
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Iron(II)-catalyzed asymmetric intramolecular olefin aminochlorination using chloride ion†
Cheng-Liang
Zhu‡
a,
Jun-Shan
Tian‡
a,
Zhen-Yuan
Gu
ab,
Guo-Wen
Xing
b and
Hao
Xu
*a
aDepartment of Chemistry, Georgia State University, 100 Piedmont Avenue SE, Atlanta, Georgia 30303, USA. E-mail: hxu@gsu.edu; Fax: +1-404-413-5505; Tel: +1-404-413-5553
bDepartment of Chemistry, Beijing Normal University, Beijing, 100875, China
First published on 13th March 2015
Abstract
An iron-catalyzed enantioselective and diastereoselective intramolecular olefin aminochlorination reaction is reported (ee up to 92%, dr up to 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). In this reaction, a functionalized hydroxylamine and chloride ion are utilized as nitrogen and chlorine sources, respectively. This new method tolerates a range of synthetically valuable internal olefins that are all incompatible with existing asymmetric olefin aminochlorination methods.
:1). In this reaction, a functionalized hydroxylamine and chloride ion are utilized as nitrogen and chlorine sources, respectively. This new method tolerates a range of synthetically valuable internal olefins that are all incompatible with existing asymmetric olefin aminochlorination methods.
Introduction
Enantioselective olefin halo-functionalization reactions constitute a range of synthetically valuable yet challenging transformations.1 Although a variety of excellent asymmetric olefin halo-oxygenation reactions have been discovered,2 there are much fewer asymmetric olefin aminohalogenation methods available.3 In particular, there have been just a few reported catalytic asymmetric olefin aminochlorination reactions.4 In one instance, Feng discovered the chiral Lewis acid-catalyzed aminochlorination of chalconic and other α,β-unsaturated olefins.4a,c Also, Chemler reported copper-catalyzed aminochlorination of terminal olefins with chlorine radical donors in the presence of MnO2 (Scheme 1A).4b Despite these and other important discoveries, catalytic asymmetric aminochlorination methods for internal, non-chalconic olefins have yet to be developed. These methods would be synthetically valuable because they would readily provide vicinal amino chlorides, a class of important chiral building blocks. Moreover, asymmetric olefin aminochlorination that proceeds through an iron-nitrenoid intermediate has not yet been reported.5 | ||
| Scheme 1 Catalytic asymmetric olefin aminochlorination: summary of this work and other existing asymmetric methods. | ||
We previously discovered Fe(BF4)2-based catalysts for both diastereoselective and enantioselective intramolecular olefin aminofluorination reactions.6 Our initial attempts to apply these catalysts to olefin aminochlorination reactions led to either low diastereoselectivity or low yield, presumably due to the reason that chlorine and fluorine atom-transfer may proceed through distinct mechanisms. Therefore, we explored a range of activating group–ligand combinations and discovered entirely new catalytic conditions for asymmetric olefin aminochlorination. Herein, we describe iron-catalyzed enantioselective and diastereoselective intramolecular aminochlorination for a range of internal, non-chalconic olefins (ee up to 92%, dr up to 15:1). In these reactions, a functionalized hydroxylamine and chloride ion were utilized as nitrogen and chlorine sources, respectively. This method tolerates a range of synthetically valuable internal olefins that are all incompatible with existing asymmetric olefin aminochlorination approaches; it also provides a new approach that is complementary to known methods for the asymmetric synthesis of amino chlorides with contiguous stereogenic centers.
Prior to this research, Bach reported an FeCl2-catalyzed racemic intramolecular olefin aminochlorination method using acyl azides, TMSCl, and EtOH under ligand-free conditions.7 Excellent syn-selectivity was observed with styrenyl olefins (dr up to > 20:1). However, poor diastereoselectivity was recorded with non-styrenyl acyclic olefins (dr: 1:1). The new method presented here has a few unique features which complement the existing iron-catalyzed olefin aminochlorination method. First, excellent anti-selectivity has been observed across a wide range of styrenyl and non-styrenyl olefins. Second, good to excellent enantioselectivity has been achieved with a variety of internal, non-chalconic olefins (ee up to 92%). Finally, acyl azides are non-reactive under the described reaction conditions (vide infra), which suggests that iron-nitrenoid generation may proceed via different pathways compared with the known azide activation pathway.
