
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNickel-catalyzed reductive cleavage of aryl alkyl ethers to arenes in absence of external reductant†
Mamoru
Tobisu
*abc,
Toshifumi
Morioka
a,
Akimichi
Ohtsuki
b and
Naoto
Chatani
*a
aDepartment of Applied Chemistry, Faculty of Engineering, Osaka University, Suita, Osaka 565-0871, Japan. E-mail: tobisu@chem.eng.osaka-u.ac.jp; chatani@chem.eng.osaka-u.ac.jp
bCenter for Atomic and Molecular Technologies, Graduate School of Engineering, Osaka University, Suita, Osaka 565-0871, Japan
cESICB, Kyoto University, Katsura, Kyoto 615-8510, Japan
First published on 27th March 2015
Abstract
The reductive cleavage of the C–O bonds of aryl ethers has great potential in organic synthesis. Although several catalysts that can promote the reductive cleavage of aryl ethers have been reported, all such systems require the use of an external reductant, e.g., hydrosilane or hydrogen. Here, we report the development of a new nickel-based catalytic system that can cleave the C–O bonds of ethers in the absence of an external reductant. The hydrogen atom required in this new reductive cleavage reaction is provided by the alkoxy group of the substrate, which serves as an internal reductant. The absence of an external reductant enables the unique chemoselectivity, i.e., the selective reduction of an alkoxy group over alkenes and ketones.
Introduction
The reductive deoxygenation of phenols and their derivatives is a fundamental synthetic transformation that has been widely used in various areas of organic synthesis; for example, reductive deoxygenation enables the use of oxygen-based functionalities as removable activating and directing groups in arene functionalization reactions.1 Reductive deoxygenation is also a potentially useful strategy for the synthesis of deoxygenated analogs of phenol-based natural products.2 The reductive deoxygenation of phenols has traditionally been achieved by converting them to activated sulfonates such as triflates, followed by catalytic reduction of the activated species in the presence of a hydride donor.3 Recent progress in nickel-catalyzed4 C–O bond activation reactions5 has enabled the direct use of unactivated phenol derivatives. One of the most important achievements in this area has been the development of a catalytic process for the reduction of inert aryl ethers;6,7 this not only expands the substrate scope of reductive deoxygenation reactions, but also provides a platform for the development of new catalytic methods for the conversion of aryl ether-containing biomass to alternative sources of fuel and chemicals, including the depolymerization of lignin.8 However, it is worth noting that these nickel-catalyzed reductive cleavage reactions of aryl ethers only proceed in the presence of a stoichiometric amount of an external reductant, e.g., hydrosilane6a,6b,6d or hydrogen.6c Here, we report the development of a nickel-catalyzed reductive cleavage reaction for the conversion of aryl alkyl ethers to the parent arenes in the absence of an external reductant.Results and discussion
