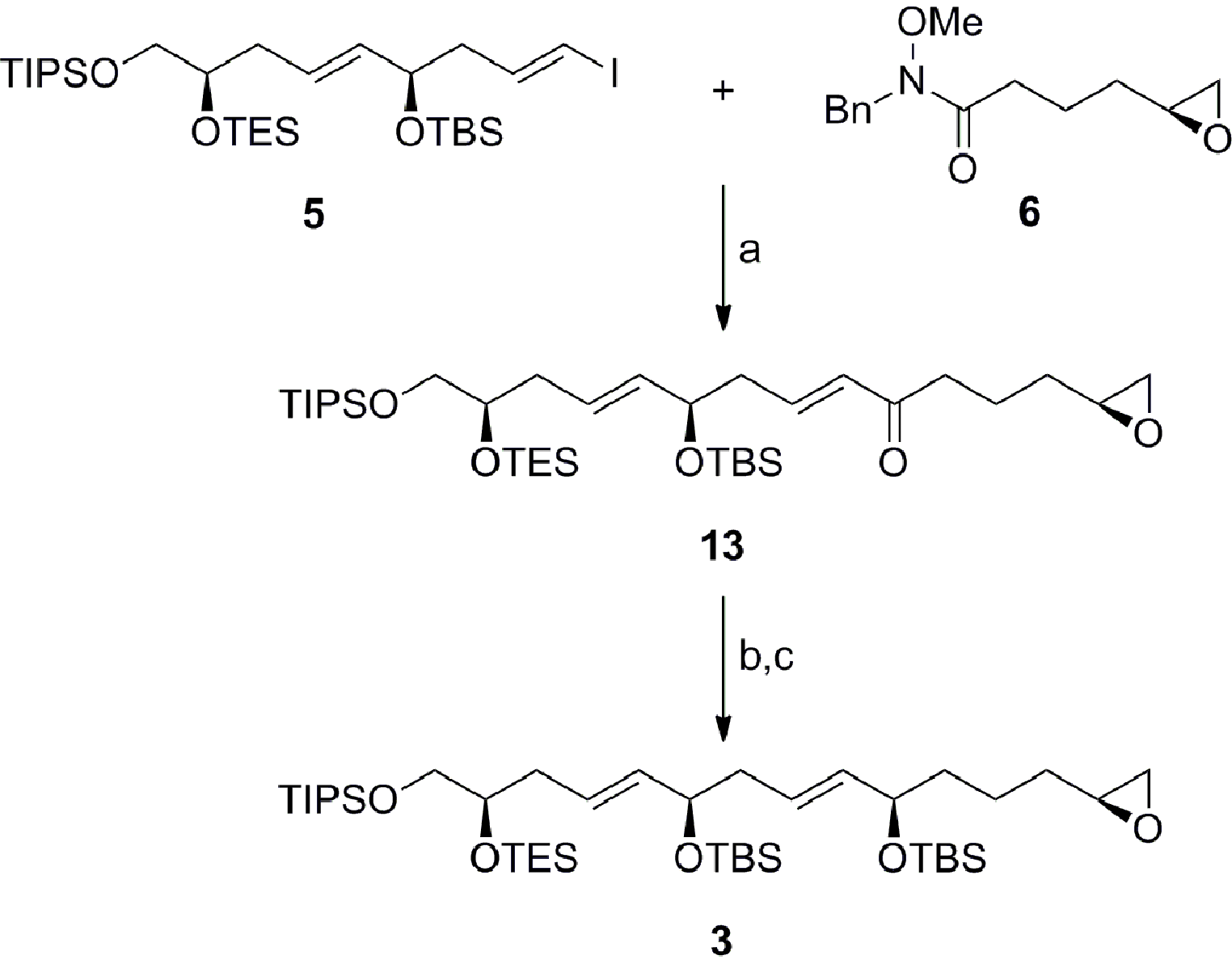
Scheme 3 outlines the coupling of the vinyl iodide 5 with the Weinreb amide 6 and elaboration to the terminal epoxide 3. Preliminary attempts to facilitate the coupling with the vinyl lithium reagent derived from 5 proceeded with moderate success, due to the reduction of the intermediary organometallic reagent. Gratifyingly, treatment of the vinyl iodide 5 with iPrMgCl·LiCl in the presence of 15-crown-5 followed by the addition of the Weinreb amide 6 furnished the α,β-unsaturated ketone 13 in 64% yield without erosion of olefin geometry.19 The fragment was then completed with the enantioselective CBS reduction of ketone 13 (ds ≥ 19:1 by NMR) and protection of the allylic alcohol to afford the C1–C15 fragment 3 in excellent overall yield.20
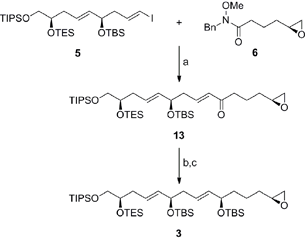 |
| | Scheme 3 Preparation of the C1–C15 fragment 3. Conditions: (a) iPrMgCl·LiCl, 15-crown-5, THF, −10 °C, 64%; (b) (S)-Me-CBS, BH3·DMS, THF, −40 °C, 99%, ds ≥ 19:1; (c) MTBSTFA, DMAP, MeCN, RT, 99%; (R)-Me-CBS = (R)-methyl oxazaborolidine, DMS = dimethyl sulfide, MTBSTFA = N-tert-butyldimethylsilyl-N-methyltrifluoroacetamide, DMAP = 4-(dimethylamino)pyridine. | |
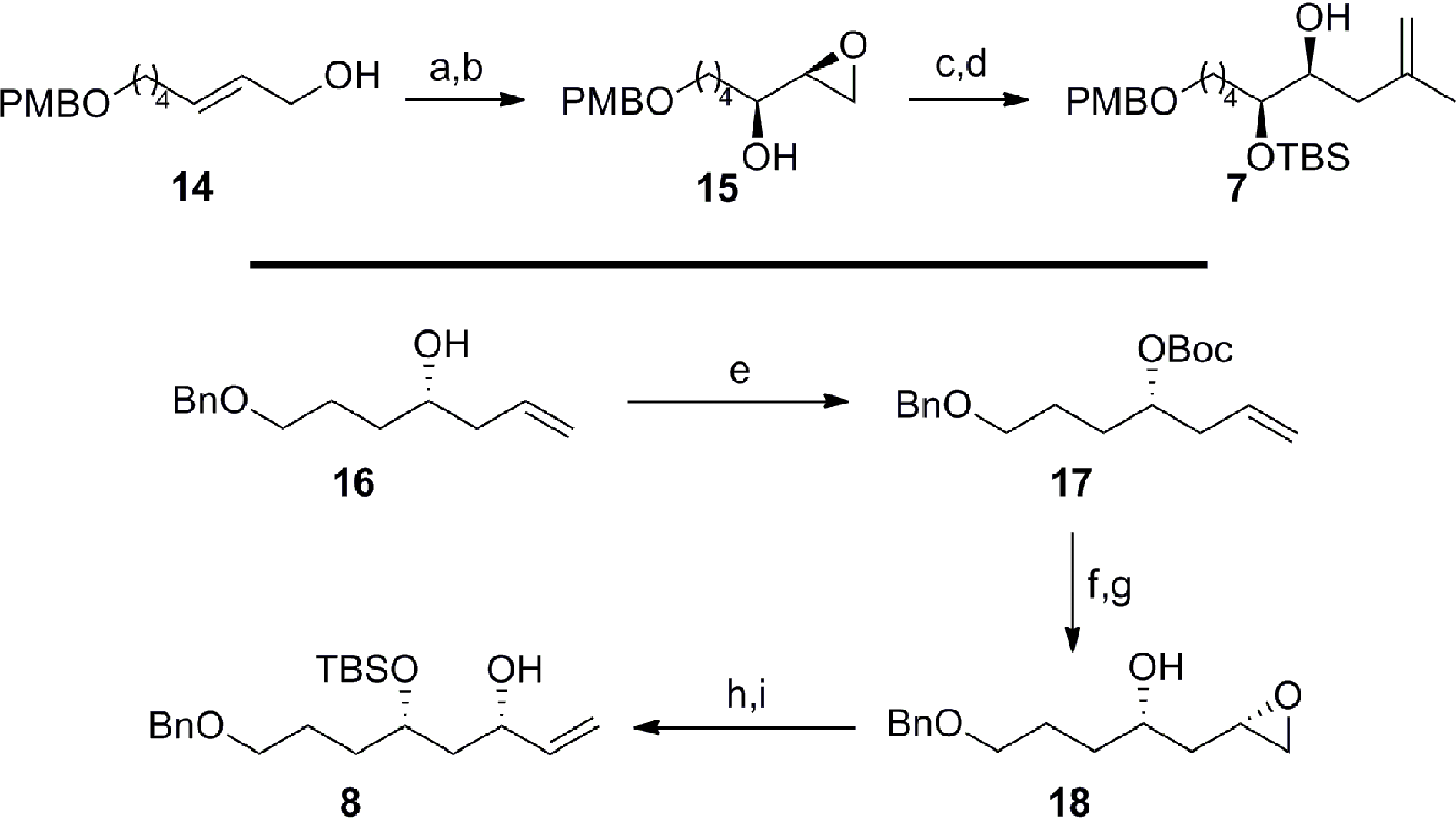