Results and discussion
A cinnamyl alcohol-derived acyloxyl carbamate 1 was selected as the model substrate for catalyst discovery (Table 1).8 In the presence of tetra-n-butylammonium chloride (TBAC), we observed that FeCl2 alone catalyzed a sluggish reaction under ligand-free conditions (entry 1, 45% yield, dr: 2:1).9 However, the FeCl2–phenanthroline (L1) complex catalyzed the anti-aminochlorination with significantly improved yield and dr (entry 2, 80% yield, dr > 20:1). We also noted that the Fe(NTf2)2–L1 complex provided essentially the same reactivity and diastereoselectivity (entry 3, 86% yield, dr > 20:1). Interestingly, the Fe(NTf2)2–bisoxazoline (L2) complex resulted in a loss of diastereoselectivity (entry 4, 82% yield, dr: 0.83:1). Furthermore, the Fe(NTf2)2–L3 complex promoted the syn-aminochlorination with moderate yield and dr (entry 5, 34% yield, dr: 0.25:1). We also observed that the Fe(NTf2)2–L4 complex catalyzed the anti-aminochlorination with a modest dr (entry 6, 75% yield, dr: 1.8:1). Notably, an iron–L4 complex resulted in high dr and reaction rate in the previously reported olefin aminofluorination reaction.6 These observations suggest that ligands are involved in the diastereoselectivity-determining step and provide excellent opportunities for diastereo-control.
|
|
|||||
|---|---|---|---|---|---|
| Entrya | Fe(X)2 | Ligand (mol%) | Conversionb | Yieldc | drb (anti:syn) |
| a Unless stated otherwise, the reactions were carried out under a nitrogen atmosphere. TBAC: tetra-n-butylammonium chloride. b Conversion and dr were determined by 1H NMR. c Isolated yield. | |||||
| 1 | FeCl2 | None | 62% | 45% | 2:1 |
| 2 | FeCl2 | L1 (20) | >95% | 80% | >20:1 |
| 3 | Fe(NTf2)2 | L1 (20) | >95% | 86% | >20:1 |
| 4 | Fe(NTf2)2 | L2 (10) | >95% | 82% | 0.83:1 |
| 5 | Fe(NTf2)2 | L3 (10) | 61% | 34% | 0.25:1 |
| 6 | Fe(NTf2)2 | L4 (20) | >95% | 75% | 1.8:1 |
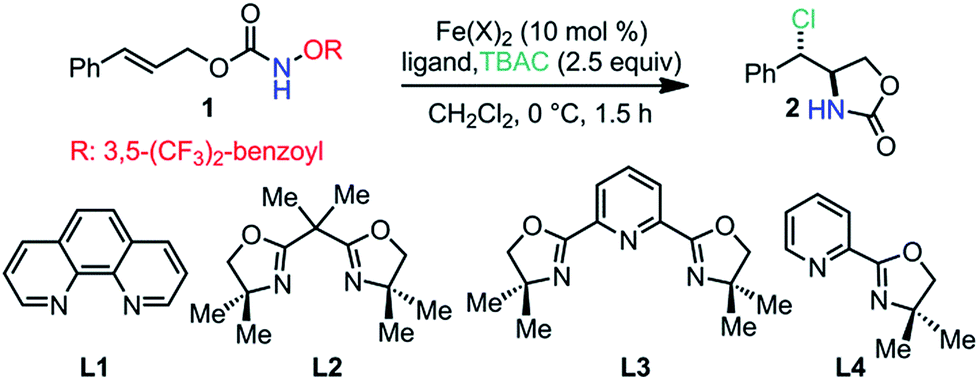
The observed ligand-enabled diastereo-control with trans-olefin 1 prompted us to evaluate cis-olefin 1′ (Scheme 2). To our surprise, the Fe(NTf2)2–L1 complex catalyzed syn-aminochlorination, while the Fe(NTf2)2–L4 complex promoted anti-aminochlorination with essentially the same dr (Scheme 2). The different reaction profiles for isomeric olefins 1 and 1′ suggest that the aminochlorination reaction is neither stereospecific nor fully stereo-convergent, which is significantly different from the iron-catalyzed olefin aminofluorination reaction.6
 | ||
| Scheme 2 Iron-catalyzed aminochlorination with a cis olefin and an acyl azide. aReaction conditions: Fe(NTf2)2 (10 mol%), L1 (20 mol%), TBAC (2.5 equiv.), CH2Cl2, 0 °C, 2 h. bReaction conditions: Fe(NTf2)2 (10 mol%), L4 (20 mol%), TBAC (2.5 equiv.), CH2Cl2, 0 °C, 2 h. | ||