One possible mechanism for the reductive cleavage of aryl methyl ethers with an external hydride reagent involves oxidative addition of the C(aryl)–O bond to form nickel(II)-methoxide A, followed by reduction of an aryl–nickel bond (Scheme 1a). Although this mechanism seems plausible in view of the reports of nickel-catalyzed cross-couplings of aryl ethers with other nucleophiles,5 the key oxidative addition of a C(aryl)–OMe bond has never been verified experimentally.9 Gómez-Bengoa and Martin proposed a new mechanism for the nickel-catalyzed reductive cleavage of aryl ethers by hydrosilane (Scheme 1b).6d Spectroscopic and computational studies indicate that silylnickel(I) species can be generated under these conditions and mediate C(aryl)–OMe bond cleavage via migratory insertion of an arene to form benzylnickel species B, followed by concerted elimination of MeOSiR3. Polyaromatic compounds such as naphthyl ethers serve as good substrates, but anisole derivatives do not react unless they contain a suitable directing group; these results are consistent with the proposed mechanism involving a dearomatized intermediate B. It was envisaged that if oxidative addition of a C(aryl)–O bond occurs to form intermediate A, β-hydrogen elimination would lead to the formation of the same reductive cleavage product viaC (Scheme 1c). This mechanistic variant therefore enables the reductive cleavage to proceed without an external reductant. Although the relevance of β-hydrogen elimination of nickel-alkoxide species in catalytic reductive cleavage of aryl ethers has been suggested before,6d,9b catalytic systems that can effect such a reaction in the absence of an external reductant have not been reported.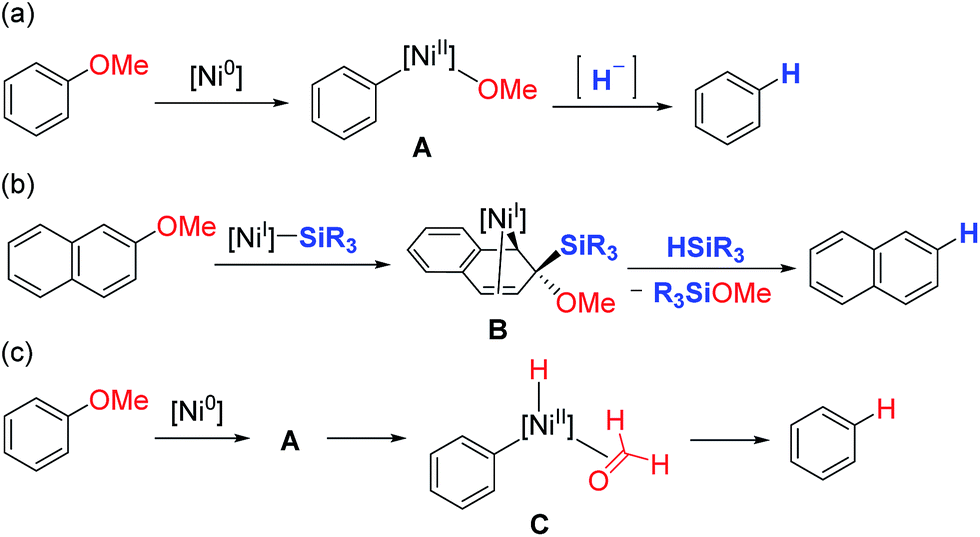 | ||
| Scheme 1 Mechanistic diversity of reductive cleavage of aryl methyl ethers. | ||
To achieve the catalytic reaction shown in Scheme 1c, we evaluated the effect of ligands in the nickel-catalyzed reduction of 2-methoxynaphthalene (1a) to give naphthalene (2). As previously reported, the addition of PCy3 and SIPr, which are the most effective ligands for the reductive deoxygenation of 1a with hydrosilane6a,6b,6d and hydrogen,6c respectively, did not lead to the formation of appreciable amounts of the reduced product 2 in the absence of an external reductant (Table 1, entries 1 and 2). Systematic screening of several other N-heterocyclic carbene (NHC)-based ligands revealed that those bearing 1-adamantyl [I(1-Ad)] and cyclohexyl (ICy)10 groups on their imidazole nitrogen atoms promoted the desired reaction to provide 2 in modest yields (entries 7 and 8). The highest yield of 2 was obtained with an NHC containing 2-adamantyl groups [I(2-Ad)] (entry 9). Although it was possible to reduce the amount of NaOtBu (80% yield with 50 mol% NaOtBu), the use of 2 equivalents resulted in a better yield of 2 (see ESI† for details). When the catalyst loading was reduced to 10 mol%, the yield of 2 was decreased to 52% (the catalyst loading could be reduced with other substrates. See the ESI† for details). The reductive cleavage proceeded in the presence of excess mercury; therefore we suggest that the soluble nickel species is responsible for the catalysis.
