
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of zinc alkyl/air systems as radical initiators for organic reactions†
Marcin
Kubisiak
a,
Karolina
Zelga
a,
Wojciech
Bury
a,
Iwona
Justyniak
b,
Krzysztof
Budny-Godlewski
a,
Zbigniew
Ochal
a and
Janusz
Lewiński
*ab
aDepartment of Chemistry, Warsaw University of Technology, Noakowskiego 3, 00-664 Warsaw, Poland. E-mail: lewin@ch.pw.edu.pl; Fax: +48 22 6282741; Tel: +48 22 2347315
bInstitute of Physical Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: lewin@ch.pw.edu.pl; Tel: +48 22 3432076
First published on 19th March 2015
Abstract
This paper reports a series of comparative experiments on the activity of carbon- and oxygen-centred radical species in a model reaction of the radical addition of THF to imines mediated by a series of zinc alkyl/air reaction systems. The study strongly contradicts the notion that generally R˙ radicals are the initiating species in organic reactions mediated by RnM/air systems, and simultaneously demonstrates that oxygen-centred radical species are the key intermediates responsible for the initiation process. In addition, a new efficient RZn(L)/air initiating system for radical organic reactions exampled by a model reaction of radical addition of THF to imines is developed. Moreover, the isolation and structural characterization of the first zinc alkylperoxide supported by a carboxylate ligand, [Zn4(μ3-OOtBu)3(μ4-O)(O2CEt)3]2, as well as the novel octanuclear zinc oxo(alkoxide) aggregate with entrapped O–THF species, [Zn4(μ4-O)(μ3-2-O–THF)(O2CEt)5]2, provide clear mechanistic signatures for the mode of function of the RZn(O2CR′)/air system.
Introduction
In the last two decades remarkable advances have been made in the area of organic radical chemistry, which have been largely driven by the development of new radical precursors.1 In this regard homoleptic boron2a,b and zinc alkyls2c,d in combination with dry air have attracted much attention as they provide a source of radical initiators for various organic reactions.3 For these RnM/air systems it is commonly assumed that R˙ radicals are formed in the initial step (cf.eqn (1)),4 and are thus considered as the initiating species in radical organic reactions.2| R2Zn + O2 → RZnOO˙ + R˙ | (1) |
We also note that the character of the radical species formed in the reactions of R2Zn with O2 has been studied by EPR using nitrones8 and stable nitroxyl radicals such as DEPMPO,4b as radical traps. These stable radicals have been assumed to be inert towards R2Zn (ref. 9) or to have no influence on the oxygenation process.4a However, very recently it has been convincingly shown that TEMPO reacts with Et2Zn smoothly affording both EtZn(TEMPO) and Zn(TEMPO)2 nitroxides with concomitant evolution of Et˙ radicals.10,11 Thus, the latter observations clearly indicate that for reactions involving alkylzinc/O2 systems the use of nitroxyl radicals as spin traps can lead to incorrect assignments in the determination of the character of the radicals present in the reaction mixture.
Building on our continuous interest and expertise in radicals' chemistry and dioxygen activation,5,7,10,12 we herein report comparative experiments on the activity of carbon- and oxygen-centred radical species (derived from a series of zinc alkyl/air reaction systems with a clear mechanistic picture, see Scheme 1), in a model reaction of radical addition of THF to imines as a selected model reaction. The study of these model systems not only provides better understanding of how the oxygenated products participate in radical organic reactions but also assists in the development of new types of radical initiators.