Furthermore, an acyl azide 3 was evaluated under the reaction conditions as a control experiment. Interestingly, the acyl azide 3 was fully recovered and no aminochlorination product was detected. These results suggest that the activation of acyloxyl carbamates (1 and 1′) may proceed via different pathways compared with the known azide activation pathway.7
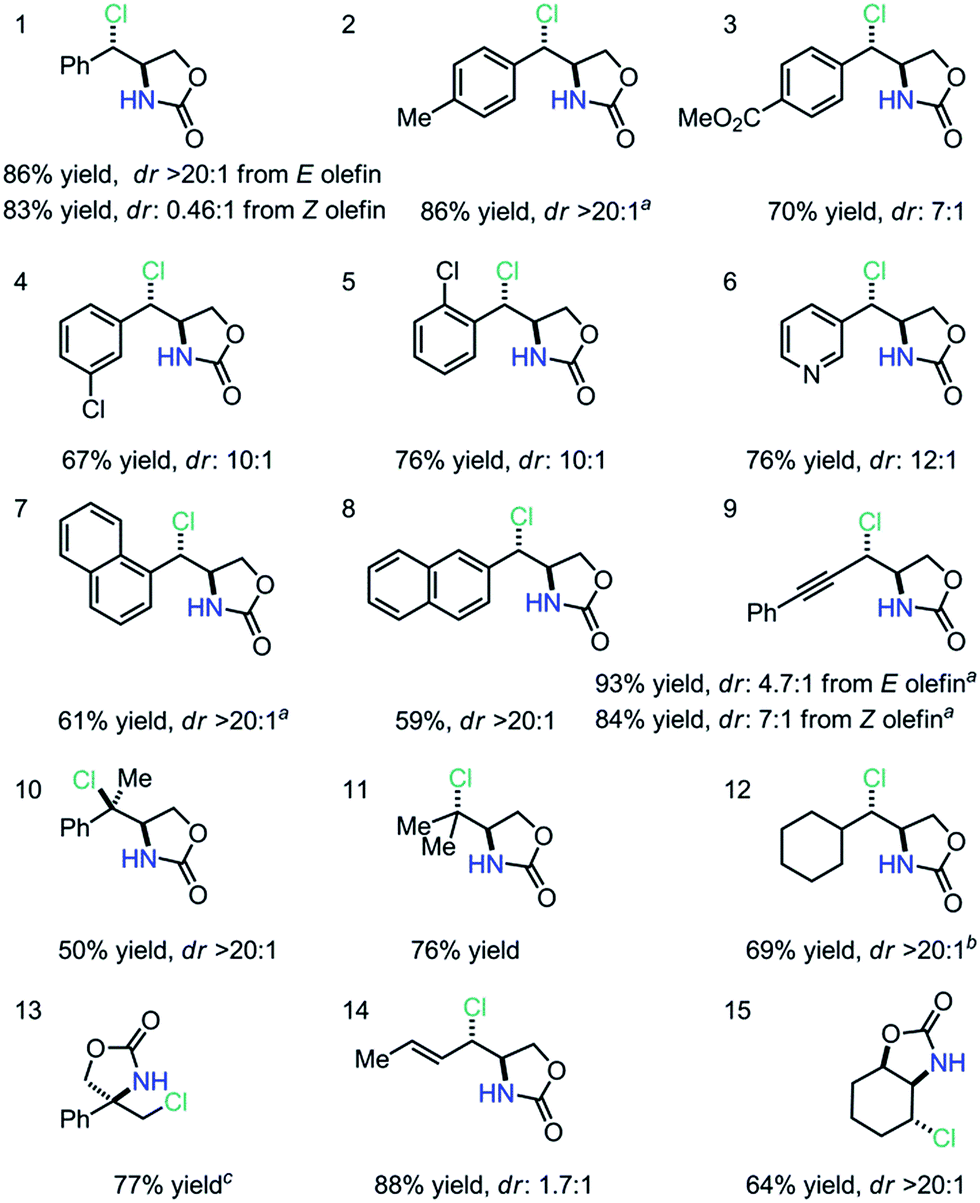
We subsequently explored a range of olefins under the optimized conditions to evaluate the scope and limitations of this anti-aminochlorination method (Table 2). We discovered that di-substituted styrenyl olefins are generally good substrates; both electron-donating and electron-withdrawing substituents are compatible with this method (entries 1–4). Importantly, ortho-substituents and pyridyl groups are both tolerated (entries 5–6). Furthermore, extended aromatics, including naphthyl olefins, are reasonable substrates (entries 7–8). Moreover, isomeric ene–ynes are both excellent substrates for the stereo-convergent and anti-selective method (entry 9). Additionally, we observed that both styrenyl and non-styrenyl tri-substituted olefins undergo aminochlorination smoothly with excellent dr (entries 10–11).10 We also discovered that a cyclohexyl-substituted olefin was an excellent substrate (entry 12, dr > 20:1). Further exploration revealed that both 1,1-disubstituted olefins and dienes are viable substrates with excellent regioselectivity (entries 13–14). Most notably, a cyclic olefin could also undergo highly diastereoselective anti-aminochlorination (entry 15, dr > 20:1), yielding a product which is difficult to obtain with known methods.11 Since the FeCl2–L1 complex provides essentially the same dr and yield in these diastereoselective reactions, FeCl2 can be a convenient substitute for Fe(NTf2)2 in racemic reactions.

In order to fulfil the need for catalytic asymmetric olefin aminochlorination, we further explored asymmetric induction for internal, non-chalconic olefins with a variety of iron–chiral ligand complexes (Table 3).12 First, we discovered that the iron–L5 complex induced diastereoselective and enantioselective anti-aminochlorination, albeit with a low yield, mostly due to the competing aminohydroxylation reaction (entry 1, 53% yield, dr: 9.9:1). Interestingly, the anti-addition product 2a was obtained with excellent ee (84% ee), while the syn-addition product 2b was obtained essentially as a racemate (<5% ee).13 Additionally, a two-step procedure can convert 2a to a chlorinated amino alcohol triad 4 without ee erosion.14 Next, we observed that the