In concurrent work, we focused on the preparation of the fragments required for the key TST-RCM cross-coupling reaction (Scheme 4).21 Conversion of the allylic alcohol 1422 to the corresponding primary allylic bromide and concomitant Sharpless asymmetric dihydroxylation,23 afforded the required α-hydroxy epoxide 15 in 75% overall yield and with 92% enantiomeric excess (by 1H NMR analysis of the Mosher's ester). Protection of the secondary alcohol 15 as the tert-butyldimethylsilyl ether and regioselective ring-opening of the terminal epoxide with isopropenylmagnesium cuprate at −78 °C furnished 7 in 79% yield over two steps. The preparation of the allylic alcohol 8 commenced with Boc protection of the homoallylic alcohol 1624 to afford carbonate 17 in 95% yield. This substrate provided the necessary functionalization to affect the strategic 1,3-syn stereoinduction using IBr at low temperature to install the C25 stereocenter with good diastereocontrol (ds = 15:1 by NMR).25 Hydrolysis of the intermediate cyclic iodocarbonate with potassium carbonate in methanol furnished the β-hydroxy epoxide 18 in 81% yield over two steps. The allylic alcohol 8 was then completed in 89% overall yield with the protection of the secondary alcohol 18 as the tert-butyldimethylsilyl ether and ring-opening of the terminal epoxide with the sulfonium ylide, generated in situ from Me3SOTf.
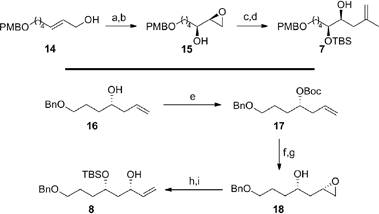 |
| | Scheme 4 Preparation of the C16–C23 fragment 7 and the C24–C30 fragment 8. Conditions: (a) Br2, PPh3, imid, 2-methyl-2-butene, CH2Cl2, 0 °C; (b) AD-mix-α, tBuOH/H2O (1:1), 0 °C, 75% (over 2 steps), 92% ee; (c) TBSCl, imid, CH2Cl2, 0 °C to RT, 80%; (d) Isopropenylmagnesium bromide, Li2[CuCl4], Et2O, −78 °C to RT, 99%; (e) Boc-ON, LiHMDS, THF, 0 °C, 95%; (f) IBr, PhMe, −85 °C, ds = 15:1; (g) K2CO3, MeOH, RT, 81% (over 2 steps); (h) TBSCl, TMEDA, DMF, 0 °C to RT, 97%; (i) Me3SOTf, nBuLi, THF, −10 °C to 0 °C, 92%; imid = imidazole, AD = asymmetric dihydroxylation, Boc-ON = 2-(tert-butoxycarbonyloxyimino)-2-phenylacetonitrile, HMDS = hexamethyldisilazane, PhMe = toluene, TMEDA = tetramethylethylenediamine, DMF = dimethylformamide. | |
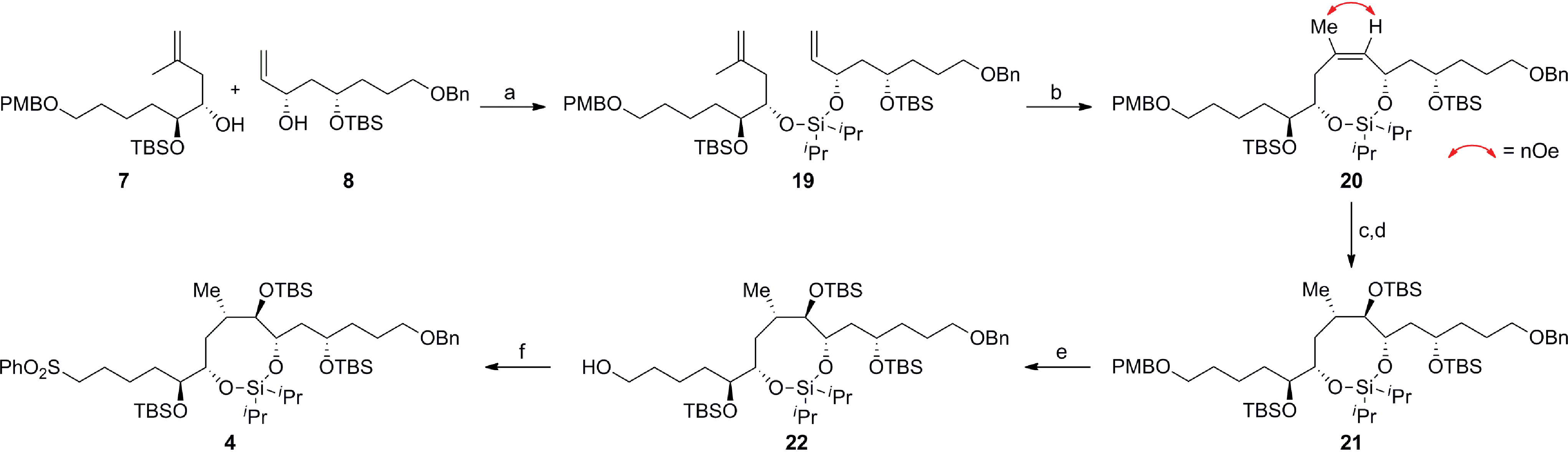
Scheme 5 delineates the TST-RCM coupling of the fragments 7 and 8 and subsequent elaboration to afford 4. Treatment of the homoallylic alcohol 7 with excess iPr2SiCl2 to afford the mono-alkoxychlorosilane, followed by removal of the excess tethering reagent and addition of the allylic alcohol 8, furnished the diene 19 in 84% yield,11,12 thereby setting the stage for the ring-closing metathesis reaction. Although preliminary studies demonstrated that the cyclization of 19 was particularly challenging, Grubbs' second-generation catalyst provided the optimal catalyst to afford the silaketal 20 in 97% yield with excellent Z/E selectivity (≥19:1 by NMR).26,27 Furthermore, this transformation was highly scalable and reproducible (>1 g scale). Diastereoselective hydroboration of the trisubstituted olefin in 20 provided the required anti-vic-alcohol using medium-ring stereocontrol (Fig. 2). Although the transformation was accompanied by the cleavage of a tert-butyldimethylsilyl ether group, this was inconsequential since the crude diol was silylated to afford the fully protected silaketal 21 in good overall yield as a single diastereoisomer (ds ≥ 19:1 by NMR). The origin of stereocontrol in the hydroboration is evident from the inspection of the molecular model of 20, which demonstrates the approach of the electrophile is favored from the convex face of the silaketal (Fig. 2).
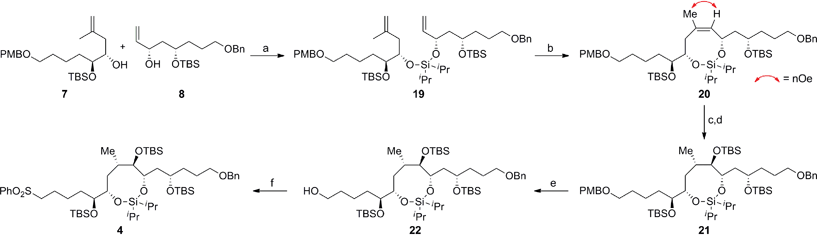 |
| | Scheme 5 Construction of the C16–C30 fragment 4 using the TST-RCM/hydroboration reaction. Conditions: (a) 7, iPr2SiCl2, imid, CH2Cl2, 0 °C to RT, then 8, imid, CH2Cl2, 0 °C to RT, 84%; (b) 2 × 15 mol% G-II, CH2Cl2, 40 °C, 97%, Z/E ≥ 19:1; (c) BH3·THF, THF, RT, then H2O2, NaOH, 0 °C to RT; (d) TBSOTf, Et3N, CH2Cl2, −40 °C, 72% (over 2 steps), ds ≥ 19:1; (e) DDQ, CH2Cl2/pH 7 buffer (20:1), 0 °C, 87%; (f) PhSSPh, PBu3, MeCN, RT, then TPAP, NMO, 40 °C, CH2Cl2, 76%; DDQ = 2,3-dichloro-5,6-dicyano-1,4-benzoquinone, imid = imidazole, TPAP = tetra-n-propylammonium perruthenate, NMO = 4-methylmorpholine N-oxide. | |
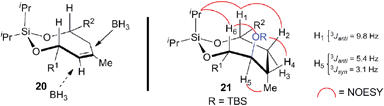 |
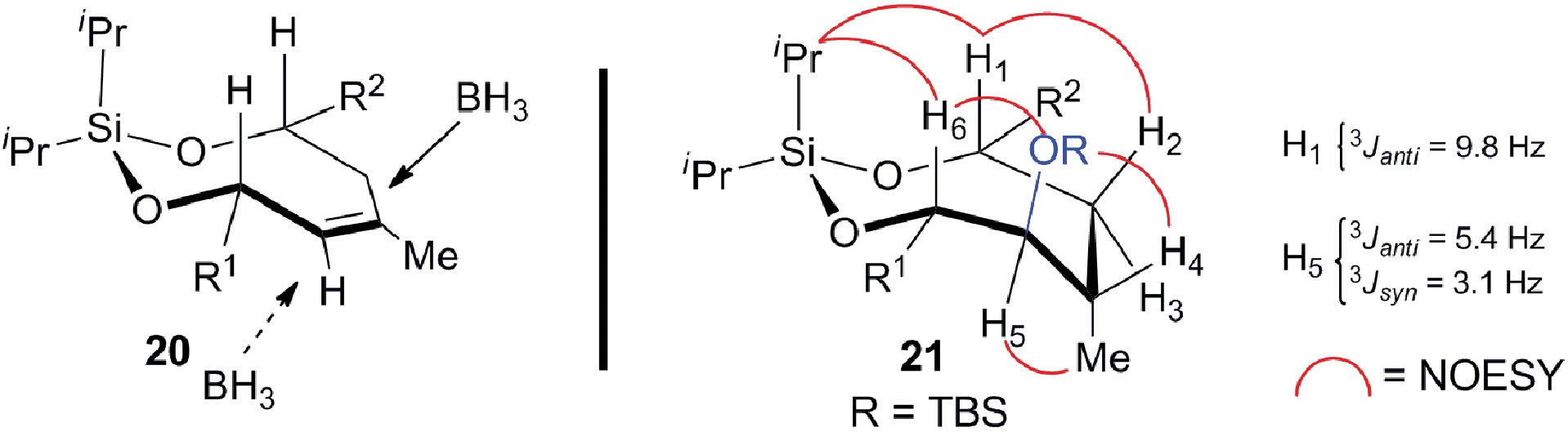
| | Fig. 2 Model for the stereocontrol in the hydroboration and the NMR analysis of the stereochemical outcome. | |
The relative stereochemistry of the hydroboration product 21 was assigned using a series of 2D NMR experiments in conjunction with coupling constant analysis, as outlined in Fig. 2. The observed spectroscopic data indicates that the silaketal 21 adopts a boat-chair conformation. The 1,2-diequatorial (gauche) coupling constant between H5 and H4 (3Jsyn = 3.1 Hz) and NOE correlation between H2–H1–iPr–H6–OR–H4, which reside on the same face of the molecule support this assignment. Furthermore, the pseudo-1,2-diaxial JH1,H3 (9.8 Hz) and 1,2-axial-equatorial JH5,H6 (5.4 Hz) coupling constants provide additional support for this connectivity. The sulfone 4 was completed in 66% overall yield by the chemoselective cleavage of the primary PMB ether 21 followed by a one-pot Mitsunobu/oxidation sequence on the primary alcohol 22.
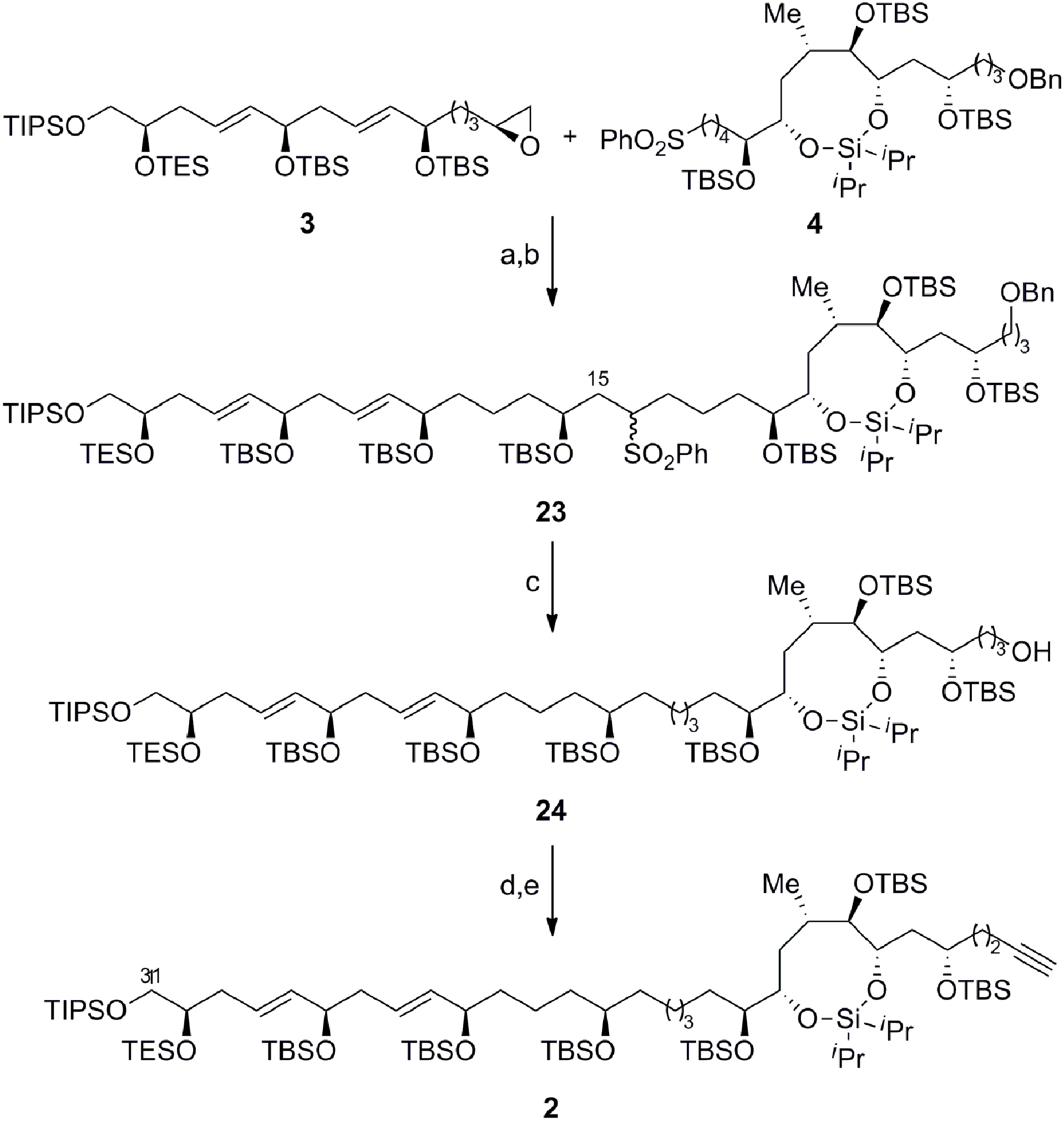
Scheme 6 outlines the union of the C1–C15 and C16–C30 fragments to complete the construction of the masked polyol 2. Following our initial plan, regioselective epoxide opening was achieved by lithiation of the phenyl sulfone 4 with nBuLi, followed by addition of the terminal epoxide 3 and BF3·Et2O at −78 °C to furnish the requisite β-hydroxysulfone intermediate. The silylation of the latter afforded the C15–C16 coupling product 23 in excellent overall yield as an inconsequential mixture of diastereoisomers at C16.29,30 The selective removal of the sulfone and the primary benzyl ether groups in 23 was achieved using a single-electron reduction with freshly prepared lithium di-tert-butylbiphenylide complex in THF at −78 °C to afford 24 in 64% yield. The resulting primary alcohol was oxidized to the aldehyde using TPAP31 and converted to the alkyne 2via Seyferth–Gilbert homologation with the Bestmann–Ohira reagent in 89% yield over 2 steps to complete the stereoselective construction of the C1–C31 fragment of AM3 (1).32 Chemoselective desulfonylation and deprotection of the silyl ethers in 23 afforded the polyol fragment to facilitate a direct comparison of the spectroscopic data (1H and 13C NMR) with the natural product to confirm the reassigned relative configuration of amphidinol 3 (1) as outlined in Fig. 3.
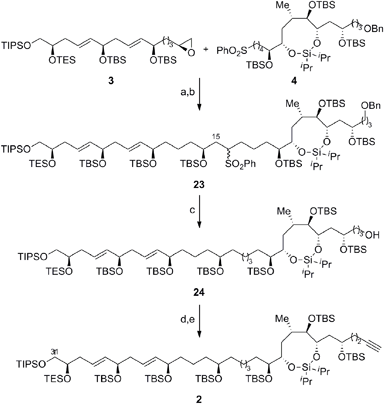 |
| | Scheme 6 Completion of the C1–C31 fragment of amphidinol 3. Conditions: (a) 4, nBuLi, THF, −78 °C, then 3, BF3·Et2O; (b) TBSOTf, Et3N, CH2Cl2, 0 °C, 90% (over 2 steps); (c) LiDBB, THF, −78 °C, 64%; (d) TPAP, NMO, molecular sieves (4 Å), CH2Cl2, 0 °C; (e) Me(CO)C(N2)P(O) (OMe)2, K2CO3, THF/MeOH (1:1), 0 °C to RT, 89% (over 2 steps); LiDBB = lithium di-tert-butylbiphenylide. | |
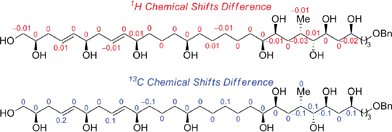 |
| | Fig. 3 Comparison of 1H and 13C NMR data of synthetic and natural polyol fragment of AM3. | |
Conclusion
In conclusion, we have developed an expeditious synthesis of the reassigned C1–C31 fragment of polyketide amphidinol 3 (1) in 12.8% overall yield using a 16-step longest linear sequence from 16. The strategy encompasses a high degree of convergence and allows for the convenient preparation of this intermediate for completion of the natural product. Our approach features the allylboration of an electron-deficient α,β-unsaturated aldehyde, mild iodine–magnesium exchange and chemoselective Weinreb amide coupling. Furthermore, the synthesis highlights the utility of the TST-RCM methodology for the non-aldol preparation of the polypropionate portion of AM3 (1) via the cross-coupling of advanced intermediates and a highly regio- and stereoselective electrophilic functionalization using medium-ring stereocontrol. Overall, this route provides the most expeditious approach to the polyol fragment of AM3 (1) developed to date and confirms the revised structure for the polyol domain of the natural product.
Acknowledgements
We sincerely thank NSERC for a Discovery Grant and a Tier 1 Canada Research Chair (PAE), in addition to a Royal Society for a Wolfson Research Merit Award (PAE). We acknowledge Alen Cusak for his preliminary efforts on the synthesis of this fragment and Dr. Rebecca L. Grange for assistance with obtaining mass spectral data for compounds 23, 24 and 2. We also recognize the EPSRC National Mass Spectrometry Service Centre (Swansea, UK) for high-resolution mass spectrometry.
Notes and references
- For isolation, structural characterization and bioactivity evaluation of the amphidinol family, see:
(a) M. Satake, M. Murata, T. Yasumoto, T. Fujita and H. Naoki, J. Am. Chem. Soc., 1991, 113, 9859 CrossRef CAS;
(b) G. K. Paul, N. Matsumori, M. Murata and K. Tachibana, Tetrahedron Lett., 1995, 36, 6279 CrossRef CAS;
(c)
G. K. Paul, N. Matsumori, K. Konoki, M. Sasaki, M. Murata and K. Tachibana, in Harmful and Toxic Algal Blooms, Proceedings of the Seventh International Conference on Toxic Phytoplankton, ed. T. Yasumoto, Y. Oshima and Y. Fukuyo, UNESCO, Sendai, Japan, 1996, p. 503 Search PubMed;
(d) G. K. Paul, N. Matsumori, K. Konoki, M. Murata and K. Tachibana, J. Mar. Biotechnol., 1997, 5, 124 CAS;
(e) R. Echigoya, L. Rhodes, Y. Oshima and M. Satake, Harmful Algae, 2005, 4, 383 CrossRef;
(f) N. Morsy, S. Matsuoka, T. Houdai, N. Matsumori, S. Adachi, M. Murata, T. Iwashita and T. Fujita, Tetrahedron, 2005, 61, 8606 CrossRef CAS;
(g) N. Morsy, T. Houdai, S. Matsuoka, N. Matsumori, S. Adachi, T. Oishi, M. Murata, T. Iwashita and T. Fujita, Bioorg. Med. Chem., 2006, 14, 6548 CrossRef CAS PubMed;
(h) Y. Meng, R. M. Van Wagoner, I. Misner, C. Tomas and J. L. C. Wright, J. Nat. Prod., 2010, 73, 409 CrossRef CAS PubMed;
(i) G. Nuzzo, A. Cutignano, A. Sardo and A. Fontana, J. Nat. Prod., 2014, 77, 1524 CrossRef CAS PubMed and pertinent references therein.
- For a recent report that proposes a barrel-stave pore model, see: R. A. Espiritu, N. Matsumori, M. Tsuda and M. Murata, Biochemistry, 2014, 53, 3287 CrossRef CAS PubMed and pertinent references cited therein.
-
(a) B. De Kruijff and R. A. Demel, Biochim. Biophys. Acta, 1974, 339, 57 CrossRef CAS PubMed;
(b) T. E. Andreoli, Ann. N. Y. Acad. Sci., 1974, 235, 448 CrossRef CAS PubMed.
- For the original structural elucidation of amphidinol 3 (1), see: M. Murata, S. Matsuoka, N. Matsumori, G. K. Paul and K. Tachibana, J. Am. Chem. Soc., 1999, 121, 870 CrossRef CAS.
- For structural revision of amphidinol 3 (1), see:
(a) T. Oishi, M. Kanemoto, R. Swasono, N. Matsumori and M. Murata, Org. Lett., 2008, 10, 5203 CrossRef CAS PubMed;
(b) R. T. Swasono, M. Kanemoto, N. Matsumori, T. Oishi and M. Murata, Heterocycles, 2011, 82, 1359 CAS;
(c) Y. Manabe, M. Ebine, N. Matsumori, M. Murata and T. Oishi, J. Nat. Prod., 2012, 75, 2003 CrossRef CAS PubMed;
(d) M. Ebine, M. Kanemoto, Y. Manabe, Y. Konno, K. Sakai, N. Matsumori, M. Murata and T. Oishi, Org. Lett., 2013, 15, 2846 CrossRef CAS PubMed.
- For a review of synthetic approaches towards AM3 (1), see: C. Bensoussan, N. Rival, G. Hanquet, F. Colobert, S. Reymond and J. Cossy, Nat. Prod. Rep., 2014, 31, 468 RSC.
- For approaches towards the polyol domain of AM3 (1), see:
(a) S. BouzBouz and J. Cossy, Org. Lett., 2001, 3, 1451 CrossRef CAS PubMed;
(b) E. M. Flamme and W. R. Roush, Org. Lett., 2005, 7, 1411 CrossRef CAS PubMed;
(c) L. A. Paquette and S.-K. Chang, Org. Lett., 2005, 7, 3111 CrossRef CAS PubMed;
(d) J. R. Huckins, J. de Vicente and S. D. Rychnovsky, Org. Lett., 2007, 9, 4757 CrossRef CAS PubMed;
(e) J. Cossy, T. Tsuchiya, S. Reymond, T. Kreuzer, F. Colobert and I. E. Markó, Synlett, 2009, 2706 CrossRef CAS;
(f) N. Rival, D. Hazelard, G. Hanquet, T. Kreuzer, C. Bensoussan, S. Reymond, J. Cossy and F. Colobert, Org. Biomol. Chem., 2012, 10, 9418 RSC;
(g) N. Rival, G. Hanquet, C. Bensoussan, S. Reymond, J. Cossy and F. Colobert, Org. Biomol. Chem., 2013, 11, 6829 RSC;
(h) T. Tsuruda, M. Ebine, A. Umeda and T. Oishi, J. Org. Chem., 2015, 80, 859. CrossRef PubMed
5a
.
- For approaches towards the tetrahydropyran domain of AM3 (1), see:
(a) J. de Vicente, B. Betzemeier and S. D. Rychnovsky, Org. Lett., 2005, 7, 1853 CrossRef CAS PubMed;
(b) C. Dubost, I. E. Markó and J. Bryans, Tetrahedron Lett., 2005, 46, 4005 CrossRef CAS;
(c) J. D. Hicks, E. M. Flamme and W. R. Roush, Org. Lett., 2005, 7, 5509 CrossRef CAS PubMed;
(d) S.-K. Chang and L. A. Paquette, Synlett, 2005, 2915 CAS;
(e) J. de Vicente, J. R. Huckins and S. D. Rychnovsky, Angew. Chem., Int. Ed., 2006, 45, 7258 CrossRef CAS PubMed;
(f) M. W. Bedore, S.-K. Chang and L. A. Paquette, Org. Lett., 2007, 9, 513 CrossRef CAS PubMed;
(g) J. D. Hicks and W. R. Roush, Org. Lett., 2008, 10, 681 CrossRef CAS PubMed;
(h) M. Kanemoto, M. Murata and T. Oishi, J. Org. Chem., 2009, 74, 8810 CrossRef CAS PubMed;
(i) M. T. Crimmins, T. J. Martin and T. A. Martinot, Org. Lett., 2010, 12, 3890 CrossRef CAS PubMed;
(j) E. Gálvez, M. Sau, P. Romea, F. Urpí and M. Font-Bardia, Tetrahedron Lett., 2013, 54, 1467 CrossRef;
(k) C. Bensoussan, N. Rival, G. Hanquet, F. Colobert, S. Reymond and J. Cossy, Tetrahedron, 2013, 69, 7759 CrossRef CAS.
- For approaches towards the polyene domain of AM3 (1), see: F. Colobert, T. Kreuzer, J. Cossy, S. Reymond, T. Tsuchiya, L. Ferrié, I. E. Marko and P. Jourdain, Synlett, 2007, 2351. CrossRef
8c,d,e,g
.
-
(a) N. Okukado and E. Negishi, Tetrahedron Lett., 1978, 19, 2357 CrossRef;
(b) D. A. Evans, D. M. Fitch, T. E. Smith and V. J. Cee, J. Am. Chem. Soc., 2000, 122, 10033 CrossRef CAS.
- For a review on the development of temporary silicon-tethered ring-closing metathesis reactions and application in synthesis, see: P. A. Evans, in Metathesis in Natural Product Synthesis, ed. J. Cossy, S. Arseniyadis and C. Meyer, Wiley-VCH, Weinheim, Germany, 2010, ch. 8, p. 225 Search PubMed.
-
(a) P. A. Evans and V. S. Murthy, J. Org. Chem., 1998, 63, 6768 CrossRef CAS PubMed;
(b) P. A. Evans, J. Cui and G. P. Buffone, Angew. Chem., Int. Ed., 2003, 42, 1734 CrossRef CAS PubMed;
(c) P. A. Evans, J. Cui, S. J. Gharpure, A. Polosukhin and H.-R. Zhang, J. Am. Chem. Soc., 2003, 125, 14702 CrossRef CAS PubMed.
- L. G. Quan, S.-H. Kim, J. C. Lee and J. K. Cha, Angew. Chem., Int. Ed., 2002, 41, 2160 CrossRef CAS PubMed.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360 CrossRef CAS PubMed.
- M. Chen, D. H. Ess and W. R. Roush, J. Am. Chem. Soc., 2010, 132, 7881 CrossRef CAS PubMed.
-
(a) S. Nahm and S. M. Weinreb, Tetrahedron Lett., 1981, 22, 3815 CrossRef CAS;
(b) C. Bernhart and C.-G. Wermuth, Tetrahedron Lett., 1974, 15, 2493 CrossRef.
- N. Hashimoto and A. Kanda, Org. Process Res. Dev., 2002, 6, 405 CrossRef CAS.
- S. E. Schaus, B. D. Brandes, J. F. Larrow, M. Tokunaga, K. B. Hansen, A. E. Gould, M. E. Furrow and E. N. Jacobsen, J. Am. Chem. Soc., 2002, 124, 1307 CrossRef CAS PubMed.
-
(a) H. Ren, A. Krasovskiy and P. Knochel, Org. Lett., 2004, 6, 4215 CrossRef CAS PubMed;
(b) A. Krasovskiy, B. F. Straub and P. Knochel, Angew. Chem., Int. Ed., 2006, 45, 159 CrossRef CAS PubMed;
(c) F. Li and S. L. Castle, Org. Lett., 2007, 9, 4033 CrossRef CAS PubMed.
- E. J. Corey and C. J. Helal, Angew. Chem., Int. Ed., 1998, 37, 1986 CrossRef CAS.
-
P. A. Evans, A. Cusak, A. Grisin and M. J. Lawler, submitted.
- M. E. Jung, J. A. Berliner, D. Angst, D. Yue, L. Koroniak, A. D. Watson and R. Li, Org. Lett., 2005, 7, 3933 CrossRef CAS PubMed.
- H. C. Kolb, M. S. VanNieuwenhze and K. B. Sharpless, Chem. Rev., 1994, 94, 2483 CrossRef CAS.
- G. K. Packard, Y. Hu, A. Vescovi and S. D. Rychnovsky, Angew. Chem., Int. Ed., 2004, 43, 2822 CrossRef CAS PubMed.
-
(a) P. A. Bartlett, J. D. Meadows, E. G. Brown, A. Morimoto and K. K. Jernstedt, J. Org. Chem., 1982, 47, 4013 CrossRef CAS;
(b) J. J. W. Duan and A. B. Smith III, J. Org. Chem., 1993, 58, 3703 CrossRef CAS.
- For the challenges associated with the formation of eight-membered rings containing di- and trisubstituted alkenes using RCM, see:
(a) S. J. Miller, S.-H. Kim, Z.-R. Chen and R. H. Grubbs, J. Am. Chem. Soc., 1995, 117, 2108 CrossRef CAS;
(b) A. Michaut and J. Rodriguez, Angew. Chem., Int. Ed., 2006, 45, 5740 CrossRef CAS PubMed.
- Interestingly, the ring-closing metathesis reaction with Hoveyda–Grubbs’ second-generation catalyst, which has been utilized to form both E- and Z-silaketals was completely ineffective for this process.28.
- R. Matsui, K. Seto, K. Fujita, T. Suzuki, A. Nakazaki and S. Kobayashi, Angew. Chem., Int. Ed., 2010, 49, 10068 CrossRef CAS PubMed.
- C. Greck, P. Grice, S. V. Ley and A. Wonnacott, Tetrahedron Lett., 1986, 27, 5277 CrossRef CAS.
- Although excess phenyl sulfone 4 is required in the coupling reaction, the unreacted material was recovered in almost quantitative yield.
- W. P. Griffith, S. V. Ley, G. P. Whitcombe and A. D. White, J. Chem. Soc., Chem. Commun., 1987, 1625 RSC.
- S. Muller, B. Liepold, G. J. Roth and H. J. Bestmann, Synlett, 1996, 521 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, spectral data, correlation with the natural product and copies of spectra. See DOI: 10.1039/c5sc00814j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a