 | ||
| Scheme 1 Selected organozinc systems generating either alkyl radicals or oxygen-centred radicals. | ||
Results
Control experiments on the addition of THF to imines mediated by systems generating solely R˙ radicals
In order to verify the hypothesis that R˙ radicals are the reactive species mediating the addition of ethers to imines,2d we have attempted to create an environment in which only alkyl radicals are liberated. For this purpose, two well-characterized reaction systems were selected as the control experiments: (i) the reaction of Me2Zn with tBuDAB previously studied by van Koten where the initially formed adduct Me2Zn(tBuDAB) decomposes slowly at ambient temperature to a C–C coupling product [MeZn(tBuDABC)]2 with concomitant liberation of Me˙ radicals (Scheme 1a),13 and (ii) the equimolar reaction of Et2Zn and TEMPO generating a nitroxide EtZn(TEMPO) complex along with an Et˙ radical (Scheme 1b).10,14 These two systems were employed as sources of R˙ radicals in the model reaction of THF addition to N-(4-methoxybenzylidene)aniline (1a) (Scheme 2, path A; see ESI for details†). Strikingly, no reactions between THF and 1a were observed after 48 h at room temperature in either case. These findings strongly contradict the notion that R˙ radicals are the key intermediates responsible for the initiation process in organic reactions mediated by RnM/air systems. The examples also highlight that there is much to be clarified with respect to the character of the radical initiators in the reaction systems considered. Therefore in the subsequent experiments, we turned our attention to the reaction systems capable of producing non-carbon centred radicals. | ||
| Scheme 2 The model reaction of the radical addition of THF to imines. | ||
Control experiments on the addition of THF to imines mediated by RZn(L)/air systems
From amongst several zinc alkyl/O2 systems affording oxygen-centred radicals as primary radical species and described recently by our group,5d–g,7 we have selected two reaction systems (Scheme 1c and d). The first one involves a trinuclear ethyl aryloxide (EtZn)2Zn(OAr)4 and O2, where a range of carbon and oxygen centred radical species, including Et˙, EtO˙ and HO˙, are generated in the course of formation of an aryloxide(hydroxide) cluster Zn8(OAr)8(OH)6(O2) (ArOH = o-hydroxybiphenyl) with an encapsulated peroxide moiety (Scheme 1c).7 However, this system proved to be inefficient, when used as a mediator of the model reaction, probably due to interception of the forming radicals in the inner sphere of the zinc complex.15Then, building upon our experience with ZnO–OR bond homolysis,5d–g we turned our attention to the alkylzinc carboxylate/O2 system. Previously, we showed that the ZnO–OR bond homolysis proceeds effectively for the ethylzinc benzoate/O2 system generating ZnO˙ and EtO˙ radicals in the course of formation of an oxo complex Zn4(μ4-O)(O2CPh)6 (Scheme 1d).5g Thus, we have anticipated that the readily prepared alkylzinc carboxylates subjected to O2 can be interesting experimental probes for determining the character of initiating species in the model reaction. Therefore, a series of alkylzinc carboxylates, RZn(O2CR′) (where R = Me, Et or tBu and R′ = Ph and Et), was prepared according to procedures described in the literature.5e It was then investigated how the character of the carboxylate ligand, and the alkyl group bonded to the metal, influence the rate of conversion of 1a with THF (Fig. 1; for experimental details also see ESI†). To the best of our knowledge, this is the first example of a heteroleptic RxM(L)y-type complex mediating an organic radical reaction.
 | ||
| Fig. 1 Conversion versus time plot for the reactions of 1a with THF conducted at 20 °C and mediated by: MeZn(O2CPh)/air (I), MeZn(O2CEt)/air (II), EtZn(O2CPh)/air (III), EtZn(O2CEt)/air (IV), Me2Zn/air (V), and tBuZn(O2CEt)/air (VI). | ||
The investigated alkylzinc carboxylate complexes were mixed with 1a or 1b (1b = N-benzylidene-4-methoxyaniline, the imine commonly used with the Me2Zn/air initiating system3a) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 molar ratio in THF (Scheme 2, path B). The reactions mediated by EtZn(O2CPh) under O2 atmosphere were found to stop after ca. 15 minutes at approximately 70% conversions. When, on the other hand, pure dioxygen was replaced by dry air, the model reactions slowed down and conversions of 95% and 93% for 1a and 1b, respectively, were observed after 2 h (90% and 87% isolated yield of 2a and 2b, respectively). It seems reasonable that the observed dependence of the conversion of 1a and 1b on the gas phase composition (O2vs. air), i.e. on oxygen partial pressure, is related to the transient concentration of free radicals (in the presence of air, a lower radical concentration disfavours their nonproductive recombination). Following this reasoning, dry air was used in the subsequent experiments. When 1a was used as the substrate in the model reaction, the MeZn(O2CPh)/air and the MeZn(O2CEt)/air systems both allowed us to obtain 2a with 98% conversion after 2 h (92% and 95% isolated yield, respectively; Fig. 1, curves I and II). The observed reaction rates were slightly higher than those for the analogous ethylzinc complexes (Fig. 1, curves III and IV). The comparison of the data for the EtZn(O2CPh)/air and EtZn(O2CEt)/air initiating systems indicates that the identity of the carboxylic group has essentially no influence on the conversion and kinetic profile (Fig. 1, curves III and IV); the addition of THF to 1a proceeded relatively quickly and reached full conversion after 2 h.