iron–L6 complex induced moderately diastereoselective syn-aminochlorination (entry 2, 68% yield, dr: 0.48:1). To our surprise, the anti-addition product 2a was obtained with moderate ee (24% ee), while the syn-addition product 2b was isolated with significant ee (79% ee). Furthermore, we evaluated chiral ligands L7 and L8 and determined that they are less effective for asymmetric induction (entries 3–4). Additionally, chiral ligand L9 induced fast yet non-selective aminochlorination with a high overall yield (entry 5).15 With the iron–L5 complex in hand, we subsequently explored other reaction parameters. First, a decreased reaction temperature was found to benefit both dr and ee (entry 6, dr: 11:1 and 90% ee for 2a at −60 °C). Next, replacing the 3,5-bis(trifluoromethyl)benzoyl activating group with a smaller acetyl group further enhanced the ee (entry 7, 97% ee for 2a); however, much lower dr and yield were obtained (entry 7, dr: 1.1:1, 42% yield). Finally, a chloroacetyl activating group induced an effective balance between overall yield and stereoselectivity (entry 8, 67% yield, dr: 9.6:1 and 89% ee for 2a). We also observed that the FeCl2–L5 complex induced a slightly less selective reaction with a lower yield (entry 9, 58% yield, dr: 9.0:1 and 83% ee for 2a).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entrya | R | Ligand | Conversionc | Yieldd | drc (anti:syn) |
eee (anti) | eee (syn) |
| a Unless stated otherwise, the reactions were carried out under a nitrogen atmosphere with 4 Å molecular sieves. b Reaction conditions: Boc2O, Et3N, DMAP; then Cs2CO3, MeOH, 85% over two steps; see ESI for details. c Conversion and dr were determined by 1H NMR. d Isolated yield. e Enantiomeric excess (ee) was measured by HPLC with chiral columns; the absolute stereochemistry was determined by X-ray crystallographic analysis of an analog of 2a. f The reaction was carried out at −60 °C for 12 h. g The FeCl2–L5 complex was used. | |||||||
| 1 | 3,5-(CF3)2-Ph | L5 | >95% | 53% | 9.9:1 |
84% | <5% |
| 2 | 3,5-(CF3)2-Ph | L6 | >95% | 68% | 0.5:1 |
24% | 79% |
| 3 | 3,5-(CF3)2-Ph | L7 | 88% | 61% | 1.7:1 |
<5% | <5% |
| 4 | 3,5-(CF3)2-Ph | L8 | >95% | 32% | 2.5:1 |
47% | 30% |
| 5 | 3,5-(CF3)2-Ph | L9 | >95% | 82% | 0.5:1 |
8% | 24% |
| 6f | 3,5-(CF3)2-Ph | L5 | >95% | 51% | 11.0:1 |
90% | <5% |
| 7f | CH3 | L5 | >95% | 42% | 1.1:1 |
97% | <5% |
| 8f | CH2Cl | L5 | >95% | 67% | 9.6:1 |
89% | <5% |
| 9f,g | CH2Cl | L5 | >95% | 58% | 9.0:1 |
83% | <5% |