|
|
||
|---|---|---|
| Entry | Ligand | GC yield of 2 (%) |
| a Reaction conditions: 1a (0.25 mmol), Ni(cod)2 (0.050 mmol), ligand (0.050 mmol), NaOtBu (0.50 mmol) in toluene (0.50 mL) at 160 °C for 18 h. | ||
| 1 | PCy3 | 0 |
| 2 | SIPr·HCl | 5 |
| 3 | IPr·HCl (R = 2,6-iPr2C6H4) | 31 |
| 4 | IMes·HCl (R = 2,4,6-Me3C6H2) | 38 |
| 5 | IjPr·HCl (R = jPr) | 2 |
| 6 | ItBu·HCl (R = tBu) | 3 |
| 7 | ICy·HCl (R = cyclohexyl) | 50 |
| 8 | I(1-Ad)·HCl (R = 1-adamantyl) | 45 |
| 9 | I(2-Ad)·HCl (R = 2-adamantyl) | 84 |
|
||
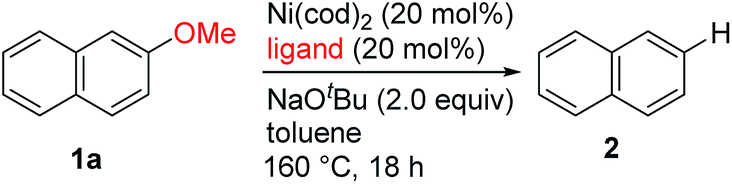
These newly developed conditions were also used with the labeled substrate 2-CD3O-naphthalene (1a-d3). The results of this experiment showed that the hydrogen delivered to the reduced product was primarily derived from the methoxy group of the substrate (eqn (1)); this supports our proposed mechanism shown in Scheme 1c.
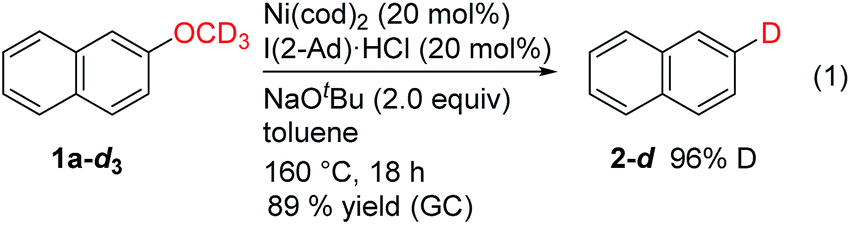 | (1) |
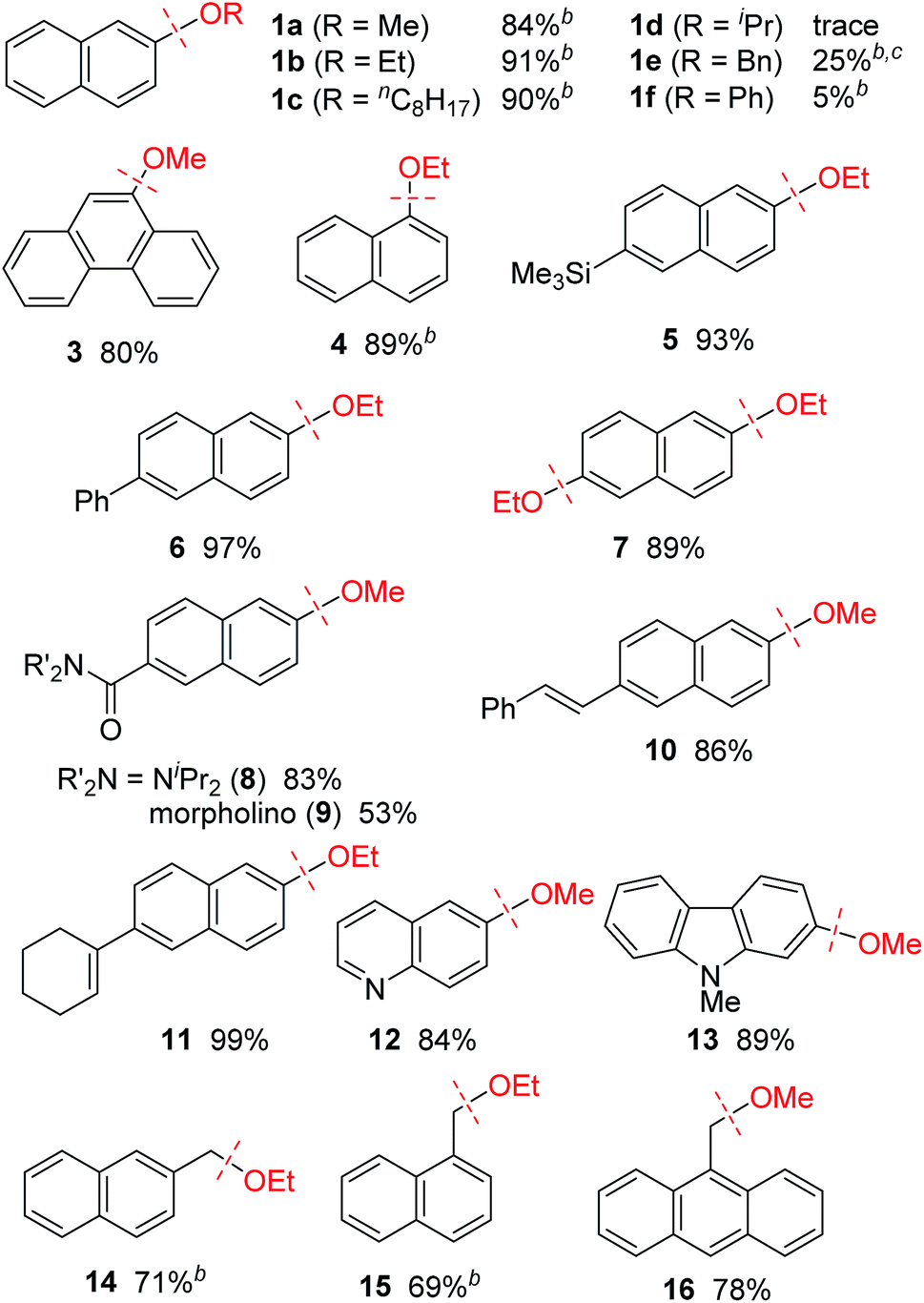
The effect of the alkoxy group on the reductive cleavage reaction was then examined using an I(2-Ad) ligand (Table 2). Primary alkoxy groups, including EtO and the longer nC8H17O groups, were efficiently removed under these condition, as in the reactions of 1b and 1c, but the bulkier iPrO group was completely unreactive. When a substrate bearing a PhCH2O group (i.e., 1e) was used, C(aryl)–O bond cleavage was accompanied by cleavage of C(benzyl)–O bonds. A PhO group remained virtually intact under these conditions, which is consistent with our proposed mechanism (Scheme 1c). Alkoxy groups on polyaromatic scaffolds such as