:4 molar ratio in THF (Scheme 2, path B). The reactions mediated by EtZn(O2CPh) under O2 atmosphere were found to stop after ca. 15 minutes at approximately 70% conversions. When, on the other hand, pure dioxygen was replaced by dry air, the model reactions slowed down and conversions of 95% and 93% for 1a and 1b, respectively, were observed after 2 h (90% and 87% isolated yield of 2a and 2b, respectively). It seems reasonable that the observed dependence of the conversion of 1a and 1b on the gas phase composition (O2vs. air), i.e. on oxygen partial pressure, is related to the transient concentration of free radicals (in the presence of air, a lower radical concentration disfavours their nonproductive recombination). Following this reasoning, dry air was used in the subsequent experiments. When 1a was used as the substrate in the model reaction, the MeZn(O2CPh)/air and the MeZn(O2CEt)/air systems both allowed us to obtain 2a with 98% conversion after 2 h (92% and 95% isolated yield, respectively; Fig. 1, curves I and II). The observed reaction rates were slightly higher than those for the analogous ethylzinc complexes (Fig. 1, curves III and IV). The comparison of the data for the EtZn(O2CPh)/air and EtZn(O2CEt)/air initiating systems indicates that the identity of the carboxylic group has essentially no influence on the conversion and kinetic profile (Fig. 1, curves III and IV); the addition of THF to 1a proceeded relatively quickly and reached full conversion after 2 h.
Strikingly, the tert-butyl derivative, tBuZn(O2CEt), appeared to be entirely ineffective for initiating THF radical addition (Fig. 1, curve VI). Comparison of the initiation effectiveness of the developed alkylzinc carboxylate/air system with the classic one, i.e. the Me2Zn/air system, showed superiority of the carboxylate-based system. In the reaction mediated by Me2Zn/air (4 equiv. of Me2Zn) at 20 °C, conversion of 1a reached only 76% after 2 h (isolated yield of 2a was 72%) and no further reaction progress was observed (Fig. 1, curve V), while 1b was obtained with 80% yield only after 18 hours, according to Tomioka et al.3a This result shows that the Me2Zn/air system becomes deactivated in the course of the reaction or the radical species are produced too rapidly and undergo recombination reactions, while no such observations can be made in the case of our RZn(O2CR′)/air systems (R = Me, Et; R′ = Ph, Et).
The described findings convincingly demonstrate that the readily prepared alkylzinc carboxylates in combination with molecular oxygen, afford oxygen-centred radical species and can act as very efficient initiators of the radical addition of THF to imines.
Detailed investigations on the oxygenation of alkylzinc carboxylates
 | ||
| Fig. 2 Molecular structure of 3; hydrogen atoms, Me groups of the tBu groups and of the carboxylate ligand were omitted for clarity. | ||
 | ||
| Scheme 3 Reaction pathways in the radical addition of THF to the imines 1a or 1b mediated by the RZn(O2CEt)/air system (R = Me, Et). | ||
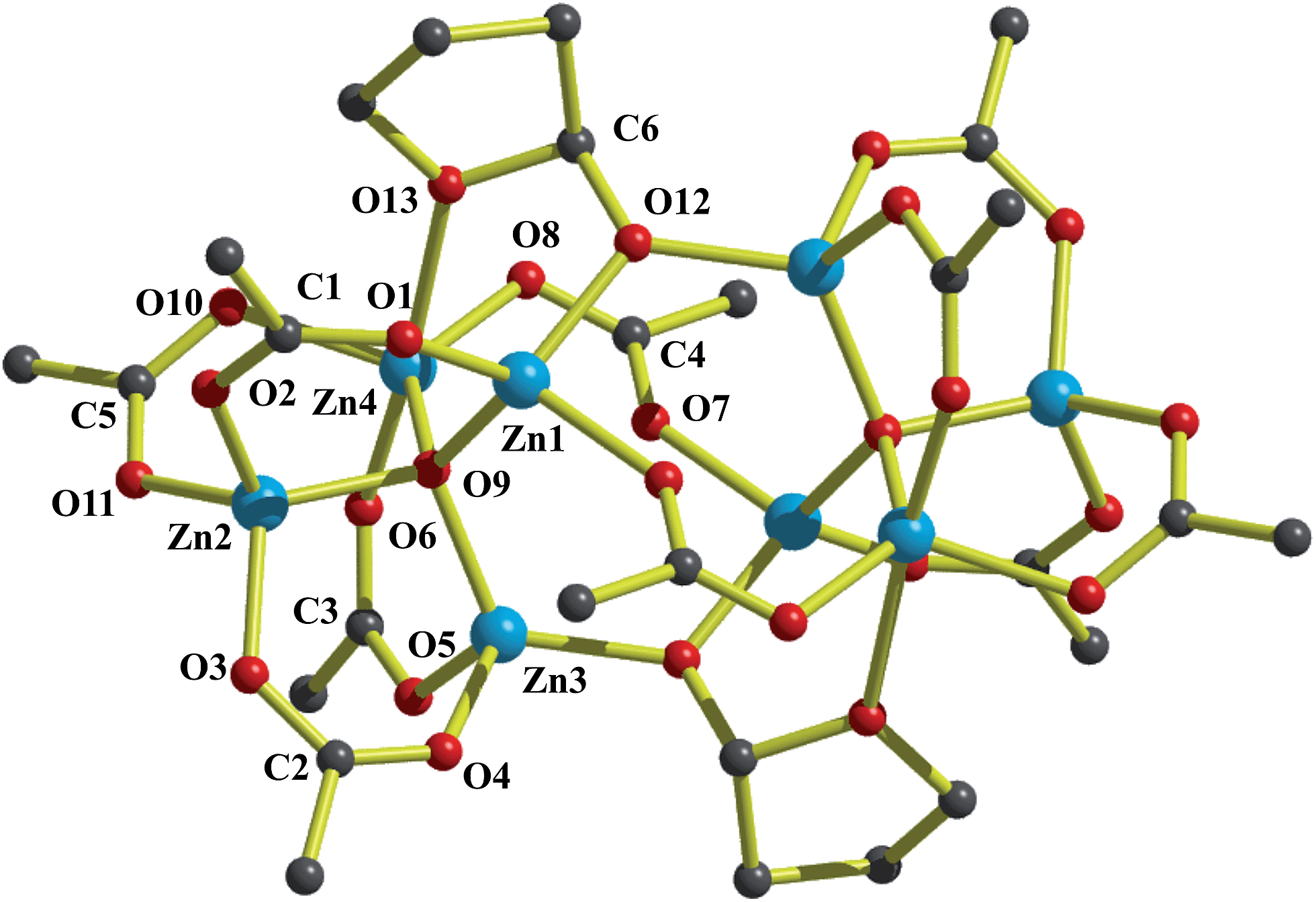 | ||
| Fig. 3 Molecular structure of 4; hydrogen atoms and Me groups of the carboxylate ligand were omitted for clarity. | ||