In order to evaluate the scope of this asymmetric method, we explored the asymmetric induction with a range of internal olefins (Table 4). The chiral catalyst provides excellent asymmetric induction with styrenyl olefins. A range of para-substituted styrenyl olefins with different electronic properties were converted to the corresponding aminochlorination products with high dr and ee (entries 1–6, dr: 9.6–15:1, ee: 86–91%). Additionally, meta-substituted styrenyl olefins are also good substrates but with slightly decreased ee (entries 7–9, dr: 10–15:1, ee: 80–87%). However, we discovered that ortho-substitution of styrenes has a deleterious effect on ee (entries 10–11, dr: 4.5–12:1, ee: 77–79%). Interestingly, both α- and β-naphthyl olefins are excellent substrates (entries 12–13, dr: 4.5–10:1, ee: 89–92%). To our delight, a 3-pyridyl olefin with a basic nitrogen atom is a reasonable substrate for the asymmetric aminochlorination (entry 14, dr: 1.8:1, ee: 70% for the anti-diastereomer). Moreover, we observed that the iron–L5 complex can induce significant ee in the aminochlorination with non-styrenyl olefins (entry 15, dr: 2:1, ee: 54% for the anti-diastereomer). To our surprise, the iron–L6 complex proved to be uniquely effective for the asymmetric induction with tri-substituted olefins, while the iron–L5 complex was less effective (entry 16, dr: 2.3:1, ee: 86% for the anti-diastereomer).16
|
|
|---|
| a Unless stated otherwise, mono-chloroacetyl was selected as the activating group for asymmetric catalysis; the ee for all syn-aminochlorination products was less than 5%. b Bis(trifluoromethyl)-benzoyl was selected as the activating group. c The ee for the syn-addition product was 12%. d L6 was used as the ligand for asymmetric induction; the ee for the syn-addition product was 50%. |

|

During the exploration of substrate scope, it was surprising to observe completely different ee values for anti- and syn-diastereomers (e.g.2a and 2b). In contrast, exactly the same ee for both diastereomeric products was observed in the iron-catalyzed aminofluorination of 1.6 In order to obtain greater mechanistic insights, we carried out ee analysis for all isolable products using several control experiments (Scheme 3). First, in an Fe(NTf2)2-catalyzed reaction with trans-olefin 1, two aminochlorination products were obtained (Scheme 3A, 90% ee for 2a, <5% ee for 2b, dr: 11:1).17 Simultaneously, diastereomers 5a and 5b were also isolated with the same ee as two competing olefin aminohydroxylation products (Scheme 3A, 88% ee for 5a and 5b, dr: 4:1). However, completely different selectivity (both dr and ee) was observed in an Fe(NTf2)2-catalyzed reaction with cis-olefin 1′ (Scheme 3A, 85% ee for 2a and 31% ee for 2b, dr: 6:1; 93% ee for 5a and 83% ee for 5b, dr: 7:1). In both cases, 5a and 5b cannot be converted to 2a under the reaction conditions.