naphthalenes 4–11 and phenanthrene 3 successfully underwent nickel-catalyzed reductive cleavage under these conditions to provide the parent arenes. Functional groups such as silyl (5), and amide (8 and 9) were compatible. Notably, a pendant alkene moiety, which could be reduced under the previously reported conditions using hydrosilane6a,b,d or hydrogen,6c remained intact under the current conditions, as shown by the reactions of 10 and 11. Heteroarenes, namely the electron-deficient quinoline 12 and electron-rich carbazole 13, were good substrates. In addition, this catalytic reaction could also be applied to benzylic C–O bonds (14–16). Unfortunately, benzylic C–O bond activation is limited to polyaromatic substrates, and simple benzyl alkyl ethers were unreactive under the current conditions.
|
|
|---|
| a Reaction conditions: aryl or benzyl ether (0.25 mmol), Ni(cod)2 (0.050 mmol), I(2-Ad)·HCl (0.050 mmol), NaOtBu (0.50 mmol) in toluene (0.50 mL) at 160 °C for 18 h. Yields shown are the isolated yields, unless otherwise noted. b Yield was determined by GC analysis because of the volatility of the product. c 2-Naphthol was obtained in 16%. |
|
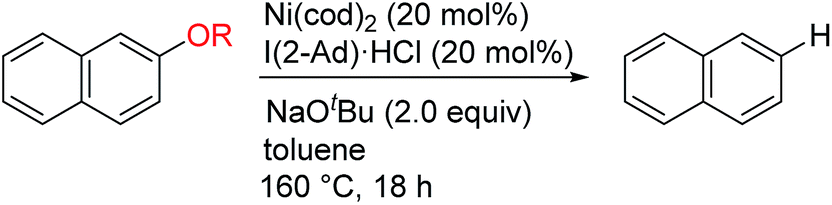
For comparison, the reductive cleavage of 14 was also examined in the presence of an external reductant (Scheme 2). The reported method using hydrosilane6b was far less effective (4% yield). The protocol using dihydrogen6c was also less effective in the presence or absence of AlMe3. These results highlight the superior catalytic activity of the Ni/I(2-Ad) system for reductive cleavage reactions.
