
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNatural products triptolide, celastrol, and withaferin A inhibit the chaperone activity of peroxiredoxin I†
Qian
Zhao
a,
Yu
Ding
b,
Zhangshuang
Deng
c,
On-Yi
Lee
a,
Peng
Gao
d,
Pin
Chen
a,
Rebecca J.
Rose
e,
Hong
Zhao
f,
Zhehao
Zhang
a,
Xin-Pei
Tao
a,
Albert J. R.
Heck
e,
Richard
Kao
d and
Dan
Yang
*a
aMorningside Laboratory for Chemical Biology, Department of Chemistry, The University of Hong Kong, Hong Kong, China. E-mail: yangdan@hku.hk
bSchool of Life Sciences, Fudan University, Shanghai, China
cHubei Key Laboratory of Natural Product Research and Development, College of Biological and Pharmaceutical Sciences, China Three Gorges University, Yichang, China
dDepartment of Microbiology, The University of Hong Kong, Hong Kong, China
eBijvoet Center for Biomolecular Research and Utrecht Institute for Pharmaceutical Sciences, Utrecht University, Netherlands Proteomics Centre, The Netherlands
fDepartment of Chemistry, The Chinese University of Hong Kong, Hong Kong, China
First published on 19th May 2015
Abstract
Peroxiredoxin I (Prx I) plays an important role in cancer development and inflammation. It is a dual-functional protein which acts as both an antioxidant enzyme and a molecular chaperone. While there have been intensive studies on its peroxidase activity, Prx I's chaperone activity remains elusive, likely due to the lack of chaperone inhibitors. Here we report that natural product triptolide selectively inhibits the chaperone activity of Prx I, but not its peroxidase activity. Through direct interaction with corresponding cysteines, triptolide triggers dissociation of high-molecular-weight oligomers of Prx I, and thereby inhibits its chaperone activity in a dose-dependent manner. We have also identified celastrol and withaferin A as novel Prx I chaperone inhibitors that are even more potent than triptolide in the chaperone activity assay. By revealing the exact molecular mechanisms of interaction and inhibition, the current study provides the first Prx I chaperone inhibitors as promising pharmacological tools for modulating and dissecting the chaperone function of Prx I.
Introduction
Essentially every cellular process relies on close coordination of complex protein networks, in which a single protein can be involved in various pathways by playing different functional roles.1,2 To elucidate the contributions of a protein with multiple functions in regulating diverse cellular processes, methods are required to study each function of the protein independently and orthogonally. Compared with genetic manipulation, such as the knockdown approach, small-molecule inhibitors offer several advantages by modulating their target proteins with rapid onset, allowing precise temporal control over protein functions.3–5 More importantly, it is feasible that one function of a protein is inhibited while all other functions remain unaltered providing that selective inhibitors are available.6 This is particularly valuable in exploring complex cellular mechanisms of proteins with multiple functions.Peroxiredoxin I (Prx I) is a dual-functional protein that can act as both an antioxidant enzyme and molecular chaperone, depending on its oligomeric states.7 In the form of homo-dimers, Prx I functions as a peroxidase, catalysing removal of H2O2.7,8 Prx I is also present in the form of homodecamers or higher-order oligomers to function as a molecular chaperone that prevents client proteins from stress-induced aggregation.9,10 Recent studies reveal that, depending on its oligomeric structure or chaperone activity, Prx I interacts with essential signalling molecules, such as p53,11 NF-κB12 and TLR4,13 and thus plays an important role in normal cell physiology and disease pathology. However, the understanding of Prx I as a molecular chaperone and its potential to become a therapeutic target is limited by a lack of chaperone inhibitors. While adenanthin has been identified as an inhibitor of the peroxidase activity of Prx I,14 there is an unmet need for specific modulators of the Prx I chaperone activity.
Here we report the identification of triptolide (TL), a bioactive natural product, as a selective inhibitor of the chaperone activity of Prx I. TL specifically binds to Cys83 and Cys173, which play crucial roles in maintaining Prx I's decameric structure and chaperone activity. Using a competition assay based on a fluorescent triptolide derivative, the natural products celastrol (Cel) and withaferin A (WA) have also been found to interact with Prx I. Mass spectrometry analyses indicate that Cel and WA share the same mechanism with TL in binding to Prx I. The present study not only identifies three novel chaperone inhibitors of Prx I, but also demonstrates that small molecules, as promising pharmacological tools, can modulate a multi-functional protein.
Results
Chemical synthesis of probes Biotin-TL and Cy3-TL
We have been working on the chemical synthesis and biological mechanism of TL,15,16 a bioactive natural product, whose derivatives have entered human clinical trials for cancers and autosomal kidney diseases.17,18 Meanwhile, TL's cellular mechanism has also attracted much attention.19–22 To further elucidate its cellular mechanism, we synthesized a biotinylated triptolide (Biotin-TL) and a fluorescent cyanine-labelled triptolide (Cy3-TL) to enrich and visualize triptolide-binding proteins, respectively (Scheme 1). More details on the synthesis can be found in the ESI.†
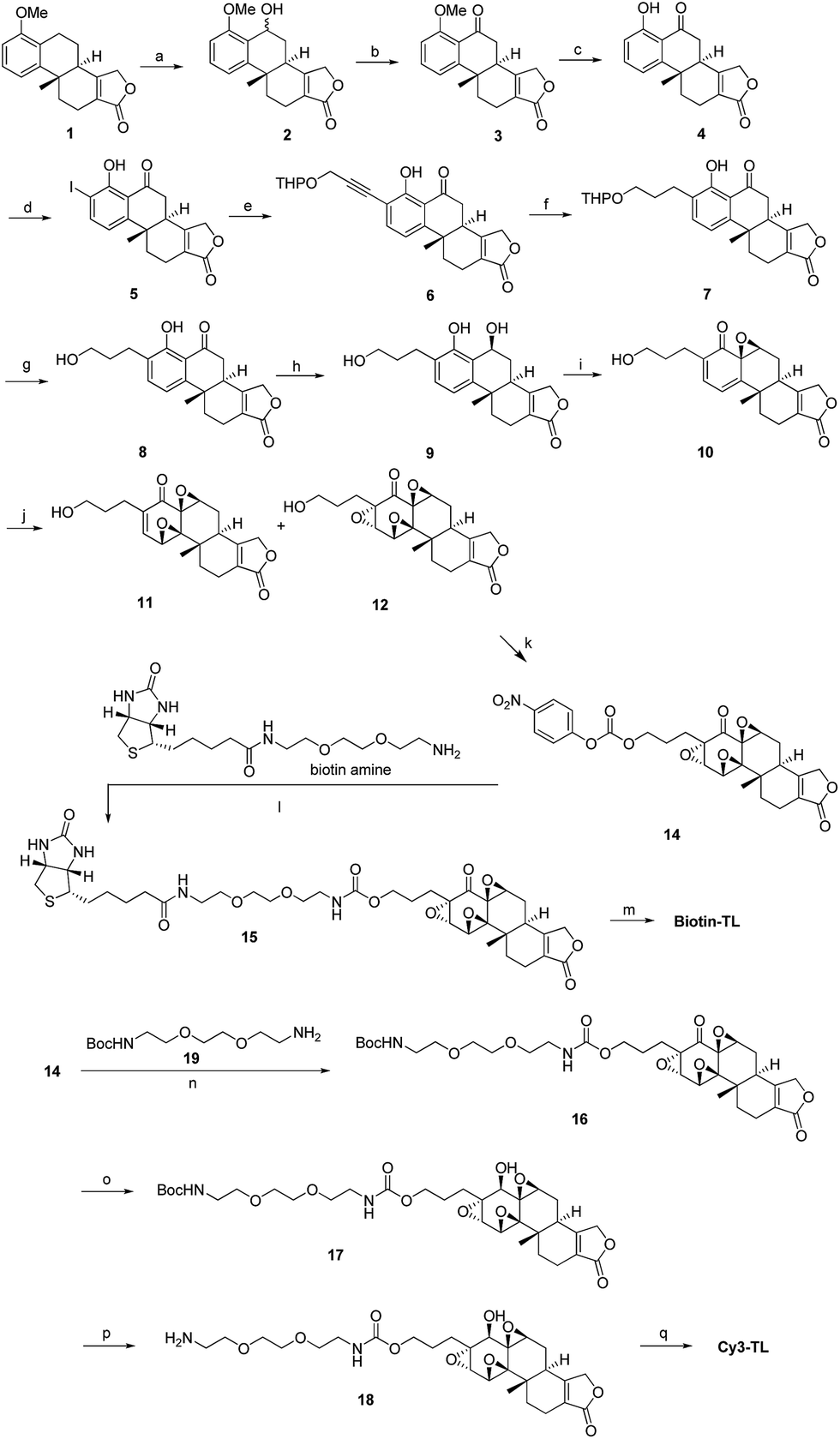 | ||
Scheme 1 General synthetic routes to Biotin-TL and Cy3-TL. Reagents and conditions: (a) cerium(VI) ammonium nitrate, MeCN, 0 °C, 99%; (b) IBX, acetone, reflux, 98%; (c) AlCl3, MeCN, reflux, 100%; (d) Sc(OTf)3, NIS, AcOH, rt, 82%; (e) THPOCH2C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, PdCl2(PPh3)2, CuI, Et3N, THF, 35 °C, 97%; (f) H2, Pd/C, EtOH, 40–50 °C, 100%; (g) TsOH·H2O, MeOH, rt, 95%; (h) NaBH4, EtOH, CH2Cl2, 0 °C, 99%; (i) NaIO4, MeOH, H2O, rt, 80%; (j) CF3COCH3, OXONE, NaHCO3, CH3CN, Na2(EDTA), 0 °C; (k) p-nitrophenylchloroformate, pyridine, CH2Cl2, rt, the yield of 14 was 13% from 10; (l) DMAP, Et3N, CH2Cl2/MeOH, rt, 24%; (m) K-selectride, THF, 40 °C, 39%; (n) DMAP, Et3N, CH2Cl2, rt, 85%; (o) K-selectride, anhydrous THF, −78 °C; (p) 30% TFA in CH2Cl2 (v/v), rt; (q) Cy3-NHS, CH2Cl2, DMAP, rt, 42%. CH, PdCl2(PPh3)2, CuI, Et3N, THF, 35 °C, 97%; (f) H2, Pd/C, EtOH, 40–50 °C, 100%; (g) TsOH·H2O, MeOH, rt, 95%; (h) NaBH4, EtOH, CH2Cl2, 0 °C, 99%; (i) NaIO4, MeOH, H2O, rt, 80%; (j) CF3COCH3, OXONE, NaHCO3, CH3CN, Na2(EDTA), 0 °C; (k) p-nitrophenylchloroformate, pyridine, CH2Cl2, rt, the yield of 14 was 13% from 10; (l) DMAP, Et3N, CH2Cl2/MeOH, rt, 24%; (m) K-selectride, THF, 40 °C, 39%; (n) DMAP, Et3N, CH2Cl2, rt, 85%; (o) K-selectride, anhydrous THF, −78 °C; (p) 30% TFA in CH2Cl2 (v/v), rt; (q) Cy3-NHS, CH2Cl2, DMAP, rt, 42%. | ||
Identification of peroxiredoxin I as a direct binding protein of triptolide
We first carried out an experiment to search for binding protein(s) of triptolide using the chemical probes synthesized. Cell lysates were incubated with Biotin-TL, and proteins were purified with streptactin-conjugated agarose beads, followed by SDS-PAGE analysis and silver staining (Fig. 1a). One band with a molecular mass of around 23 kDa was observed in the protein sample enriched by Biotin-TL, but not by biotin. This band was observed reproducibly using different cell lines (Fig. S1†). Furthermore, the protein band can be competed away by excess triptolide prior to enrichment by Biotin-TL (Fig. S2†). MALDI-TOF MS analysis of the protein band revealed the identity of this protein as Prx I (Fig. S3†).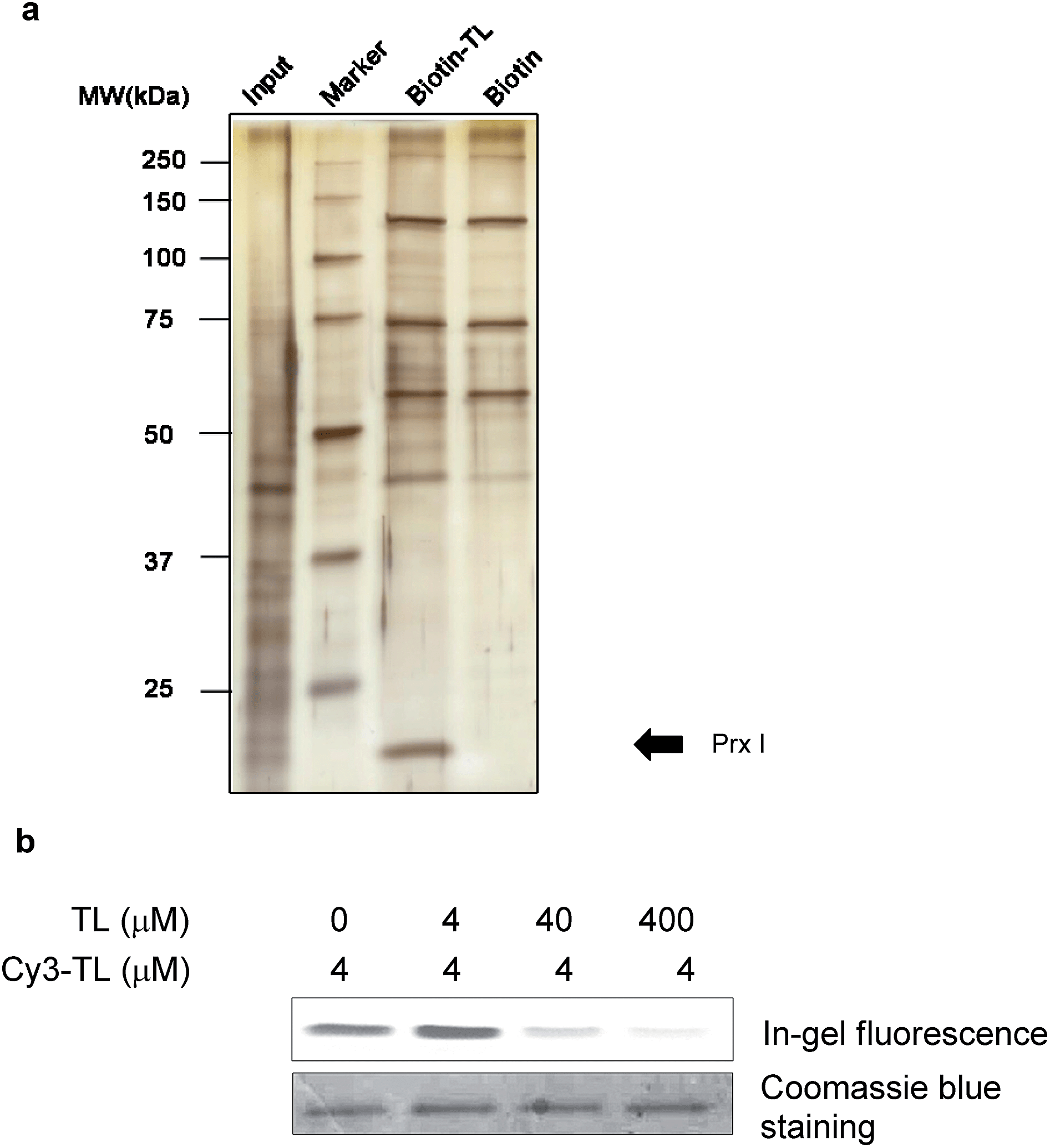 | ||
| Fig. 1 Identification of peroxiredoxin I as a direct binding protein of triptolide. (a) A silver-stained gel of pull-down products of Biotin-TL or biotin. MDCK cell lysate was incubated with 5 μM biotin or Biotin-TL. Proteins were then purified using streptactin-conjugated agarose beads, followed by SDS-PAGE analysis. The arrow indicates a band confirmed as Prx I. (b) In-gel fluorescence analysis of recombinant Prx I after incubation with Cy3-TL in the absence or presence of TL pretreatment. | ||
To validate the specificity of the interaction between Prx I and triptolide, a competition binding assay was performed. Recombinant Prx I was first incubated with the indicated concentration of TL and then with fluorescent probe Cy3-TL. The resultant mixture was subjected to SDS-PAGE and in-gel fluorescence scanning. Co-migration of Cy3-TL and Prx I protein on a denaturing gel suggests a covalent linkage between them. As shown by the relative fluorescence intensity, the Prx I–TL interaction was sensitive to competition with excess TL (Fig. 1b).
As a negative control, recombinant thioredoxin and thioredoxin reductase incubated with Cy3-TL did not give any fluorescence, suggesting that they cannot form a covalent linkage with Cy3-TL, even though they possess more reactive cysteine residues (Fig. S4†).23
Structural characterization of Prx I–TL and confirmation of the binding sites
To further pin down the exact TL-binding peptides or binding site(s), recombinant Prx I was incubated with TL, and the resulting mixture was subjected to LC-MS/MS analysis. The m/z of the Cys173-containing peptide HGEVCPAGWKPGSDTIKPDVQK had a mass increase of 360.2 Da, which is consistent with the calculated value for the addition of one TL molecule to this peptide. Further fragmentation of this peptide produced a series of b/y-ion fragments. According to the MS/MS spectrum, only the Cys173-containing fragments (Fig. 2a, b5-b10 and b14-b16, marked in red) display a 360.2 Da mass shift, suggesting Cys173 is the covalently modified residue.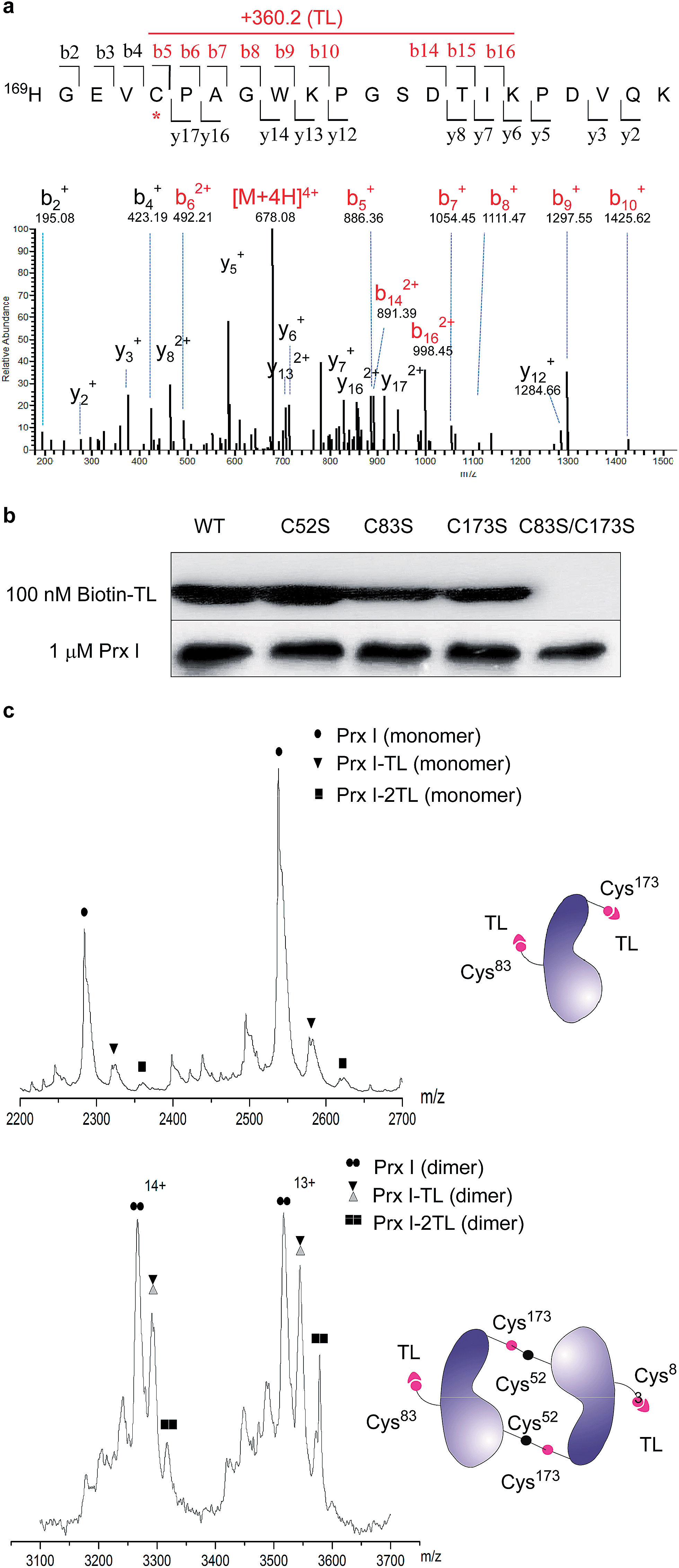 | ||
| Fig. 2 Structural characterization of Prx I–TL and confirmation of the binding sites. (a) MS/MS analysis of the Cys173-containing tryptic peptide after recombinant Prx I was incubated with TL, followed by trypsin digestion. The asterisk indicates the residue bound by TL; red labels show ions that display a mass shift corresponding to TL. (b) 1 μM recombinant Prx I and its mutants were incubated with 100 nM Biotin-TL at 4 °C for 3 h, followed by Western blotting for biotin or Prx I. (c) Native ESI-MS analysis of the monomer (upper) and dimer (lower) of the Prx I–TL complex. 10 μM recombinant Prx I was incubated with vehicle or 40 μM TL at 4 °C overnight before analysis. Schematic diagrams show interactions between TL and the Prx I monomer/dimer. | ||
Given that Cys173 can be modified by triptolide, we next examined whether other cysteine residues are involved in the Prx I–triptolide interaction. In our experiment, wild-type and mutant Prx I recombinant proteins were incubated with Biotin-TL and subsequently resolved by SDS-PAGE. Co-migration of proteins and Biotin-TL was detected by immunoblot with streptavidin-HRP. When Cys83 and Cys173 were simultaneously replaced with serine residues, the interactions between Prx I and Biotin-TL were totally abolished (Fig. 2b and S5†). This result illustrates that both Cys83 and Cys173 play important roles in the formation of the Prx I–TL complex.
This conclusion was further supported by the investigation of the Prx I–TL complex in different oligomeric statuses using native mass spectrometry (MS). Native MS allows large intact protein homo/hetero-complexes to be analyzed with high sensitivity and resolution. As shown in Fig. 2c, there were two TL-binding sites in monomeric Prx I, whereas the dimeric form of Prx I bound to two TL molecules instead of four. Considering that a Prx I dimer is formed through an intermolecular disulphide bond between Cys173 and Cys52,24 it is reasonable that Cys173 is no longer available to bind to TL (Fig. 2c), leading to the loss of two reactive sites in a Prx I dimer. This is consistent with our aforementioned observation that Cys173 is one of the two reactive sites for TL. Meanwhile, the other binding site, Cys83, should not differ significantly in its monomeric or dimeric forms, and thereby becomes the sole site for the TL interaction.
Functional characterization of the interaction between Prx I and TL
We next examined the functional consequence of this TL-Prx I interaction. First, we measured the peroxidase activity of Prx I in the presence or absence of triptolide. Adenanthin (AND) was reported to inhibit Prx I's peroxidase activity at 1 μM.14 In our assay, we used 80 μM AND as a positive control to significantly inhibit the Prx I peroxidase activity. However, treatment with TL at the same concentration (80 μM) did not suppress the peroxidase activity of recombinant Prx I (Fig. 3a and S6†).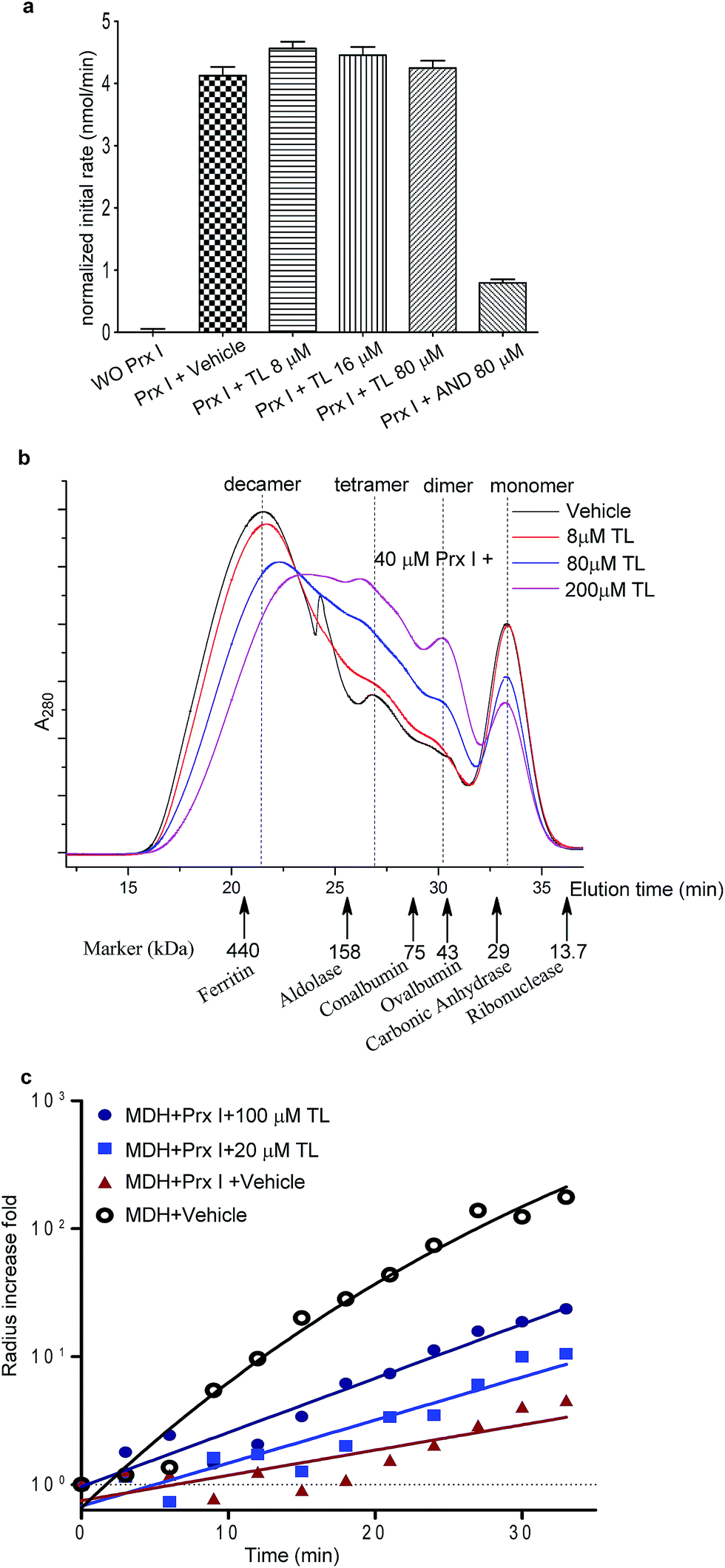 | ||
| Fig. 3 Effects of TL on the biological activity of Prx I. (a) Peroxidase activity assay of 1 μM Prx I with indicated treatment. (b) Gel filtration analysis of 40 μM Prx I after overnight incubation with the indicated concentration of TL at 4 °C. (c) Chaperone activity assay of Prx I in the presence of TL. 2.5 μM recombinant Prx I was incubated with TL overnight at 4 °C. Light scattering of 10 μM client protein MDH was monitored under heating at 45 °C for 30 min. | ||
The knowledge of the binding residues of TL on Prx I guided us to investigate Prx I's oligomeric state because both Cys83 and Cys173 play important roles in regulating the oligomeric structures of Prx I. 40 μM recombinant Prx I was incubated with TL at protein-compound molar ratios of 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, 1:2, and 1:5. Subsequent size-exclusion chromatographic analyses were performed and revealed that TL induced dramatic Prx I decamer dissociation, and the proportion of tetramers and dimers increased in a dose-dependent manner (Fig. 3b). This observation is consistent with a previous report that glutathionylation of Cys83 and Cys173 induces dissociation of Prx I decamers.25
:1, 1:2, and 1:5. Subsequent size-exclusion chromatographic analyses were performed and revealed that TL induced dramatic Prx I decamer dissociation, and the proportion of tetramers and dimers increased in a dose-dependent manner (Fig. 3b). This observation is consistent with a previous report that glutathionylation of Cys83 and Cys173 induces dissociation of Prx I decamers.25
There is a growing body of evidence suggesting that higher-ordered Prx I oligomers function as crucial molecular chaperones to protect client molecules from stress-induced aggregation.26,27 Hence, we employed a chaperone assay to investigate whether TL influences the chaperone activity of Prx I. As previously described, Prx I is able to protect its client protein malate dehydrogenase (MDH) from thermal aggregation.28 To test TL's effect, recombinant Prx I was incubated with TL overnight at 4 °C. Then, MDH was added to the mixture immediately before the assay. The radii of the particles were monitored by light scattering, which serves as an indicator of MDH aggregation, during heating at 45 °C for 30 min. In one control experiment, aggregation of MDH remained the same in the absence or presence of TL, suggesting that TL does not influence MDH directly (Fig. S9†). In the other control experiment, TL-treated Prx I did not exhibit obvious aggregation under heating, indicating that TL does not directly induce Prx I aggregation (Fig. S10†). However, when MDH was mixed with TL-treated Prx I, instead of DMSO-treated Prx I, enhanced aggregation was observed, suggesting suppression of Prx I's chaperone activity by TL in a dose-dependent manner (Fig. 3c). We noticed that TL's inhibition of the Prx I chaperone activity was partial. Actually, this observation is consistent with the partial engagement of TL with Prx I, as shown by native MS analysis (Fig. 2c). This became the initial motivation for us to screen for other potent inhibitors of Prx I.
Identification and validation of novel Prx I inhibitors
Finally, we carried out a competition experiment, employing Cy3-TL as a reporter to screen TL derivatives and other natural products as Prx I inhibitors (Fig. 4a). Recombinant Prx I was first incubated with an excess of the individual compounds, and then incubated with Cy3-TL before being subjected to SDS-PAGE and an in-gel fluorescence scan. While triptonide (TN), a naturally occurring TL analogue, competed away Cy3-TL, the other analogue 12-epitriptriolide (eTL) could not. Among those natural products that we tested, WA and Cel competed away Cy3-TL (Fig. 4b). This result indicates that TN, WA, and Cel covalently modified Prx I, while eTL failed to interact with Prx I. Tandem mass spectrometry analysis of the WA-PrxI complex demonstrated that WA binds to Prx I also at residues Cys83 and Cys173 (Fig. 4d and S8†).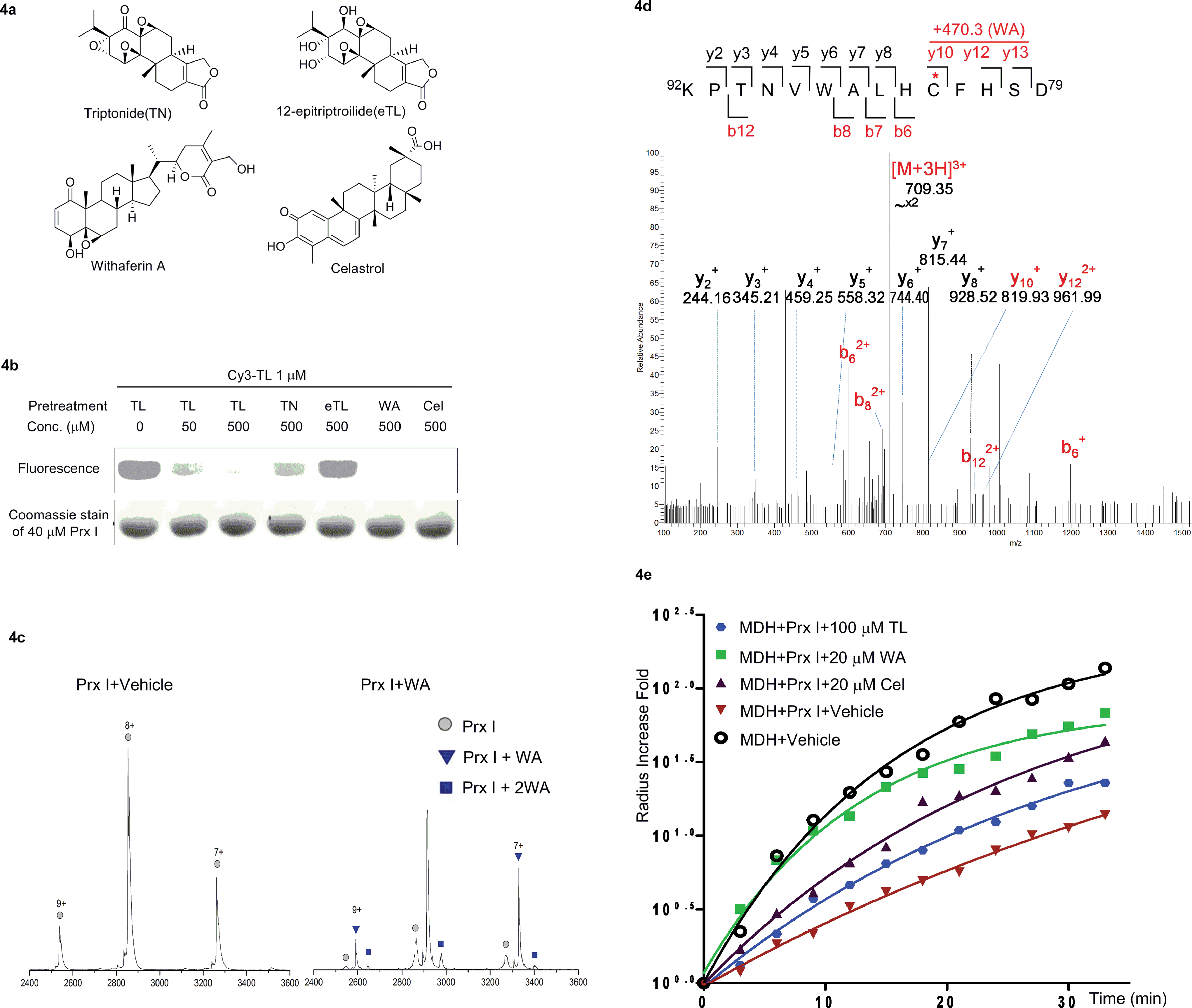 | ||
| Fig. 4 Identification and validation of novel Prx I chaperone inhibitors. (a) Chemical structures of withaferin A and celastrol. (b) In-gel fluorescence analysis of recombinant Prx I after incubation with Cy3-TL in the absence or presence of the indicated treatment. (c) Native ESI-MS analysis of the monomer of (left) Prx I and (right) the Prx I–WA complex. 10 μM recombinant Prx I was incubated with vehicle or 10 μM WA at 4 °C overnight before analysis. (d) LC-MS/MS analysis of the Cys173-containing tryptic peptide after recombinant Prx I was incubated with WA at 4 °C overnight followed by trypsin digestion. The asterisk indicates the cysteine bound by WA; red labels show the Cys173-containing ions that are identified to bind to WA. (e) Chaperone activity assay of Prx I in the presence of the indicated compounds. 2.5 μM recombinant Prx I was incubated with TL overnight at 4 °C. Light scattering of 10 μM client protein MDH was monitored under heating at 45 °C for 30 min. | ||
Next, we investigated Cel and WA's effect on the chaperone activity of Prx I. As with TL, Cel and WA did not directly increase the aggregation of MDH or Prx I under heating, as shown in control experiments (Fig. S9 and S10†). Incubation of Prx I with Cel and WA enhanced MDH thermal aggregation significantly (Fig. 4e), suggesting suppression of Prx I's chaperone activity. Interestingly, Cel and WA exhibited even stronger inhibition than TL in the chaperone assay, consistent with their higher binding occupancy in Prx I than TL as observed by native MS (Fig. 4c and S7†).
Discussion
Several triptolide-binding proteins have been identified through biochemical and fractionation approaches. Recently, it was reported that triptolide binds XPB selectively at its Cys342 despite having 14 cysteine residues. The proposed molecular mechanism involves cysteine reacting with triptolide's C12/C13 epoxide,29 which may also apply to the present study. Another interesting comparison can be drawn between Prx I and dCTPP1, which was also found to bind triptolide using biotinylated triptolide probes.21 To identify dCTPP1, Crew's group synthesized a biotinylated triptolide by modifying the free β hydroxyl group at C14 of triptolide, which is a known key functional group for its antitumor activity.30–32 In our study, we chose to modify triptolide at its C13 site because it showed minimal influence on triptolide's anti-cancer activities (unpublished data). Although the affinity pull-down approach was used in both cases, it is interesting to note that chemical derivatization at different sites of small molecules can lead to identification of different target proteins.As a member of the peroxiredoxin family (Prxs) which catalyzes H2O2 removal, Prx I is unique in that it can also function as a molecular chaperone. The switching of the oligomeric status and corresponding dual functions of Prx I are modulated by modifications at its cysteines. It is reported that simultaneous glutathionylation of Cys83 and Cys173 induces dissociation of Prx I decamers. The same event occurs when triptolide modifies these two residues, as observed in our study. On the contrary, adenanthin, which does not bind to Cys83, does not show a significant influence on the oligomeric status of Prx I.14 As chemical tools, these two natural products with different effects on Prx I validate the importance of Cys83 in regulating Prx I's oligomeric status. Given that Cys83 is not conserved in all Prxs, it constitutes a promising and practical target site for the selective modulation of Prx I’s function. It is worth mentioning that since triptolide modifies Cys83 in Prx I, its fluorescent analogue Cy3-TL has potential as an activity-based probe for screening Prx I ligands or qualitative/quantitative assay development in future studies.
In addition to triptolide, we have discovered that the natural products withaferin A and celastrol can also interact with Prx I at the same sites. It is likely that triptolide, withaferin A, and celastrol interact with Prx I through the same mechanism, i.e. by cysteine alkylation. In contrast to triptonide and triptolide with a C12/C13-epoxide group as an electrophilic center for cysteine attack, 12-epitriptriolide has a cis-diol structure at the C12/C13 positions, and hence cannot be alkylated by cysteine residues of Prx I. Interestingly, withaferin A and celastrol, two bioactive natural terpenoids, also contain electrophilic carbon centers which can be attacked by a well-positioned cysteine residue within a binding pocket. This result indicates the importance of electrophilic groups of the natural products for the interactions with Prx I.
In the present study, we have identified and validated that natural products, including triptolide, celastrol, and withaferin A directly bind to Prx I in a covalent manner. This is, to the best of our knowledge, the first identification of natural products that trigger dissociation of the Prx I decamer and selectively inhibit its chaperone activity. Although several targets of celastrol and withaferin A have been reported in the literature, it is the first time that Prx I has been identified as a novel target of celastrol and withaferin A, which may shed light on the understanding of their biological action.33,34 Our study provides new chemical probes to perturb Prx I's chaperone activity, particularly through interaction with Cys83. Taken together, we demonstrate that natural products can serve as powerful chemical probes to precisely modulate their target proteins, and thus possess great potential to facilitate the biological study of multifunctional proteins. Considering that triptolide, withaferin A, and celastrol share anti-inflammatory,35–37 pro-apoptotic,38–40 and anti-angiogenic activities in common, further dissection of the correlation between their alkylation of Prx I and common bioactivities at the cellular level is an important next step and will be reported in due course.
Acknowledgements
We thank Prof. Chi Wu (Chinese University of Hong Kong) for generous support on the light scattering experiment, Dr. Xiang. D. Li for manuscript revision, and Prof. Chi-Ming Che and Dr. Yi Man Eva Fung (University of Hong Kong) for support on the high resolution MS analysis of compounds. We also thank Dr. Arjan Barendregt and Dr. Houjiang Zhou (Netherlands Proteomics Centre) for discussions on protein mass spectrometry methods, and Mr. Ben Ho and Ms. Levina Lam (HKU Genome Research Centre) for fluorescence scans. We acknowledge the financial support from the Hong Kong Research Grants Council (HKU no. 705213), the University of Hong Kong, and the Morningside Group. LC-MS/MS analysis was partially supported by the Special Equipment Grant from the University Grants Committee of the Hong Kong Special Administrative Region, China (Project Code: SEG_HKU02).Notes and references
- S. J. Gardai, Y.-Q. Xiao, M. Dickinson, J. A. Nick, D. R. Voelker, K. E. Greene and P. M. Henson, Cell, 2003, 115, 13–23 CrossRef CAS.
- J.-W. Chen, C. Dodia, S. I. Feinstein, M. K. Jain and A. B. Fisher, J. Biol. Chem., 2000, 275, 28421–28427 CrossRef CAS PubMed.
- S. Kamisuki, Q. Mao, L. Abu-Elheiga, Z. Gu, A. Kugimiya, Y. Kwon, T. Shinohara, Y. Kawazoe, S.-i. Sato, K. Asakura, H.-Y. P. Choo, J. Sakai, S. J. Wakil and M. Uesugi, Chem. Biol., 2009, 16, 882–892 CrossRef CAS PubMed.
- J. Ge, C.-J. Zhang, L. Li, L. M. Chong, X. Wu, P. Hao, S. K. Sze and S. Q. Yao, ACS Chem. Biol., 2013, 8, 2577–2585 CrossRef CAS PubMed.
- G. Zimmermann, B. Papke, S. Ismail, N. Vartak, A. Chandra, M. Hoffmann, S. A. Hahn, G. Triola, A. Wittinghofer, P. I. H. Bastiaens and H. Waldmann, Nature, 2013, 497, 638–642 CrossRef CAS PubMed.
- R. E. Moellering and B. F. Cravatt, Chem. Biol., 2012, 19, 11–22 CrossRef CAS PubMed.
- H. H. Jang, K. O. Lee, Y. H. Chi, B. G. Jung, S. K. Park, J. H. Park, J. R. Lee, S. S. Lee, J. C. Moon, J. W. Yun, Y. O. Choi, W. Y. Kim, J. S. Kang, G.-W. Cheong, D.-J. Yun, S. G. Rhee, M. J. Cho and S. Y. Lee, Cell, 2004, 117, 625–635 CrossRef CAS PubMed.
- H. A. Woo, H. Z. Chae, S. C. Hwang, K.-S. Yang, S. W. Kang, K. Kim and S. G. Rhee, Science, 2003, 300, 653–656 CrossRef CAS PubMed.
- F. Saccoccia, P. Di Micco, G. Boumis, M. Brunori, I. Koutris, A. E. Miele, V. Morea, P. Sriratana, D. L. Williams, A. Bellelli and F. Angelucci, Structure, 2012, 20, 429–439 CrossRef CAS PubMed.
- T. Matsumura, K. Okamoto, S.-i. Iwahara, H. Hori, Y. Takahashi, T. Nishino and Y. Abe, J. Biol. Chem., 2008, 283, 284–293 CrossRef CAS PubMed.
- A. Morinaka, Y. Funato, K. Uesugi and H. Miki, Oncogene, 2011, 30, 4208–4218 CrossRef CAS PubMed.
- X. Wang, S. He, J.-M. Sun, G. P. Delcuve and J. R. Davie, Mol. Biol. Cell, 2010, 21, 2987–2995 CrossRef CAS PubMed.
- J. R. Riddell, X.-Y. Wang, H. Minderman and S. O. Gollnick, J. Immunol., 2010, 184, 1022–1030 CrossRef CAS PubMed.
- C.-X. Liu, Q.-Q. Yin, H.-C. Zhou, Y.-L. Wu, J.-X. Pu, L. Xia, W. Liu, X. Huang, T. Jiang, M.-X. Wu, L.-C. He, Y.-X. Zhao, X.-L. Wang, W.-L. Xiao, H.-Z. Chen, Q. Zhao, A.-W. Zhou, L.-S. Wang, H.-D. Sun and G.-Q. Chen, Nat. Chem. Biol., 2012, 8, 486–493 CrossRef CAS PubMed.
- D. Yang, X. Y. Ye, S. Gu and M. Xu, J. Am. Chem. Soc., 1999, 121, 5579–5580 CrossRef CAS.
- X.-H. Jiang, B. C.-Y. Wong, M. C.-M. Lin, G.-H. Zhu, H.-F. Kung, S.-H. Jiang, D. Yang and S.-K. Lam, Oncogene, 2001, 20, 8009–8018 CrossRef CAS PubMed.
- Z. L. Zhou, Y. X. Yang, J. Ding, Y. C. Li and Z. H. Miao, Nat. Prod. Rep., 2012, 29, 457–475 RSC.
- R. Chugh, V. Sangwan, S. P. Patil, V. Dudeja, R. K. Dawra, S. Banerjee, R. J. Schumacher, B. R. Blazar, G. I. Georg, S. M. Vickers and A. K. Saluja, Sci. Transl. Med., 2012, 4, 156ra139 Search PubMed.
- S. J. Leuenroth, D. Okuhara, J. D. Shotwell, G. S. Markowitz, Z. Yu, S. Somlo and C. M. Crews, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 4389–4394 CrossRef CAS PubMed.
- D. V. Titov, B. Gilman, Q.-L. He, S. Bhat, W.-K. Low, Y. Dang, M. Smeaton, A. L. Demain, P. S. Miller, J. F. Kugel, J. A. Goodrich and J. O. Liu, Nat. Chem. Biol., 2011, 7, 182–188 CrossRef CAS PubMed.
- T. W. Corson, H. Cavga, N. Aberle and C. M. Crews, ChemBioChem, 2011, 12, 1767–1883 CrossRef CAS PubMed.
- Y. Smurnyy, M. Cai, H. Wu, E. McWhinnie, J. A. Tallarico, Y. Yang and Y. Feng, Nat. Chem. Biol., 2014, 10, 623–625 CrossRef CAS PubMed.
- E. Weerapana, C. Wang, G. M. Simon, F. Richter, S. Khare, M. B. D. Dillon, D. A. Bachovchin, K. Mowen, D. Baker and B. F. Cravatt, Nature, 2010, 468, 790 CrossRef CAS PubMed.
- Z. A. Wood, L. B. Poole and P. A. Karplus, Science, 2003, 300, 650–653 CrossRef CAS PubMed.
- J. W. Park, G. Piszczek, S. G. Rhee and P. B. Chock, Biochemistry, 2011, 50, 3204–3210 CrossRef CAS PubMed.
- S. Y. Kim, H. H. Jang, J. R. Lee, N. R. Sung, H. B. Lee, D. H. Lee, D.-J. Park, C. H. Kang, W. S. Chung, C. O. Lim, D.-J. Yun, W. Y. Kim, K. O. Lee and S. Y. Lee, Plant. Sci., 2009, 177, 227–232 CrossRef CAS PubMed.
- Q. Y. Liu, T. Y. Chen, G. Y. Chen, N. Li, J. L. Wang, P. C. Ma and X. T. Cao, Biochem. Biophys. Res. Commun., 2006, 345, 1122–1130 CrossRef CAS PubMed.
- W. Lee, K.-S. Choi, J. Riddell, C. Ip, D. Ghosh, J.-H. Park and Y.-M. Park, J. Biol. Chem., 2007, 282, 22011–22022 CrossRef CAS PubMed.
- Q.-L. He, D. V. Titov, J. Li, M. Tan, Z. Ye, Y. Zhao, D. Romo and J. O. Liu, Angew. Chem., Int. Ed., 2015, 54, 1859–1863 CrossRef CAS PubMed.
- F. Xu, X. Shi, S. Li, J. Cui, Z. Lu, Y. Jin, Y. Lin, J. Pang and J. Pan, Bioorg. Med. Chem., 2010, 18, 1806–1815 CrossRef CAS PubMed.
- Z. Li, Z.-L. Zhou, Z.-H. Miao, L.-P. Lin, H.-J. Feng, L.-J. Tong, J. Ding and Y.-C. Li, J. Med. Chem., 2009, 52, 5115–5123 CrossRef CAS PubMed.
- S. M. Kupchan, W. A. Court, R. G. Dailey, C. J. Gilmore and R. F. Bryan, J. Am. Chem. Soc., 1972, 94, 7194–7195 CrossRef CAS.
- W. Vanden Berghe, L. Sabbe, M. Kaileh, G. Haegeman and K. Heyninck, Biochem. Pharmacol., 2012, 84, 1282–1291 CrossRef CAS PubMed.
- R. Kannaiyan, M. K. Shanmugam and G. Sethi, Cancer Lett., 2011, 303, 9–20 CrossRef CAS PubMed.
- X. Wang, L. Zhang, W. Duan, B. Liu, P. Gong, Y. Ding and X. Wu, Mol. Med. Rep., 2014, 10, 447–452 CAS.
- R. Maitra, M. Porter, S. Huang and B. Gilmour, J. Inflammation, 2009, 6, 15 CrossRef PubMed.
- A. C. Allison, R. Cacabelos, V. R. M. Lombardi, X. A. Álvarez and C. Vigo, Prog. Neuro-Psychopharmacol. Biol. Psychiatry, 2001, 25, 1341–1357 CrossRef CAS.
- T. M. Kiviharju, P. S. Lecane, R. G. Sellers and D. M. Peehl, Clin. Cancer Res., 2002, 8, 2666–2674 CAS.
- R. Munagala, H. Kausar, C. Munjal and R. C. Gupta, Carcinogenesis, 2011, 32, 1697–1705 CrossRef CAS PubMed.
- R. Kannaiyan, K. Manu, L. Chen, F. Li, P. Rajendran, A. Subramaniam, P. Lam, A. Kumar and G. Sethi, Apoptosis, 2011, 16, 1028–1041 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Chemical synthesis and characterization of triptolide probes; Fig. S1–S10; Table S1–S3. See DOI: 10.1039/c5sc00633c |
| This journal is © The Royal Society of Chemistry 2015 |