 | ||
| Scheme 3 Control experiments to probe the mechanism. aReaction conditions: Fe(NTf2)2 (15 mol%), L1 (15 mol%), TBAC (2.5 equiv.), CHCl3, −60 °C, 12 h. bReaction conditions: Fe(NTf2)2 (15 mol%), L1 (15 mol%), CHCl3, −60 °C, 12 h. | ||
These observations provide several important mechanistic insights. First, the non-stereospecificity observed in the iron-catalyzed olefin aminochlorination suggests that the formation of C–N and C–Cl bonds occurs in a stepwise fashion.18 Second, the lack of complete stereo-convergence between the reaction profiles of isomeric olefins (1 and 1′) suggests that C–N bond formation may be the rate- and ee-determining step.18 Furthermore, since essentially the same ee was observed for 2a, 5a, and 5b from the reaction with trans-olefin 1, it is likely that these products are derived from the same intermediate after the ee-determining step. Additionally, the fact that the syn-aminochlorination product 2b was isolated as a racemate suggests that 2b may be derived from non-stereoselective pathways which are distinct from the one leading to the formation of 2a, 5a, and 5b.
The product divergence (2avs.5a/b) after the ee-determining step is mechanistically interesting. Therefore, we studied the effect of external chloride ion. To our surprise, in the absence of TBAC, the Fe(NTf2)2–L5 complex alone was ineffective for the nitrogen atom-transfer at −60 °C; 1 and 1′ were both fully recovered (Scheme 3B). However, aminochlorination occurred as soon as a stoichiometric amount of TBAC was introduced. This observation suggests that the Fe(NTf2)2–L5 complex may serve as a pre-catalyst and it may be activated by chloride ion in situ.
In order to test this hypothesis, we further carried out the FeCl2-catalyzed reaction in the presence of TBAC (Scheme 3C). Notably, 2a was isolated with essentially the same ee as that obtained under the standard conditions (88% ee for 2a and <5% ee for 2b). This result suggests that the catalytically relevant species may also be generated from the FeCl2–L5 complex.
To probe for more mechanistic details, we subsequently carried out the FeCl2-promoted olefin aminochlorination in the absence of TBAC (100 mol% FeCl2, 100 mol% L5, Scheme 3C). Under these conditions, FeCl2 is the only available chlorine source. Surprisingly, we discovered that 2a was obtained with essentially the same ee compared with the two previous control experiments (88% ee for 2a). Furthermore, a syn-aminohydroxylation product 5a was isolated with excellent dr and ee (dr > 20:1, 88% ee). These observations suggest that Fe–Cl bond cleavage may be relevant for the chlorine atom-transfer step during the enantioselective anti-aminochlorination.19 In addition, we also identified a small amount of aziridine 6 (15% yield, 82% ee), and further discovered that it could not be converted to either 2a or 5a under the reaction conditions.
With the accumulated mechanistic evidence, we propose a plausible mechanistic working hypothesis for the iron-catalyzed asymmetric aminochlorination of trans-olefin 1 (Scheme 4). First, the iron catalyst reversibly cleaves the N–O bond in the acyloxyl carbamate 1, generating iron-nitrenoid A with chloride as a counter ion. From there, A may participate in enantioselective and diastereoselective aminochlorination and aminohydroxylation to afford 2a and 5a, respectively. Since the aminochlorination–aminohydroxylation competition occurs after the ee-determining step, 2a is obtained with essentially the same ee as 5a. At the same time, 1 may be converted to 2bvia a non-stereoselective pathway which is distinct from the one leading to the formation of 2a and 5a. Further mechanistic studies are required to elucidate the details.