 | ||
| Scheme 2 Comparison with reported catalytic systems. | ||
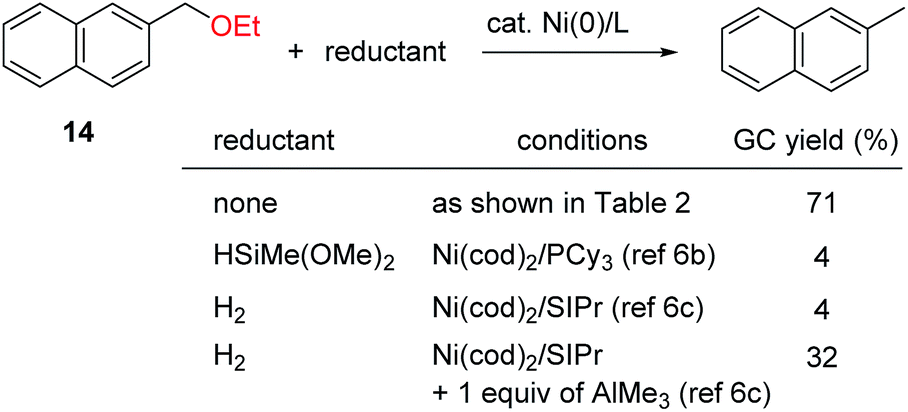
The lower reactivity of substrates lacking a fused aromatic ring is a common problem in nickel-catalyzed transformations of inert phenol derivatives.11 Similarly, 4-tert-butylanisole (15) was found to be much less reactive under the current conditions (Table 3). However, the introduction of a phenyl group, as in 16, significantly enhanced the reactivity, and the reductive cleavage product was obtained in 51% yield. Note that compound 16 is completely unreactive under previously reported nickel-catalyzed reductive cleavage conditions using hydrosilane.6a,b,d Replacement of the methoxy group with an ethoxy group further improved the efficiency of this catalysis. Ethoxybenzenes bearing a phenyl group at the para and meta positions, i.e., 17 and 18, respectively, therefore underwent the reaction to give good yields. In the case of methoxy arene substrates, β-hydrogen elimination of the postulated intermediate A generates formaldehyde complex C (see Scheme 1c), which readily decomposes to give an inactive Ni(CO) species via decarbonylation.6d Although some Ni(CO) species can generate an active Ni(0) species by the dissociation of a CO ligand under the current conditions, Ni(CO) formation would be expected to decrease the efficiency of catalyst turnover. The higher reactivities of ethoxyarenes compared with methoxyarenes can be attributed to the lower tendency of the acetaldehyde complex, which is generated from the nickel-ethoxide intermediate, to undergo the undesired decarbonylation.12 As well as a phenyl group, several electron-withdrawing groups can accelerate the present reductive cleavage reaction. Particularly important examples include anisoles bearing a ketone functionality, e.g., 19 and 20, because previous methods using an external reductant reduce the pendant ketone moiety. The current nickel catalyst system also enables the selective conversion of aryl ethers bearing amide 21, alkene 22, and pyridine 23 groups to the corresponding arenes. ortho-Substituted anisoles, such as 2-ethoxy-1,1′-biphenyl, failed to react under the current conditions.
|
|
|---|
| a Reaction conditions: aryl or benzyl ether (0.25 mmol), Ni(cod)2 (0.050 mmol), I(2-Ad)·HCl (0.050 mmol), NaOtBu (0.50 mmol), in toluene (0.50 mL) at 160 °C for 18 h. The yields shown are isolated yields unless otherwise noted. b Yield was determined by GC analysis because of the volatility of the product. c NaOtBu (0.13 mmol) was used. |
|
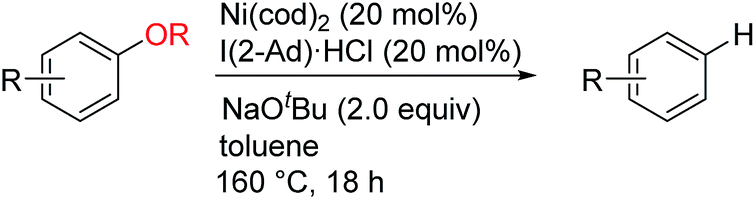
The key to a successful reaction is the use of a new NHC ligand I(2-Ad). The electronic and steric properties of I(2-Ad) were investigated to gain insights that will help in the future design of better ligands for catalytic C–O bond activation processes (Table 4). In terms of the electronic properties, the Tolman electronic parameter (TEP)13 for the new I(2-Ad) ligand was determined to be 2049, by synthesizing [IrCl(I(2-Ad))(CO)2]. This value is similar to the TEP values obtained for ICy10 and I(1-Ad), and I(2-Ad) is a significantly stronger σ-donor than PCy3, the commonest ligand used in nickel-mediated C–O bond activation.5 The steric properties were determined by calculating the buried volume (%Vbur),14 based on the crystal structure of [IrCl(I(2-Ad))(cod)]. The %Vbur of I(2-Ad) (33.5) was found to be close to that for IMes (33.8), exerting a significantly larger steric demand than ICy,10 but not as large as those of I(1-Ad) and IPr. The catalytic activity in this study clearly cannot be explained solely by these two static parameters, and other factors such as dynamic behavior of the substituents15 should be considered for a complete understanding of the structure–activity relationships.
| Entry | Ligand | TEPa | %Vburb |
|---|---|---|---|
| a Tolman electronic parameter. Values are taken from ref. 13 unless otherwise noted. b Buried volume (ref. 14). Values are calculated based on crystal structures of the corresponding [IrCl(NHC)(cod)] unless otherwise noted. See ESI for details. c Determined by synthesizing [IrCl(I(2-Ad))(CO)2]. See ESI† for details. d Calculated based on the crystal structure of [Ir(PCy3)(cod)(pyridine)]PF6. | |||
| 1 | PCy3 | 2060 | (35.7)d |
| 2 | IMes | 2050.7 | 33.8 |
| 3 | IPr | 2051.5 | 36.9 |
| 4 | ICy | 2049.6 | 28.1 |
| 5 | I(1-Ad) | 2048.3 | 37.3 |
| 6 | I(2-Ad) | 2049.4c | 33.5 |
Conclusions
In summary, we have developed a novel procedure for the nickel-catalyzed reductive deoxygenation of aryl ethers. This new reaction proceeds in the absence of an external stoichiometric reductant via a mechanism involving oxidative addition of the C–O bond followed by β-hydrogen elimination, which has not previously been exploited in a catalytic setting. The present catalytic system not only provides an atomically economic process by avoiding the use of an external reductant, but also provides a significantly wider substrate scope. These results also show that the elusive oxidative addition of a C(aryl)–OMe bond is a reliable elementary step in nickel-catalyzed C–O bond transformations of aryl ethers, particularly when an NHC-based ligand is used. Further studies of nickel-catalyzed transformations of aryl ethers using a Ni/NHC system are currently underway in our laboratory.Acknowledgements
This work was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Molecular Activation Directed toward Straightforward Synthesis” from MEXT, Japan. M. T. was also supported by the “Elements Strategy Initiative to Form Core Research Center” from MEXT. T. M. thanks the Program for Leading Graduate Schools: “Interactive Materials Science Cadet Program.” for their support. We also thank the Instrumental Analysis Center, Faculty of Engineering, Osaka University, for assistance with HRMS. Professor Makoto Yasuda and Professor Yoshihiro Nishimoto are thanked for obtaining IR spectra, and Dr Nobuko Kanehisa and Professor Masato Ohashi for X-ray analysis.Notes and references
- Selected examples: (a) G. Arnott, H. Brice, J. Clayden and E. Blaney, Org. Lett., 2008, 10, 3089 CrossRef CAS PubMed; (b) C. J. Rohbogner, G. C. Clososki and P. Knochel, Angew. Chem., Int. Ed., 2008, 47, 1503 CrossRef CAS PubMed.
- Selected examples: (a) J. Cianci, J. B. Baell, B. L. Flynn, R. W. Gable, J. A. Mould, D. Paul and A. J. Harvey, Bioorg. Med. Chem. Lett., 2008, 18, 2055 CrossRef CAS PubMed; (b) N. Z. Burns, I. N. Krylova, R. N. Hannoush and P. S. Baran, J. Am. Chem. Soc., 2009, 131, 9172 CrossRef CAS PubMed.
- Selected examples: (a) S. Cacchi, P. G. Ciattini, E. Morera and G. Ortar, Tetrahedron Lett., 1986, 27, 5541 CrossRef CAS; (b) A. Mori, T. Mizusaki, T. Ikawa, T. Maegawa, Y. Monguchi and H. Sajiki, Chem.–Eur. J., 2007, 13, 1432 CrossRef CAS PubMed.
- A review on homogeneous nickel catalysis: S. Z. Tasker, E. A. Standley and T. F. Jamison, Nature, 2014, 509, 299 CrossRef CAS PubMed.
- Reviews: (a) D.-G. Yu, B.-J. Li and Z.-J. Shi, Acc. Chem. Res., 2010, 43, 1486 CrossRef CAS PubMed; (b) B.-J. Li, D.-G. Yu, C.-L. Sun and Z.-J. Shi, Chem.–Eur. J., 2011, 17, 1728 CrossRef CAS PubMed; (c) B. M. Rosen, K. W. Quasdorf, D. A. Wilson, N. Zhang, A.-M. Resmerita, N. K. Garg and V. Percec, Chem. Rev., 2011, 111, 1346 CrossRef CAS PubMed; (d) T. Mesganaw and N. K. Garg, Org. Process Res. Dev., 2013, 17, 29 CrossRef CAS; (e) M. Tobisu and N. Chatani, Top. Organomet. Chem., 2013, 44, 35 CrossRef CAS; (f) J. Yamaguchi, K. Muto and K. Itami, Eur. J. Org. Chem., 2013, 19 CrossRef CAS PubMed; (g) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270 RSC; (h) J. Cornella, C. Zarate and R. Martin, Chem. Soc. Rev., 2014, 43, 8081 RSC.
- (a) P. Álvarez-Bercedo and R. Martin, J. Am. Chem. Soc., 2010, 132, 17352 CrossRef PubMed; (b) M. Tobisu, K. Yamakawa, T. Shimasaki and N. Chatani, Chem. Commun., 2011, 47, 2946 RSC; (c) A. G. Sergeev and J. F. Hartwig, Science, 2011, 332, 439 CrossRef CAS PubMed; (d) J. Cornella, E. Gómez-Bengoa and R. Martin, J. Am. Chem. Soc., 2013, 135, 1997 CrossRef CAS PubMed; (e) B. Sawatlon, T. Wititsuwannakul, Y. Tantirungrotechai and P. Surawatanawong, Dalton Trans., 2014, 43, 18123 RSC. For a review, see: (f) M. Tobisu and N. Chatani, ChemCatChem, 2011, 3, 1410 CrossRef CAS PubMed.
- Selected works on reductive cleavage of aryl ethers under heterogeneous catalysis: (a) A. G. Sergeev, J. D. Webb and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 20226 CrossRef CAS PubMed; (b) Y. Ren, M. Yan, J. Wang, Z. C. Zhang and K. Yao, Angew. Chem., Int. Ed., 2013, 52, 12674 CrossRef CAS PubMed; (c) M. Chatterjee, T. Ishizaka, A. Suzuki and H. Kawanami, Chem. Commun., 2013, 49, 4567 RSC.
- (a) J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed; (b) A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 709 CrossRef CAS PubMed.
- Stoichiometric reactions using aryl ethers containing a chelating group have been reported: (a) S. Ueno, E. Mizushima, N. Chatani and F. Kakiuchi, J. Am. Chem. Soc., 2006, 128, 16516 CrossRef CAS PubMed; (b) P. Kelley, S. Lin, G. Edouard, M. W. Day and T. Agapie, J. Am. Chem. Soc., 2012, 134, 5480 CrossRef CAS PubMed.
- The use of ICy in nickel-catalyzed C–O bond activation: (a) M. Tobisu, A. Yasutome, H. Kinuta, K. Nakamura and N. Chatani, Org. Lett., 2014, 16, 5572 CrossRef CAS PubMed; (b) M. Tobisu, T. Takahira, A. Ohtsuki and N. Chatani, Org. Lett., 2015, 17, 680 CrossRef CAS PubMed.
- Selected examples: (a) E. Wenkert, E. L. Michelotti and C. S. Swindell, J. Am. Chem. Soc., 1979, 101, 2246 CrossRef CAS; (b) B.-T. Guan, S.-K. Xiang, T. Wu, Z.-P. Sun, B.-Q. Wang, K.-Q. Zhao and Z.-J. Shi, Chem. Commun., 2008, 1437 RSC; (c) M. Tobisu, T. Shimasaki and N. Chatani, Angew. Chem., Int. Ed., 2008, 47, 4866 CrossRef CAS PubMed; (d) M. Tobisu, T. Shimasaki and N. Chatani, Chem. Lett., 2009, 38, 710 CrossRef CAS; (e) D.-G. Yu, B.-J. Li, S.-F. Zheng, B.-T. Guan, B.-Q. Wang and Z.-J. Shi, Angew. Chem., Int. Ed., 2010, 49, 4566 CrossRef CAS PubMed; (f) D.-G. Yu and Z.-J. Shi, Angew. Chem., Int. Ed., 2011, 50, 7097 CrossRef CAS PubMed; (g) K. Muto, J. Yamaguchi and K. Itami, J. Am. Chem. Soc., 2012, 134, 169 CrossRef CAS PubMed; (h) A. R. Ehle, Q. Zhou and M. P. Watson, Org. Lett., 2012, 14, 1202 CrossRef CAS PubMed; (i) C. Wang, T. Ozaki, R. Takita and M. Uchiyama, Chem.–Eur. J., 2012, 18, 3482 CrossRef CAS PubMed; (j) A. Correa, T. León and R. Martin, J. Am. Chem. Soc., 2014, 136, 1062 CrossRef CAS PubMed; (k) E. Koch, R. Takise, A. Studer, J. Yamaguchi and K. Itami, Chem. Commun., 2015, 51, 855 RSC ; see also ref. 6a, b and d.
- In fact, stoichiometric studies by Martin (ref. 6d) reported successful isolation of an acetaldehyde-bound nickel(0) complex, while the attempted synthesis of the corresponding formaldehyde complex resulted in the formation of a carbonyl–nickel(0) complex via a rapid decarbonylation process.
- D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723 RSC.
- H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841 RSC.
- Selected examples for the importance of the flexibility of the NHC ligands: (a) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, Angew. Chem., Int. Ed., 2003, 42, 3690 CrossRef CAS PubMed; (b) G. Altenhoff, R. Goddard, C. W. Lehmann and F. Glorius, J. Am. Chem. Soc., 2004, 126, 15195 CrossRef CAS PubMed; (c) G. C. Fortman, A. M. Z. Slawin and S. P. Nolan, Dalton Trans., 2010, 39, 3923 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and characterization data for all new compounds. See DOI: 10.1039/c5sc00305a |
| This journal is © The Royal Society of Chemistry 2015 |