The composition of 4 provides strong evidence that homolysis of the incipient alkylperoxide O–O bond is likely responsible for the formation of oxo ions5e–h and is the source of the O−THF species. The peroxide undergoing homolytic cleavage of the O–O bond produces zinc oxyl ZnO˙ and alkoxy RO˙ radicals, i.e. the intermediates responsible for the initiation of further transformations. These oxygen centred radicals are able to abstract an α-hydrogen atom from THF and the resulting tetrahydrofuran-2-yl radical can react further with 1a or 1b to form 2a or 2b, respectively. Otherwise the forming radicals can recombine with the ZnO˙ radical leading to the alkoxide 4, which is outlined in Scheme 3.
The observed high yield of the radical addition of THF to imines indicates that this reaction is favoured over the recombination of ZnO˙ and tetrahydrofuran-2-yl radicals. Also, comparison of the inefficiency of the R˙ yielding radical initiating systems (Scheme 1a–c) with our well defined, potent alkylzinc carboxylate-based system producing RO˙ and ZnO˙ radical species leads to another conclusion – namely that alkyl radicals are rather unlikely to be involved in mediating the type of radical organic reactions we studied.
Conclusions
In conclusion, a new and efficient RZn(L)/air-type initiating system for the radical addition of THF to imines was developed that provides a new strategy to expand the scope of organozinc initiators for organic synthesis. The mode of its function was evidenced by the isolation and structural characterization of the novel zinc oxo aggregate with the entrapped oxidized THF moieties from the RZn(O2CR′)/air systems. Moreover the latter result along with additional control experiments demonstrate that the oxygen-centred radicals are the key initiating species in radical organic reactions mediated by RnM/air systems and strongly contradict the notion that R˙ radicals are the initiating species. Undoubtedly, further in-depth understanding of the various modes of reactivity of the metal alkyl complexes with O2 will be the key to developing effective systems promoting organic radical reactions.Experimental
Ethylzinc benzoate,5e [(EtZn)2Zn(OAr)4],7N-(4-methoxy-benzylidene)aniline (1a), N-benzylidene-4-methoxyaniline (1b)17 and tBu2Zn,18 were prepared according to the literature procedures under a nitrogen atmosphere using standard Schlenk techniques. All other reagents were of commercial grade obtained from Sigma-Aldrich Co. The solvents and propionic acid used for preparation of the initiators were carefully dried and distilled under a nitrogen atmosphere prior to use. TEMPO was sublimated and stored under a nitrogen atmosphere. Oxygenation reactions were carried out using atmospheric air dried by passing it through a tube filled with anhydrous CaCl2/KOH. NMR spectra were acquired on a Varian Mercury 400 Spectrometer, and IR spectra were recorded on a Perkin-Elmer System 2000 FT-IR spectrophotometer. GC-MS analysis was performed on an Agilent 6890N chromatograph coupled with an Agilent 5973N MS detector.General procedure for synthesis of alkylzinc carboxylates
R2Zn (2 mmol) was added dropwise to the solution of the appropriate carboxylic acid (2 mmol) at −78 °C. The reaction mixture was allowed to warm up to room temperature and stirred for 2 h. The resulting alkylzinc carboxylates were used in the model radical reaction without further purification.General procedure for addition of the THF to imines mediated by the RZn(L)/air and R2Zn/air systems
An excess of dry air was introduced to a stirred THF (14 mL) solution of an appropriate initiator (4.00 mmol) and 1a (0.220 g, 1.00 mmol) at 20 °C. The samples (2 mL) for measuring the conversion rates were subsequently taken via septum after 1.5 min, 5 min, 15 min, 1 h, 2 h, 4 h, 24 h, followed by hydrolysis with a saturated solution of NaHCO3. 5 mL of ethyl acetate was added and the white precipitate was filtered off and the water phase was extracted with ethyl acetate (2 × 10 mL). Combined organic phases were dried over anhydrous MgSO4 and the solvents were removed under vacuum. The product obtained after 24 h was purified by column chromatography (AcOEt/n-hexane 9/1). 2a was obtained as a mixture of diastereomers in various yields, depending on the time of sample collection and the reaction conditions (relevant conversion ratios in these samples calculated according to 1H NMR analyses are summarized in Table 1S† together with the final % yields). Product 2a is a dark-orange oily mixture of diastereomers. δH(400 MHz, CDCl3) ppm: 1.70–1.86 (m, 4H, –CH2–), 3.72–3.83 (m, 1.1H, –OCH2–, major diastereomer), 3.78 (s, 3H, –OCH3), 3.88–3.96 (m, 0.9H, –OCH2–, minor diastereomer), 4.01 (q, 0.55H, –OCH–, major diastereomer), 4.16 (d, 0.55H, –NCH–, major diastereomer), 4.23 (q, 0.45H, –OCH–, minor diastereomer), 4.37 (d, 0.45H, –NCH–, minor diastereomer), 4.44–4.96 (br s, 1H, –NH), 6.52 (m, 2H, CH(Ar)), 6.62 (m, 1H, CH(Ar)), 6.85 (m, 2H, CH(Ar)), 7.06 (m, 2H, CH(Ar)), 7.27–7.36 (m, 2H, CH(Ar)). δC (100 MHz, CDCl3) ppm: 25.7 (−CHCH2CH2−(THF)), 29.7 (−CH2CH2CH2−(THF)), 55.1 (−OCH3), 60.1 (−OCH2−(THF)), 68.7 (−NCH−), 83.0 (−OCH−(THF)), 113.6 (C(Ar)), 113.8 (C(Ar)), 117.3 (C(Ar)), 128.1 (C(Ar)), 128.7 (C(Ar)), 131.9 (CCH(Ar)), 147.8 (CN(Ar)), 158.7 (CO(Ar)). The isolation method and analytical data for the product 2b was identical to the literature report.3a:1) at −78 °C, for 5 min. The reaction mixture was then deoxygenated and stored at −20 °C. After 6 h, colourless crystals of 3 were deposited from this solution (57 mg, 30%). δH(400 MHz, C6D6) ppm: 1.03 (t, 3H, CH3CH2–), 1.43 (s, 9H, (CH3)3COO–Zn), 2.27 (q, 2H, CH3CH2–). Elemental analysis (%) found: C 32.95, H 5.46, calcd for C21H42O13Zn4: C 33.01, H 5.54.
Crystal data for 3: C42H84O26Zn8: M = 1528.05, crystal dimensions 0.42 × 0.28 × 0.22 mm3, monoclinic, space group C2/c (no. 15), a = 22.502(2) Å, b = 12.8820(12) Å, c = 21.7730(14) Å, β = 98.995(5)°, U = 6233.7(9) Å3, Z = 4, F(000) = 3136, Dc = 1.628 g cm−3, T = 100(2) K, μ(Mo-Kα) = 3.097 mm−1, θmax = 21.97°, R1 = 0.0956, wR2 = 0.1910 for 2200 reflections with Io > 2σ(Io). The residual electron density = +1.01/−0.93 eÅ−3. Crystal data for 4, C38H64O26Zn8: M = 1460.01, crystal dimensions 0.52 × 0.45 × 0.38 mm3, triclinic, space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 9.5731(4) Å, b = 10.8542(6) Å, c = 13.0771(7) Å, α = 77.371(2)°, β = 83.044(3)°, γ = 80.914(3)°, U = 1304.01(12) Å3, Z = 1, F(000) = 740, Dc = 1.859 g cm−3, T = 100(2) K, μ(Mo-Kα) = 3.696 mm−1, θmax = 24.71°, R1 = 0.0535, wR2 = 0.0831 R1 = 0.0391, wR2 = 0.0774 for 3685 reflections with Io > 2σ(Io). The residual electron density = +0.51/−0.56 eÅ−3. The structures were solved by direct methods using the SHELXS97 (ref. 19) program and refined by full matrix least–squares on F2 using the program SHELXL97.20 H-atoms were included in idealized positions and refined with a fixed isotropic displacement parameter equal to 1.2 (3) or isotropically (4). Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 746458 (3) and CCDC 753795 (4).
(no. 2), a = 9.5731(4) Å, b = 10.8542(6) Å, c = 13.0771(7) Å, α = 77.371(2)°, β = 83.044(3)°, γ = 80.914(3)°, U = 1304.01(12) Å3, Z = 1, F(000) = 740, Dc = 1.859 g cm−3, T = 100(2) K, μ(Mo-Kα) = 3.696 mm−1, θmax = 24.71°, R1 = 0.0535, wR2 = 0.0831 R1 = 0.0391, wR2 = 0.0774 for 3685 reflections with Io > 2σ(Io). The residual electron density = +0.51/−0.56 eÅ−3. The structures were solved by direct methods using the SHELXS97 (ref. 19) program and refined by full matrix least–squares on F2 using the program SHELXL97.20 H-atoms were included in idealized positions and refined with a fixed isotropic displacement parameter equal to 1.2 (3) or isotropically (4). Crystallographic data (excluding structure factors) for the structures reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 746458 (3) and CCDC 753795 (4).
Acknowledgements
The authors gratefully acknowledge the support of the National Science Centre (Grant Maestro DEC-2012/04/A/ST5/00595) and DEC-2011/01/N/ST5/05551 (MK).Notes and references
- Radicals in Organic Synthesis, ed. P. Renaud and M. P. Sibi, Wiley-VCH Verlag GmbH, Weinheim, 2001, vol. 1 and 2 Search PubMed; Radicals in Synthesis I and II, ed. A. Gansäuer, Top. Curr. Chem., Springer-Verlag, Berlin, Heidelberg, 2006, vol. 263 and 264 Search PubMed.
- For reviews, see: (a) C. Ollivier and P. Renaud, Chem. Rev., 2001, 101, 3415–3434 CrossRef CAS PubMed; (b) V. Darmency and P. Renaud, Radicals in Synthesis I, Springer-Verlag, Berlin/Heidelberg, 2006, vol. 263 Search PubMed; (c) S. Bazin, L. Feray and M. P. Bertrand, Chimia, 2006, 60, 260–265 CrossRef CAS; (d) T. Akindele, K. Yamada and K. Tomioka, Acc. Chem. Res., 2009, 42, 345–355 CrossRef CAS PubMed.
- For selected examples, see: (a) Radical addition of ethers to imines: K. Yamada, H. Fujihara, Y. Yamamoto, Y. Miwa, T. Taga and K. Tomioka, Org. Lett., 2002, 4, 3509–3511 CrossRef CAS PubMed; (b) K. Yamada, Y. Yamamoto, M. Maekawa and K. Tomioka, J. Org. Chem., 2004, 69, 3581–3584 CrossRef PubMed; (c) Radical addition of ethers to aldehydes: K.-I. Yamada, Y. Yamamoto and K. Tomioka, Org. Lett., 2003, 5, 1797–1799 CrossRef CAS PubMed; (d) Radical addition of cycloalkanes to imines: K. Yamada, Y. Yamamoto, M. Maekawa, J. Chen and K. Tomioka, Tetrahedron Lett., 2004, 45, 6595–6597 CrossRef CAS PubMed; (e) Radical addition of ethers to arylamines, alkoxyamines, dialkylhydrazines: Y. Yamamoto, M. Maekawa, T. Akindele, K. Yamada and K. Tomioka, Tetrahedron, 2005, 61, 379–384 CrossRef CAS PubMed; (f) Radical addition of primary alkyls to imines: K. Yamada, Y. Yamamoto, M. Maekawa, T. Akindele, H. Umeki and K. Tomioka, Org. Lett., 2006, 8, 87–89 CrossRef CAS PubMed; (g) Radical addition of iodomethylesters to imines: K. Yamada, M. Nakano, M. Maekawa, T. Akindele and K. Tomioka, Org. Lett., 2008, 10, 3805–3808 CrossRef CAS PubMed; (h) Radical addition of terminal alkynes to imines: Z. Chen, Y.-X. Zhang, Y. An, X.-L. Song, Y.-H. Wang, L.-L. Zhu and L. Guo, Eur. J. Org. Chem., 2009, 5146–5152 CrossRef CAS; (i) Aminomethylation of Michael acceptors: J. Maury, D. Mouysset, L. Feray, S. R. a. Marque, D. Siri and M. P. Bertrand, Chem.–Eur. J., 2012, 18, 3241–3247 CrossRef CAS PubMed; (j) Domino reactions involving 1,4-addition of alkyl radical to β-(propargyloxy)enoates followed by carbocyclization: A. Pérez-Luna, C. Botuha, F. Ferreira and F. Chemla, Chem.–Eur. J., 2008, 14, 8784–8788 CrossRef PubMed; (k) Radical addition of alkyls to imines and enones: M. P. Bertrand, L. Feray, R. Nouguier and P. Perfetti, J. Org. Chem., 1999, 64, 9189–9193 CrossRef CAS; (l) Radical Reformatsky reaction: P. G. Cozzi, Adv. Synth. Catal., 2006, 348, 2075–2079 CrossRef CAS; (m) P. G. Cozzi, F. Benfatti, M. G. Capdevila and A. Mignogna, Chem. Commun., 2008, 3317–3318 RSC; (n) Silylzincation of ynamides: E. Romain, C. Fopp, F. Chemla, F. Ferreira, O. Jackowski, M. Oestreich and A. Perez-Luna, Angew. Chem., Int. Ed., 2014, 53, 11333–11337 CrossRef CAS PubMed.
- For selected examples, see: (a) A. G. Davies and B. P. Roberts, J. Chem. Soc. B, 1968, 1074–1078 RSC; (b) J. Maury, L. Feray, S. Bazin, J.-L. Clément, S. R. A. Marque, D. Siri and M. P. Bertrand, Chem.–Eur. J., 2011, 17, 1586–1595 CrossRef CAS PubMed; (c) D. Mukherjee, A. Ellern and A. D. Sadow, J. Am. Chem. Soc., 2012, 134, 13018–13026 CrossRef CAS PubMed; (d) J. M. Grevy, in Encyclopedia of Inorganic Chemistry, ed. R. B. King, Wiley-VCH Verlag GmbH, Chichester, 2005, p. 5953 Search PubMed.
- (a) J. Lewiński, Z. Ochal, E. Bojarski, E. Tratkiewicz, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2003, 42, 4643–4646 CrossRef PubMed; (b) J. Lewiński, W. Marciniak, J. Lipkowski and I. Justyniak, J. Am. Chem. Soc., 2003, 125, 12698–12699 CrossRef PubMed; (c) J. Lewiński, W. Śliwiński, M. Dranka, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2006, 45, 4826–4829 CrossRef PubMed; (d) J. Lewiński, K. Suwała, M. Kubisiak, Z. Ochal, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2008, 47, 7888–7891 CrossRef PubMed; (e) J. Lewiński, W. Bury, M. Dutkiewicz, M. Maurin, I. Justyniak and J. Lipkowski, Angew. Chem., Int. Ed., 2008, 47, 573–576 CrossRef PubMed; (f) J. Lewiński, K. Suwała, T. Kaczorowski, M. Gałęzowski, D. T. Gryko, I. Justyniak and J. Lipkowski, Chem. Commun., 2009, 215–217 RSC; (g) J. Lewiński, M. Kościelski, K. Suwała and I. Justyniak, Angew. Chem., Int. Ed., 2009, 48, 7017–7020 CrossRef PubMed; (h) K. Zelga, M. Leszczyński, I. Justyniak, A. Kornowicz, M. Cabaj, A. E. H. Wheatley and J. Lewiński, Dalton Trans., 2012, 41, 5934–5938 RSC; (i) M. Kubisiak, K. Zelga, I. Justyniak, E. Tratkiewicz, T. Pietrzak, A. R. Keeri, Z. Ochal, L. Hartenstein, P. W. Roesky and J. Lewiński, Organometallics, 2013, 32, 5263–5265 CrossRef CAS.
- For the related studies, see: (a) S. Jana, R. J. F. Berger, R. Fröhlich, T. Pape and N. W. Mitzel, Inorg. Chem., 2007, 46, 4293–4297 CrossRef CAS PubMed; (b) N. Hollingsworth, A. L. Johnson, A. Kingsley, G. Kociok-Köhn and K. C. Molloy, Organometallics, 2010, 29, 3318–3326 CrossRef CAS; (c) S. Xu, W. C. Everett, A. Ellern, T. L. Windus and A. D. Sadow, Dalton Trans., 2014, 43, 14368–14376 RSC.
- (a) P. Sobota, R. Petrus, K. Zelga, L. Mąkolski, D. Kubicki and J. Lewiński, Chem. Commun., 2013, 49, 10477–10479 RSC; (b) Ł. Mąkolski, K. Zelga, R. Petrus, D. Kubicki, P. Zarzycki, P. Sobota and J. Lewiński, Chem.–Eur. J., 2014, 20, 14790–14799 CrossRef PubMed.
- E. Mileo, F. Benfatti, P. G. Cozzi and M. Lucarini, Chem. Commun., 2009, 469–470 RSC.
- A. Grirrane, I. Resa, A. Rodriguez, E. Carmona, E. Alvarez, E. Gutierrez-Puebla, A. Monge, A. Galindo, D. del Río and R. A. Andersen, J. Am. Chem. Soc., 2007, 129, 693–703 CrossRef CAS PubMed.
- K. Budny-Godlewski, D. Kubicki, I. Justyniak and J. Lewiński, Organometallics, 2014, 33, 5093–5096 CrossRef CAS.
- For a qualitative EPR study suggesting that TEMPO reacts smoothly with Et2Zn, see ref. 4b herein.
- I. Dranka, M. Kubisiak, I. Justyniak, M. Lesiuk, D. Kubicki and J. Lewiński, Chem.–Eur. J., 2011, 17, 12713–12721 CrossRef CAS PubMed.
- C. Van Koten and K. Vrieze, Adv. Organomet. Chem., 1982, 21, 151–239 CrossRef.
- In the reaction of TEMPO with Et2Zn the Et˙ radicals being formed are trapped by unreacted TEMPO. This process is most prominent when TEMPO is present in high concentration. When, however, it is added dropwise to the solution containing Et2Zn the radical entrapment is minimized and free Et˙ radicals are present in higher concentrations (see ref. 10 herein). Employment of the latter procedure in the model reaction did not however lead to formation of any product of reaction of 1a.
- Since products of reactions of reactive oxygen species with the ligand are observed in the reaction mixture after oxygenation of (EtZn)2Zn(OAr)4 (see ref. 7.) and there is no product in the model reaction, it is reasonable to conclude that the forming radical species are trapped within a solvent cage.
- (a) T. Matsumoto, K. Ohkubo, K. Honda, A. Yazawa, H. Furutachi, S. Fujinami, S. Fukuzumi and M. Suzuki, J. Am. Chem. Soc., 2009, 131, 9258–9267 CrossRef CAS PubMed; (b) R. F. Moreira, E. Y. Tshuva and S. J. Lippard, Inorg. Chem., 2004, 43, 4427–4434 CrossRef CAS PubMed; (c) C. X. Zhang, H.-C. Liang, E.-I. Kim, J. Shearer, M. E. Helton, E. Kim, S. Kaderli, C. D. Incarvito, A. D. Zuberbühler, A. L. Rheingold and K. D. Karlin, J. Am. Chem. Soc., 2003, 125, 634–635 CrossRef CAS PubMed; (d) H. Kelm and H.-J. Krűger, Angew. Chem., Int. Ed., 2001, 40, 2344–2348 CrossRef CAS; (e) M. T. Reetz and K. Töllner, Tetrahedron Lett., 1995, 36, 9461–9464 CrossRef CAS; (f) F. M. MacDonnell, N. L. P. Fackler, C. Stern and T. V. O'Halloran, J. Am. Chem. Soc., 1994, 116, 7431–7432 CrossRef CAS.
- L. Casarrubios, J. A. Perez, M. Brookhart and J. L. Templeton, J. Org. Chem., 1996, 61, 8358–8359 CrossRef CAS.
- J. Lewiński, M. Dranka, W. Bury, W. Śliwiński, I. Justyniak and J. Lipkowski, J. Am. Chem. Soc., 2007, 129, 3096–3098 CrossRef PubMed.
- SHELXS-97, Program for Structure Solution: G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467–473 CrossRef.
- G. M. Sheldrick, SHELXL-97, Program for Structure Refinement, Universität Göttingen, 1997 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, a table of conversions and isolated yields of 2a in the model reaction, characterization data for new products, crystallographic data. CCDC 746458 and 753795. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00600g |
| ‡ Due to a poor quality of crystals of 3 for single-crystal X-ray diffraction, it was not possible to get a fully satisfactory refinement of the molecular structure (the final R = 9.56%), this precludes also a detailed discussion of the metrical parameters. |
| This journal is © The Royal Society of Chemistry 2015 |