 | ||
| Scheme 4 Proposed mechanistic working hypothesis for the iron-catalyzed asymmetric aminochlorination of trans-olefin 1. | ||
Conclusions
In conclusion, we have described an iron-catalyzed enantioselective and diastereoselective aminochlorination method for internal, non-chalconic olefins. This method tolerates a range of synthetically valuable olefins that are all incompatible with existing asymmetric olefin aminochlorination methods. It also provides a complementary approach for the asymmetric synthesis of amino chlorides with contiguous stereogenic centers. Our preliminary mechanistic studies revealed that an FeCl2-derived nitrenoid may be a feasible reactive intermediate and that Fe–Cl bond cleavage may be relevant for stereoselective chlorine atom-transfer. Our current efforts are focused on the mechanistic investigation of this new reaction and method development for the enantioselective intermolecular olefin aminochlorination.Acknowledgements
This work was supported by the National Institutes of Health (GM110382) and Georgia State University. Z.-Y. G. was supported by NSFC (21272027) and a fellowship from China Scholarship Council.Notes and references
- For selected reviews of asymmetric olefin halofunctionalization, see:
(a) S. E. Denmark, W. E. Kuester and M. T. Burk, Angew. Chem., Int. Ed., 2012, 51, 10938 CrossRef CAS PubMed
; (b) S. R. Chemler and M. T. Bovino, ACS Catal., 2013, 3, 1076 CrossRef CAS
- For selected reports on catalytic asymmetric olefin halo-oxygenation, see:
(a) S. H. Kang, S. B. Lee and C. M. Park, J. Am. Chem. Soc., 2003, 125, 15748 CrossRef CAS PubMed
- For selected reports on catalytic asymmetric olefin aminobromination and aminoiodination, see:
(a) Y. Cai, X. Liu, Y. Hui, J. Jiang, W. Wang, W. Chen, L. Lin and X. Feng, Angew. Chem., Int. Ed., 2010, 49, 6160 CrossRef CAS PubMed
- For existing asymmetric olefin aminochlorination methods, see:
(a) Y. F. Cai, X. H. Liu, J. Jiang, W. L. Chen, L. L. Lin and X. M. Feng, J. Am. Chem. Soc., 2011, 133, 5636 CrossRef CAS PubMed
- For catalytic olefin aminohydroxylation that proceeds through an iron-nitrenoid intermediate, see:
(a) G.-S. Liu, Y.-Q. Zhang, Y.-A. Yuan and H. Xu, J. Am. Chem. Soc., 2013, 135, 3343 CrossRef CAS PubMed
- D.-F. Lu, G.-S. Liu, C.-L. Zhu, B. Yuan and H. Xu, Org. Lett., 2014, 16, 2912 CrossRef CAS PubMed
-
(a) T. Bach, B. Schlummer and K. Harms, Chem. Commun., 2000, 287 RSC
- See ESI† for details of substrate synthesis. Acyloxyl carbamates are reactive, while tosyloxyl and alkoxyl carbamates are non-reactive and fully recovered under the reaction conditions.
- The relative stereochemistry of 2a was determined by comparison of the experimental NMR data with those reported in ref. 7. It was further corroborated by 1H NMR and X-ray crystallographic analysis of a structural analog of 2a. See ESI† for details.
- The relative stereochemistry was assigned based on the 1H NMR and X-ray crystallographic analysis of a structural analog described in ref. 6; see ESI† for details.
- Complementary stereochemistry was achieved (in entry 15 of Table 2) compared with the known method reported in ref. 7, where the syn-aminochlorination product was isolated. This substrate did not undergo kinetic resolution with a chiral catalyst, the complex Fe(NTf2)2–L5. Both the starting material and product were isolated as racemates.
- For leading references on chiral BOX and related ligands, see:
(a) D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726 CrossRef CAS
- The absolute stereochemistry of 2a was determined by X-ray crystallographic analysis of a structural analog of 2a. See ESI† for details.
- For detailed procedure and HPLC traces of 4, see ESI.†.
- For the synthesis of L9, see ref. 6.
- The iron–L5 complex catalyzed the reaction favoring the syn-addition product (dr (anti/syn): 0.47:1); ee for the anti-addition product was 60% and ee for the syn-addition product was <5%. The relative stereochemistry was assigned based on the 1H NMR and X-ray crystallographic analysis of a structural analog described in ref. 6; see ESI† for details.
- When a chloroacetyl group was used as the activating group, a different result was obtained. For details, see entry 8 of Table 3.
- For an example of stepwise atom transfer reactions with different reaction profiles for cis/trans isomeric olefins, see: N. H. Lee and E. N. Jacobsen, Tetrahedron Lett., 1991, 32, 6533 CrossRef CAS
- For the oxidation of a radical species by a high-valent metal through ligand transfer or electron transfer, see:
(a) M. S. Kharasch and G. Sosnovsky, J. Am. Chem. Soc., 1958, 80, 756 CrossRef CAS
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedure, characterization data for all new compounds, selected NMR spectra and HPLC traces. CCDC 1041826. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5sc00221d |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |