
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Unexpected effect of catalyst concentration on photochemical CO2 reduction by trans(Cl)–Ru(bpy)(CO)2Cl2: new mechanistic insight into the CO/HCOO− selectivity†
Yusuke
Kuramochi
a,
Jun
Itabashi
a,
Kyohei
Fukaya
a,
Akito
Enomoto
a,
Makoto
Yoshida
a and
Hitoshi
Ishida
*ab
aDepartment of Chemistry, Graduate School of Science, Kitasato University, 1-15-1 Kitasato, Minami-ku, Sagamihara, Kanagawa 252-0373, Japan. E-mail: ishida@sci.kitasato-u.ac.jp
bPrecursory Research for Embryonic Science (PRESTO), Japan Science and Technology Agency (JST), 4-1-8 Honcho, Kawaguchi, Saitama 332-0012, Japan
First published on 12th March 2015
Abstract
Photochemical CO2 reduction catalysed by trans(Cl)–Ru(bpy)(CO)2Cl2 (bpy = 2,2′-bipyridine) efficiently produces carbon monoxide (CO) and formate (HCOO−) in N,N-dimethylacetamide (DMA)/water containing [Ru(bpy)3]2+ as a photosensitizer and 1-benzyl-1,4-dihydronicotinamide (BNAH) as an electron donor. We have unexpectedly found catalyst concentration dependence of the product ratio (CO/HCOO−) in the photochemical CO2 reduction: the ratio of CO/HCOO− decreases with increasing catalyst concentration. The result has led us to propose a new mechanism in which HCOO− is selectively produced by the formation of a Ru(I)–Ru(I) dimer as the catalyst intermediate. This reaction mechanism predicts that the Ru–Ru bond dissociates in the reaction of the dimer with CO2, and that the insufficient electron supply to the catalyst results in the dominant formation of HCOO−. The proposed mechanism is supported by the result that the time-course profiles of CO and HCOO− in the photochemical CO2 reduction catalysed by [Ru(bpy)(CO)2Cl]2 (0.05 mM) are very similar to those of the reduction catalysed by trans(Cl)–Ru(bpy)(CO)2Cl2 (0.10 mM), and that HCOO− formation becomes dominant under low-intensity light. The kinetic analyses based on the proposed mechanism could excellently reproduce the unusual catalyst concentration effect on the product ratio. The catalyst concentration effect observed in the photochemical CO2 reduction using [Ru(4dmbpy)3]2+ (4dmbpy = 4,4′-dimethyl-2,2′-bipyridine) instead of [Ru(bpy)3]2+ as the photosensitizer is also explained with the kinetic analyses, reflecting the smaller quenching rate constant of excited [Ru(4dmbpy)3]2+ by BNAH than that of excited [Ru(bpy)3]2+. We have further synthesized trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2 (6Mes-bpy = 6,6′-dimesityl-2,2′-bipyridine), which bears bulky substituents at the 6,6′-positions in the 2,2′-bipyridyl ligand, so that the ruthenium complex cannot form the dimer due to the steric hindrance. We have found that this ruthenium complex selectively produces CO, which strongly supports the catalytic mechanism proposed in this work.
Introduction
Photocatalytic CO2 reduction represents a major concern in relation to the construction of artificial photosynthetic systems and solar fuels, which are relevant to the solution of the fossil fuel shortage and the global warming problems.1–4 Until now, many metal complexes have been investigated for CO2 reduction catalysis.5–15 As metal complexes for the catalysts, manganese mono(bipyridyl) tricarbonyl,16–18 cobalt and iron porphyrin,19–21 cobalt tris(bipyridyl)22 and macrocycle,23,24 nickel cyclam,25–27 molybdenum and tungsten mono(bipyridyl) tetracarbonyl,28 rhodium bis(bipyridyl),29,30 palladium phosphine,31–33 rhenium mono(bipyridyl) tricarbonyl,34–42 osmium mono(bipyridyl) dicarbonyl,43,44 iridium poly(pyridyl) and dihydride pincer,45,46 ruthenium mono(bipyridyl) and bis(bipyridyl) dicarbonyl complexes47–70 have been investigated. Most of these yield CO and/or formate as the two-electron reduction products of CO2. Among the metal complex catalysts, ruthenium complexes (e.g., [Ru(bpy)2(CO)2]2+) have actively been studied for the CO/HCOO− selectivity. In the catalyses, the product selectivity depends on the reaction conditions: acidic conditions enhance CO production while basic conditions cause formate production.51–56,69 Photochemical reductions have mostly resulted in formate production47–49,51–53,57–62,70 while electrochemical reductions have achieved the selective formation of CO.55,63–69 The reaction mechanisms of Ru(II) complexes have been proposed as shown in Scheme 1.50–56,69 The widely accepted mechanism of CO production is as follows: (1) the Ru(II) complex accepts one electron to release CO, (2) the one-electron-reduced complex accepts another electron, and (3) the two-electron-reduced complex undergoes an electrophilic attack by CO2 along with protonation and dehydration to regenerate the starting Ru(II) complex. For formate production, two mechanisms have been proposed so far. Tanaka and co-authors have proposed a mechanism in which the equilibrium between [Ru–C(O)OH](n+1)+ and [Ru–CO](n+2)+ governs the product selectivity between CO/HCOO−: the two-electron reduction of [Ru–C(O)OH](n+1)+ causes formate production.51–56,69 This mechanism can explain well that formate is mainly produced under basic conditions where the equilibrium shifts to the hydroxycarbonyl complex. However, it is not fully accepted because the reaction requires a specific proton attack on the carbon atom of the hydroxycarbonyl group. Meyer et al. have proposed that formate is generated via insertion of CO2 into the Ru–H bond in [Ru–H]n+.50 The mechanism via the hydride complex is based on the experimental result that [Ru(bpy)2(CO)H]0 reacts with CO2 to yield [Ru(bpy)2(CO)(OC(O)H)]0, and is generally accepted as the mechanism of formate production in organometallic chemistry. However, this mechanism does not successfully elucidate why formate is selectively produced under less protic conditions and why dihydrogen originating from the hydride intermediate scarcely evolves when formate is produced, but does evolve with CO production. Even today with more than 20 years having passed since these mechanisms were proposed, consensus on the reaction mechanism of formate production has not yet been reached.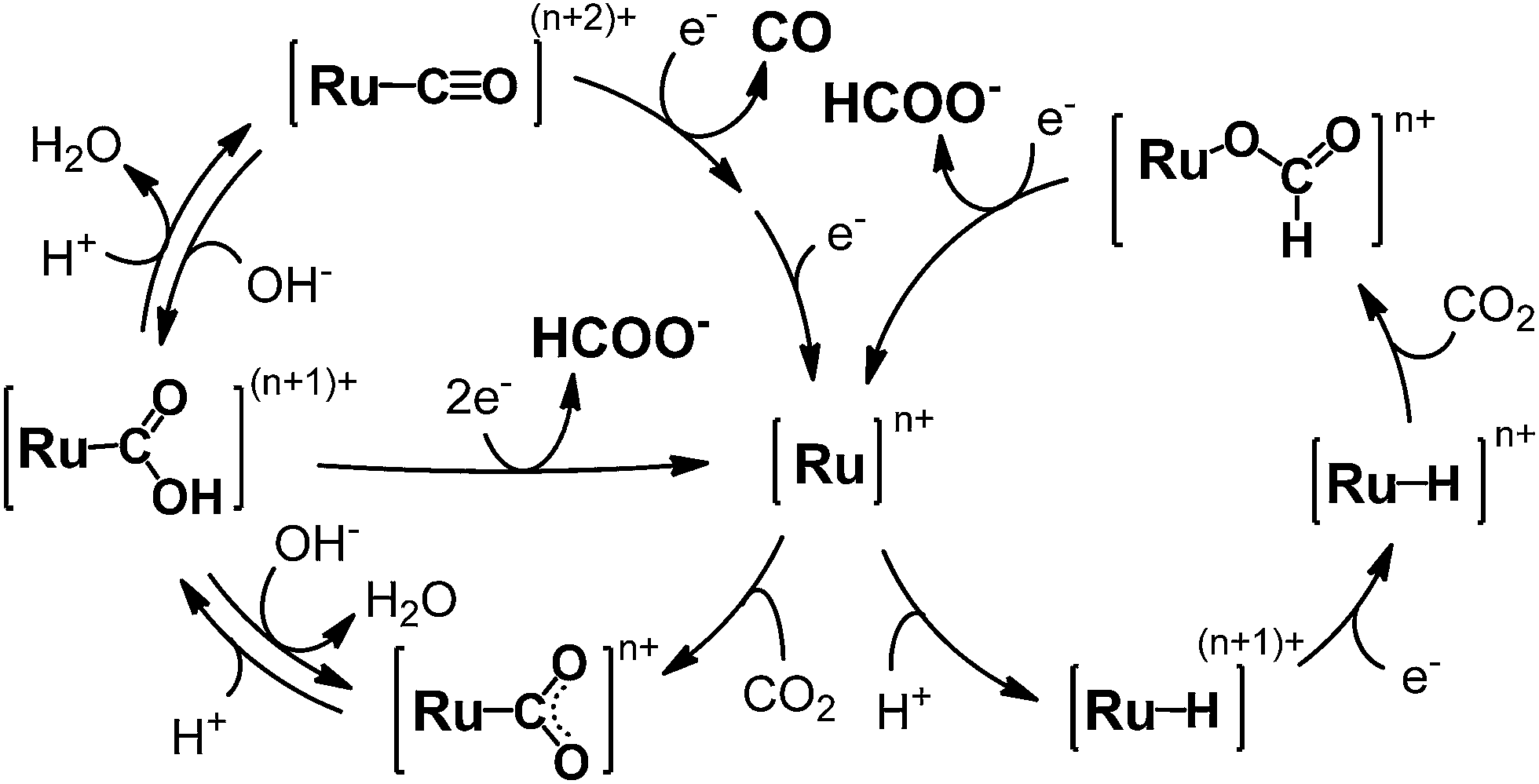
Recently, polymeric ruthenium mono(bipyridyl) dicarbonyl complexes (e.g., [Ru(L)(CO)2]n (L = bipyridyl derivatives)) have been utilized as reduction catalysts in artificial photosynthetic systems using semiconductors, which can utilize water as an electron donor.60 The system consists of TiO2 and InP modified with the ruthenium polymer. Photo-irradiation of the system induces electron transfer from the semiconductors to the ruthenium polymer, which catalyses CO2 reduction using the electrons to afford formate in a 10 mM NaHCO3 aqueous solution with CO2 bubbling. Polymeric ruthenium mono(bipyridyl) dicarbonyl complexes have been reported as catalysts in electrochemical CO2 reduction by Deronzier and Ziessel et al.63–65,67,68 The selectivity of CO vs. formate production depends essentially on the substituents which are introduced at the 4,4' position of the bipyridyl ligand in aqueous solution.64 Ruthenium mono(bipyridyl) dicarbonyl polymers and their derivatives with electron-donating substituents give mainly CO as the reduction product at pH 6. The pH values of the solution and the electrolytes (e.g., NaSO4vs. LiClO4) used for the electrolyses moderately affect the selectivity. On the contrary, polymer complexes with electron-withdrawing substituents quantitatively yield HCOO−. This difference is explained by the electronic structures of the catalyst intermediates (the hydroxycarbonyl or formato complexes) formed during the electrocatalytic process. A similar tendency has been reported for electrocatalyses by derivatives of [Ru(bpy)2(CO)2]2+.69 Thus, results in the literature indicate that the reaction mechanisms and the product selectivity strongly depend on the reaction conditions. However, to the best of our knowledge, there is no report on the effects of catalyst concentration on product selectivity in CO2 reduction.
The polymeric ruthenium mono(bipyridyl) dicarbonyl complexes are obtained by electrochemical reductions of mono(bipyridyl) dicarbonyl dichloride complexes,63–65,67,68,71,72 as well as ruthenium bis(bipyridyl) dicarbonyl complexes.66 For instance, the formation process of [Ru(bpy)(CO)2]n is shown in Scheme 2. The electrochemical reduction of Ru(bpy)(CO)2Cl2 initially dissociates the chloride ion to form a dimer, [Ru(bpy)(CO)2Cl]2, which has already been elucidated by Haukka et al. with X-ray crystallographic analysis.73 Further reduction of the ruthenium dimer causes dissociation of the chloride ions to give [Ru(bpy)(CO)2]n. The monomeric ruthenium complex, Ru(bpy)(CO)2Cl2, also shows catalytic activity for electrochemical CO2 reduction,63,65,67,69 but the complex tends to form an adherent film of the polymer on the electrode during electrochemical CO2 reduction, making it difficult to investigate the catalytic properties of the monomeric complex in detail. The photochemical CO2 reduction catalysed by Ru(bpy)(CO)2Cl2 has been reported in the presence of [Ru(bpy)3]2+ as the photosensitizer and triethanolamine (TEOA) as the electron donor,70 in which the reaction starts when [Ru(bpy)3]2+ absorbs visible light to induce electron transfer relay from TEOA to the catalyst. However, the system also causes the polymeric complex to form a black precipitate during the catalytic reaction, which probably inhibits light absorption and/or electron transfer from the photosensitizer.
 | ||
| Scheme 2 Formation of the polymeric ruthenium complex from trans(Cl)–Ru(bpy)(CO)2Cl2. | ||
We have very recently reported photochemical CO2 reduction catalysed by [Ru(bpy)2(CO)2]2+ in N,N-dimethylacetamide (DMA)/water containing [Ru(bpy)3]2+ and 1-benzyl-1,4-dihydronicotinamide (BNAH).47 DMA is used as an alternative solvent for N,N-dimethylformamide (DMF), which is the most frequently used solvent in photochemical CO2 reduction, but has been indicated to cause contamination of HCOO− by hydrolysis.74 In the DMA/water systems using BNAH as the electron donor, the black precipitate scarcely formed, and the photocatalytic CO2 reduction proceeded smoothly. The products were CO and formate, which were confirmed as the CO2 reduction products by the 13C NMR experiments. We further showed that the oxidized form of BNAH was the BNA dimer, e.g., 1,1′-dibenzyl-1,1′,4,4′-tetrahydro-4,4′-binicotinamide (4,4′-BNA2),47,49 indicating that the reduced species of the photosensitizer ([Ru(bpy)3]+), which was generated by the reductive quenching of the excited [Ru(bpy)3]2+ with BNAH, supplied the electrons to the catalyst.
In this work, we have investigated photochemical CO2 reduction catalysed by trans(Cl)–Ru(bpy)(CO)2Cl2 in a DMA/water solution containing [Ru(bpy)3]2+ and BNAH (Fig. 1). We have unexpectedly discovered that the concentration of trans(Cl)–Ru(bpy)(CO)2Cl2 affects the product selectivity (CO/HCOO−). The mechanisms reported so far48,50–56,64,69,70 cannot explain the phenomenon. This motivated us to reconsider the reaction mechanism of photochemical CO2 reduction. We propose a new reaction mechanism involving a Ru(I)–Ru(I) dimer, which catalyses CO2 reduction to selectively produce formate, because HCOO− production becomes dominant under low-intensity light, in which the Ru–Ru bond tends to form. The photochemical CO2 reduction catalysed by [Ru(bpy)(CO)2Cl]2 (0.05 mM) shows similar time–course profiles for the production of CO and HCOO− to those of trans(Cl)–Ru(bpy)(CO)2Cl2 (0.10 mM), indicating that the immediate dissociation of the dimer occurs to regenerate the monomeric complex during the CO2 reduction. We carried out kinetic analyses based on the new reaction mechanism, and the simulation curve reproduced the concentration dependence of the product selectivity (CO/HCOO−) well. In order to further verify the proposed mechanism, we synthesized trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2 (Fig. 1, 6Mes-bpy: 6,6′-dimesityl-2,2′-bipyridine) which cannot form a dimer due to the bulky substituents at 6,6′-positions in 2,2′-bipyridine. The photochemical CO2 reduction catalysed by trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2 selectively produced CO, strongly supporting our proposed mechanism for formate production.
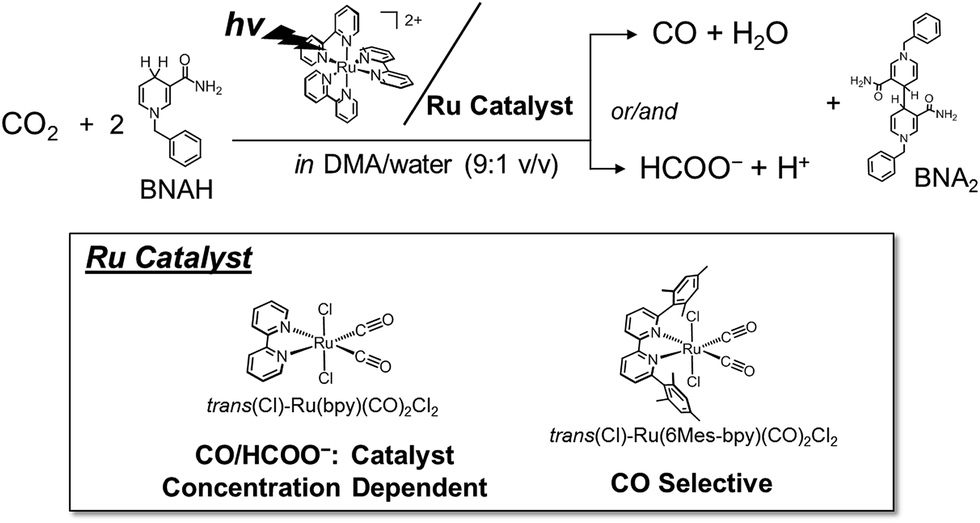 | ||
| Fig. 1 Photochemical CO2 reduction catalysed by trans(Cl)–Ru(bpy)(CO)2Cl2 or trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2 in a DMA/water solution containing [Ru(bpy)3]2+ and BNAH as the photosensitizer and the electron donor, respectively. | ||
Experimental section
General procedure
[Ru(bpy)(CO)2Cl]2, [Ru(CO)2Cl2]n, trans(Cl)–Ru(bpy)(CO)2Cl2, [Ru(bpy)3](PF6)2, [Ru(4dmbpy)3](PF6)2 (4dmbpy = 4,4′-dimethyl-2,2′-bipyridine), 6,6′-dimesityl-2,2′-bipyridine (6Mes-bpy), and BNAH were prepared according to the literature.73,75–78 DMA (Wako, dehydrate) was used as supplied. High-purity water (resistivity: 18.2 MΩ cm) was obtained from an ultrapure water system (RFU424TA, Advantec). Cyclic voltammograms (CVs) and differential pulse voltammograms (DPVs) were obtained using a Bio-Logic VSP Potentiostat using EC-Lab software. As the electrodes, a BAS glassy-carbon working electrode, a BAS Pt counter electrode, and a BAS RE-7 (Ag/AgNO3 0.01 M in acetonitrile) reference electrode were used. Absorption spectra in the spectroelectrochemical experiments were obtained on an ALS SEC2000 using an electrochemical cell of 1 mm path length incorporating the three-electrode system.Synthesis of trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2
In a 30 mL flask equipped with a reflux condenser were placed [Ru(CO)2Cl2]n (30 mg), 6,6′-dimesityl-2,2′-bipyridine (6Mes-bpy; 51 mg, 0.13 mmol) and ethanol (6 mL) under an argon atmosphere, and the solution was refluxed for 18 h. As the reaction proceeded, the starting solution became a white suspension. The precipitate was filtrated and washed with ethanol. The solid was recrystallized from CHCl3–ether to afford pale yellow crystals (45 mg, 56%): 1H NMR (400 MHz, CDCl3) δ 8.26 (d, J = 8.0 Hz, 2H), 8.09 (dd, J = 8.0 and 7.6 Hz, 2H), 7.45 (d, J = 7.6 Hz, 2H), 6.97 (s, 4H), 2.33 (s, 6H), 2.16 (s, 12H); FTIR (KBr) νCO/cm−1 1986, 2051. Anal. calcd (%) for C30H28Cl2N2O2Ru: C, 58.07; H, 4.55; N, 4.51. Found: C, 58.11; H, 4.76; N, 4.65.Photocatalytic CO2 reduction
Solutions (5 mL) of the catalyst (trans(Cl)–Ru(bpy)(CO)2Cl2 or trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2), [Ru(bpy)3](PF6)2 and BNAH in Ar-saturated DMA/water were placed in quartz tubes (23 mL volume, i.d. = 14 mm). The solutions were bubbled through septum caps with CO2 gas for 20 min, and then were irradiated using a 400 W high-pressure mercury lamp at λ > 400 nm (Riko Kagaku, L-39 cutoff filter) in a carousel irradiation apparatus (Riko Kagaku, RH400-10W). The reaction temperature was maintained at 298 ± 3 K by using a water bath. The gaseous products (CO and H2) were analyzed with GC, and formate was also quantified with GC by acidifying formate to formic acid.47Quenching experiments
Emission from the excited state of [Ru(4dmbpy)3](PF6)2 in the Ar-saturated DMA/water solution was recorded on a Hitachi F-4500 spectrometer (λex = 453 nm) in the absence and in the presence of the quencher, BNAH. The Stern–Volmer relationship (eqn (1)) was obtained from the plots of the relative emission intensity (I0/I) versus the concentration of the quencher (Q: BNAH):| I0/I = 1 + KSV[Q] = 1 + kqτ[Q] | (1) |
Light intensity dependence
In square quartz cells (l = 1.0 cm) were placed DMA/water (9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, 3 mL) solutions containing the Ru catalyst, [Ru(bpy)3](PF6)2 and BNAH, and CO2 was bubbled through the septum caps for at least 30 min before measurement. The solutions were irradiated using a 500 W superhigh-pressure mercury lamp (Ushio, USH-500D) through a Toshiba Y-43 glass filter (λ > 400 nm) with and without neutral-density (ND) filters. The absorption spectra of the solutions were measured with a Shimadzu MultiSpec-1500 Spectrometer. The gaseous products (CO and H2) were analysed with GC, and formate was also quantified with GC by acidifying formate to formic acid.47
:1 v/v, 3 mL) solutions containing the Ru catalyst, [Ru(bpy)3](PF6)2 and BNAH, and CO2 was bubbled through the septum caps for at least 30 min before measurement. The solutions were irradiated using a 500 W superhigh-pressure mercury lamp (Ushio, USH-500D) through a Toshiba Y-43 glass filter (λ > 400 nm) with and without neutral-density (ND) filters. The absorption spectra of the solutions were measured with a Shimadzu MultiSpec-1500 Spectrometer. The gaseous products (CO and H2) were analysed with GC, and formate was also quantified with GC by acidifying formate to formic acid.47
Results and discussion
Photochemical CO2 reduction catalysed by trans(Cl)–Ru(bpy)(CO)2Cl2
The photocatalytic CO2 reductions were carried out in DMA/water (9:1 v/v) solutions containing trans(Cl)–Ru(bpy)(CO)2Cl2 (the catalyst), [Ru(bpy)3]2+ (the photosensitizer) and BNAH (the electron donor) under visible light irradiation (λ > 400 nm), where [Ru(bpy)3]2+ was selectively excited and reductively quenched by BNAH to yield the reduced species of the photosensitizer, [Ru(bpy)3]+.47 The 10 vol% water content was selected for the reaction solvent because this water ratio gave the highest amount of the reduction products. The water in the reaction solution plays an important role as the transporter of the protons for the CO2 reduction, but higher contents of water decrease the quenching efficiency of BNAH toward [Ru(bpy)3]2+*.47 The reduction potential of trans(Cl)–Ru(bpy)(CO)2Cl2, which was estimated to be −1.51 V vs. Ag/Ag+ in DMA/water (9:1 v/v) with the use of the differential pulse voltammetry (Fig. S1 in ESI†), indicates that electron transfer can occur thermodynamically from the reduced photosensitizer ([Ru(bpy)3]2+/+: −1.68 V vs. Ag/Ag+)47 to trans(Cl)–Ru(bpy)(CO)2Cl2.
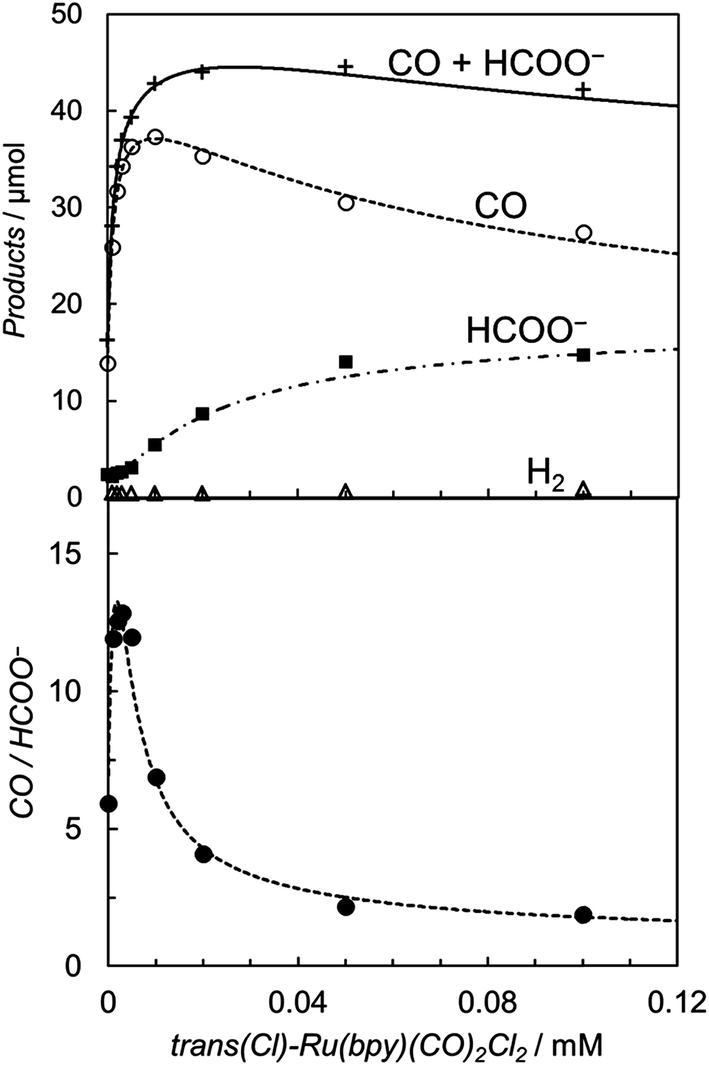
Fig. 2 shows the time-courses of the products in the photochemical CO2 reduction in the CO2-saturated DMA/water (9:1 v/v) solution containing trans(Cl)–Ru(bpy)(CO)2Cl2 ((a) 0.10 mM and (b) 5.0 μM), [Ru(bpy)3]2+ (0.50 mM) and BNAH (0.10 M). The profiles show that CO and formate are selectively yielded, with scarce accompanying H2 evolution even in the aqueous solutions, suggesting that the reduced catalyst reacts much more favourably with CO2 than H+. The turnover number (TON) for the total amount of CO and formate was ca. 300 at 0.10 mM of the catalyst after photo-irradiation for 4 h. When 5.0 μM of trans(Cl)–Ru(bpy)(CO)2Cl2 was used, the TON was dramatically improved to ca. 4000, because there was no superfluous catalyst at the lower concentration. However, the rate of the formation of the products becomes slow over 2 hours. In our previous work, we reported that these effects are mainly attributable to the decrease of BNAH and the increase of BNA2.47 The latter depresses the photochemical CO2 reduction because BNA2 reductively quenches the excited state of [Ru(bpy)3]2+ faster than BNAH, but the back electron transfer is much more efficient.
 | ||
| Fig. 2 Photo-irradiation time dependence of the products in a CO2-saturated DMA/water (9:1 v/v) solution containing (a) 0.1 mM and (b) 5.0 μM of trans(Cl)–Ru(bpy)(CO)2Cl2, [Ru(bpy)3](PF6)2 (0.50 mM) and BNAH (0.10 M): CO (○), HCOO− (■), H2 (Δ) and CO + HCOO− (+). | ||
It is worth noting that the product ratio of CO/HCOO− in Fig. 2 is notably higher at 5.0 μM of trans(Cl)–Ru(bpy)(CO)2Cl2 than that at 0.1 mM. For example, the ratio of CO/HCOO− is approximately 2 at 0.1 mM of the catalyst but ca. 7 at 5.0 μM after 30 min of photo-irradiation. Fig. 3 shows the dependence of the amounts of CO and formate and the product ratio of CO/HCOO− on the catalyst concentration after 30 min of photo-irradiation, which reflects the initial reaction rates of the CO2 reduction. In Fig. 3 (bottom), the initially increased product ratio of CO/HCOO− is lowered by increasing the catalyst concentration. The unexpected profile of the product ratio comes from the differing behaviours of the initial production rates of CO and formate: the rate of CO formation increases as the catalyst concentration increases up to 20–30 μM, then decreases as the concentration increases above 30 μM, while the rate of formate production continues to increase as the catalyst concentration increases. In Fig. 3 (bottom), the increase in the product ratio with increasing catalyst concentration from 0 to 5.0 μM comes from the contribution of the blank products, which are detected even in the absence of trans(Cl)–Ru(bpy)(CO)2Cl2. It is known that [Ru(bpy)3]2+ releases the bipyridyl ligand by photo-labilization to provide catalytically active species, resulting in the blank products.57,70 The amounts of the blank products are 11 and 4 μmol for CO and formate, respectively, and the blank product ratio of CO/HCOO− is ca. 3. Thus, if the blank products caused by [Ru(bpy)3]2+ were excluded, the selectivity of CO would continue to increase with decreasing catalyst concentration.
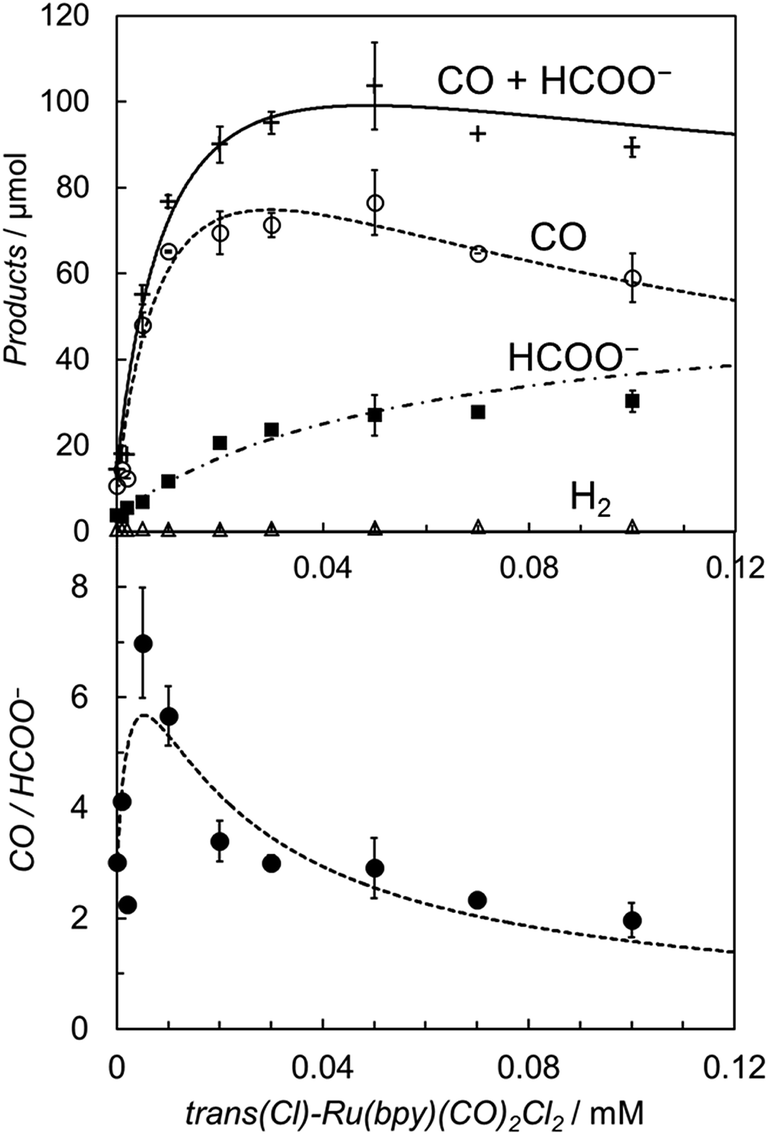 | ||
| Fig. 3 (Top) Plots of the amounts of the reduction products after 30 min of photo-irradiation (400 W Hg lamp, λ > 400 nm) versus the concentration of trans(Cl)–Ru(bpy)(CO)2Cl2 in CO2-saturated DMA/water (9:1 v/v) in the presence of [Ru(bpy)3](PF6)2 (0.50 mM) and BNAH (0.10 M): CO (○), HCOO− (■), H2 (Δ) and CO + HCOO− (+). (Bottom) Plots of the CO/HCOO− ratio versus the concentration of trans(Cl)–Ru(bpy)(CO)2Cl2. The curves represent the theoretical fittings based on the kinetic analyses (see eqn (3) and (5)). | ||
Changes in the electronic absorption spectra during the photo-irradiation and light intensity dependence of the product selectivity
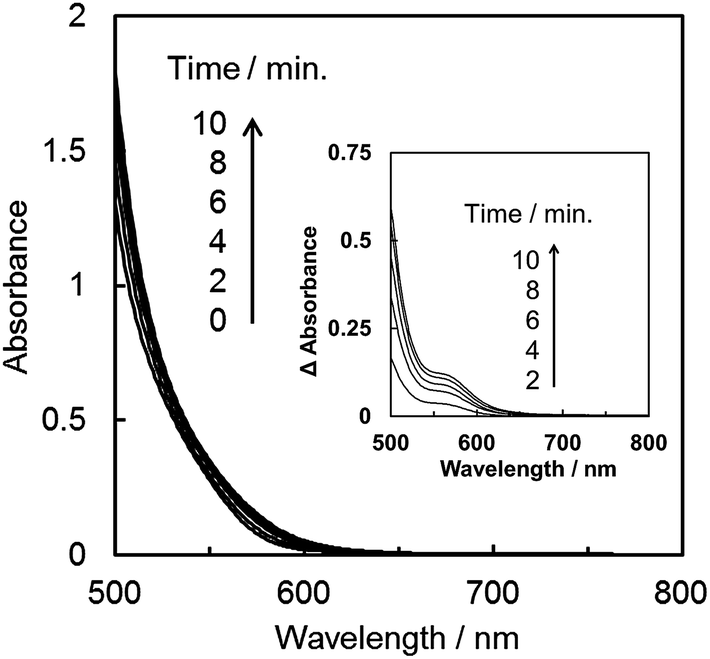
As shown in Fig. 3, the product selectivity of CO/HCOO− was affected by the catalyst concentration. The behaviour led us to consider that an association of the catalyst occurs during the CO2 reduction. The photo-irradiation of an Ar-saturated DMA/water solution containing trans(Cl)–Ru(bpy)(CO)2Cl2 (0.20 mM), [Ru(bpy)3]2+ and BNAH produced H2 instead of CO and formate. During the photo-irradiation, the solution colour changed from orange to dark red. The spectra of the Ar-saturated reaction solution showed the appearance of a characteristic broad peak at 700–800 nm (Fig. 4a), indicating that the polymeric ruthenium complex, [Ru(bpy)(CO)2]n, formed by the reduction of trans(Cl)–Ru(bpy)(CO)2Cl2.49,79,80 On the other hand, photo-irradiation (400 W high-pressure mercury lamp without the ND filter, λ > 400 nm) under a CO2 atmosphere showed no colour change of the solution, suggesting that the reduced catalyst was oxidized by coordination with CO2 to suppress the formation of the polymeric complex. It has been reported that trans(Cl)–Ru(bpy)(CO)2Cl2 is electrochemically reduced to form the polymeric ruthenium complex via the Ru(I)–Ru(I) dimer (Scheme 2).71,72 As the absorption band of the dimer overlaps with that of [Ru(bpy)3]2+ (Fig. 4b), the formation of the dimer could not be observed using the absorption spectra. Thus, if the product selectivity of CO/HCOO− is related to an association of the catalysts, and changes to the absorption spectrum of the reaction solution are not observed during the photo-irradiation, the associated species might be the ruthenium dimeric complex. | ||
| Fig. 4 (a) Absorption spectra of an Ar-saturated DMA/water (9:1 v/v) solution containing trans(Cl)–Ru(bpy)(CO)2Cl2 (0.20 mM), [Ru(bpy)3](PF6)2 (0.50 mM) and BNAH (0.10 M) during photo-irradiation with λ > 400 nm light with an intensity of 7.5 × 10−7 einstein s−1. (b) Absorption spectrum of [Ru(bpy)(CO)2Cl]2 (0.40 mM) in DMA/water (9:1 v/v). (c) Absorption spectra of CO2-saturated DMA/water (9:1 v/v) solutions containing trans(Cl)–Ru(bpy)(CO)2Cl2 (0.10 mM), [Ru(bpy)3](PF6)2 (0.50 mM) and BNAH (0.10 M) by photo-irradiation with λ > 400 nm light of 7.5 × 10−7 einstein s−1 and (d) 3.8 × 10−8 einstein s−1 (total incident light: 2.3 × 10−4 einstein). | ||
Fig. 4c and d show the spectra during photochemical CO2 reduction under high- and low-intensity light (500 W superhigh-pressure mercury lamp without and with the ND filters, λ > 400 nm). The irradiation time was adjusted for the total incident light to be 2.3 × 10−4 einstein. While no spectral change is observed in Fig. 4c, a polymeric absorption at 700–800 nm appears in Fig. 4d. This indicates that the Ru–Ru bond tends to form under the lower-intensity light.
We have further investigated the light intensity dependence of the product selectivity in the photochemical CO2 reduction. Fig. 5 shows the light intensity dependence of the product ratio at a dilute concentration of trans(Cl)–Ru(bpy)(CO)2Cl2 (20 μM). The ratio of CO/HCOO− decreases with reduction of the light intensity, that is, the formation of HCOO− becomes dominant at lower light intensity. Since the concentration of 20 μM was selected to prevent the formation of the polymer that caused a dramatic decrease of the effective catalyst concentration in the solution, we could exclude the possibility that the polymeric species contributed to the product selectivity. Considering that the low-intensity light induces the Ru–Ru bond formation, the most plausible key intermediate for forming HCOO− would be the ruthenium dimer.
 | ||
| Fig. 5 Light intensity dependence of the product ratio of CO/HCOO− in DMA/water (9:1 v/v) solutions containing trans(Cl)–Ru(bpy)(CO)2Cl2 (20 μM), [Ru(bpy)3](PF6)2 (0.50 mM) and BNAH (0.10 M) during photo-irradiation with λ > 400 nm light. The photo-irradiation times were (I) 50 min, (II) 20 min, (III) 10 min and (IV) 5 min (total incident light: 2.3 × 10−4 einstein). | ||
Mechanistic insight into product selectivity
The photochemical CO2 reduction system consists of two parts: an electron relay cycle and a catalytic cycle (Scheme 3). In the electron relay cycle, the excited state of the photosensitizer (PS*) and BNAH form the encounter complex, where electron transfer from BNAH to PS* occurs to produce the charge-separated state ([PS−⋯BNAH·+]).81,82 Dissociation of the encounter complex gives the free reduced photosensitizer (PS−), which could supply electrons to the catalyst. In Scheme 3, Iex is the rate of incident photons, kq is the quenching rate constant by BNAH, kr+nr is the sum of the radiative and non-radiative rate constants of the PS*, α is the cage escape efficiency after the electron transfer from BNAH to the PS*,82β is the fraction of back-electron transfer in the solvent cage (β = 1 − α), kb is the quenching rate constant of the PS−, ki is the electron transfer rate constant from the PS− to the ith form of the catalyst, and [cati] is the concentration of the ith form of the catalyst. In the initial stage of the reaction, the quenching by BNA2 can be ignored.47 The steady-state concentration of the PS− is evaluated as a function of the concentration of the catalyst (eqn (2)) by applying the steady state approximation to Scheme 3: | (2) |
 | ||
| Scheme 3 The electron relay cycle and the catalytic cycle in the photochemical CO2 reduction. | ||
According to eqn (2), when the light intensity and the concentration of BNAH are constant, the steady-state concentration of the PS− is affected by the catalyst concentration: [PS−] decreases as the catalyst concentration increases.
In the catalytic cycle, we have assumed that the dimeric complex produces formate while the monomeric complex produces CO through reaction with CO2 (Scheme 4). It has been reported that the one-electron-reduced catalyst (Ru+) does not react with CO2 (ref. 48–56, 69, 70 and 83) but instead forms a dimer Ru+–Ru+.71,72 Therefore the resting state in the catalytic cycle would be the Ru+ species. In the low concentration region of the catalyst, it is possible for the catalyst to accept two electrons smoothly from the PS−. On the other hand, in the high concentration region, the amount of the PS− would not be enough for the catalyst to receive two electrons smoothly. In particular, in the photochemical reaction, electron transfer to the catalyst hardly occurs, and accordingly the Ru+ remains unreacted.
 | ||
| Scheme 4 Plausible mechanism for CO2 reduction. | ||
The valence of the ruthenium centre in the dimer is possibly changed by the reaction with CO2 and H+. When the Ru(II) valence state forms, the Ru–Ru bond would dissociate into the monomeric species. In order to confirm the cleavage of the Ru–Ru bond, we examined the photocatalytic CO2 reduction by [Ru(bpy)(CO)2Cl]2, which was separately prepared.73 According to the reduction potential of [Ru(bpy)(CO)2Cl]2, which is estimated to be ca. −1.6 V vs. Ag/Ag+ in DMA/water (9:1 v/v) (Fig. S2 in ESI†), electron transfer can thermodynamically occur from the reduced photosensitizer to the dimer. The reaction catalysed by 0.05 mM of [Ru(bpy)(CO)2Cl]2 (0.10 mM of the Ru unit) showed very similar time–course profiles of CO and HCOO− to those of 0.10 mM of trans(Cl)–Ru(bpy)(CO)2Cl2 (Fig. S3 in ESI†). This result supports formate production being accompanied with the cleavage of the Ru–Ru bond to regenerate the monomeric species. We carried out the photo-irradiation of a concentrated solution of [Ru(bpy)(CO)2Cl]2 (0.40 mM) in Ar-saturated DMA/water without either the photosensitizer or the electron donor. The direct photo-irradiation of the dimer results in the appearance of a broad absorption corresponding to the polymeric species, suggesting that disproportionation of the Ru(I)–Ru(I) dimer occurs (Scheme 4). The disproportionation of the dimer has been proposed in the isomerization from trans(Cl) to cis(Cl)–Ru(bpy)(CO)2Cl2, which is induced by the addition of NaBH4.84 However, in the reaction conditions in the presence of [Ru(bpy)3]2+ (0.50 mM) as the photosensitizer, direct photo-excitation of the dimer scarcely occurs because most of the light is absorbed by the photosensitizer when the concentration of the dimer is low. In addition, we have observed that [Ru(bpy)(CO)2Cl]2 is stable against CO2 in DMA/water in the dark. Thus, it is thought that formate production would start with the electrical reduction of Ru+–Ru+.
The relationship between the concentration of the catalyst and the initial rates for the formation of CO and formate was evaluated by applying the steady state approximation to Scheme 4 and using eqn (2). The equations for CO and formate are expressed as the following eqn (3) and (4), respectively (See ESI†):
 | (3) |
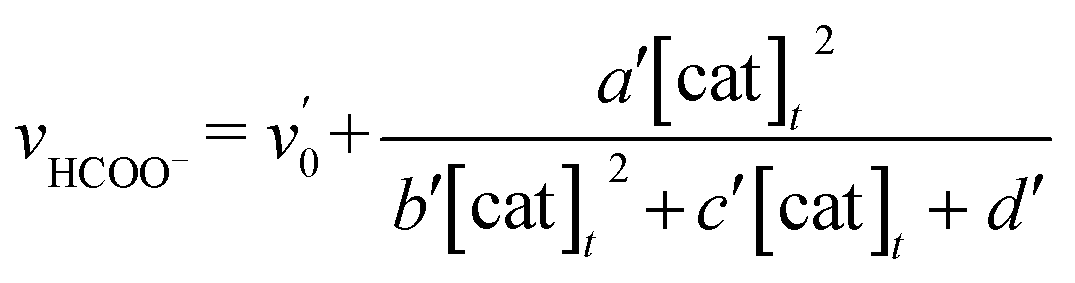 | (4) |
In Fig. 3 top, curve fitting according to eqn (3) gives the parameters: v0 = 1.2 × 10−6 M s−1, a/d = 1.1 s−1, b/d = 1.1 × 109 M−2, c/d = 9.1 × 104 M−1. The simulation curve well reproduces the experimental behaviour, where the rate increases as the catalyst concentration increases up to 20–30 μM then decreases as the concentration increases above 30 μM. Eqn (4) can be simplified when the d′ term is negligible (Fig. S9 in ESI†):
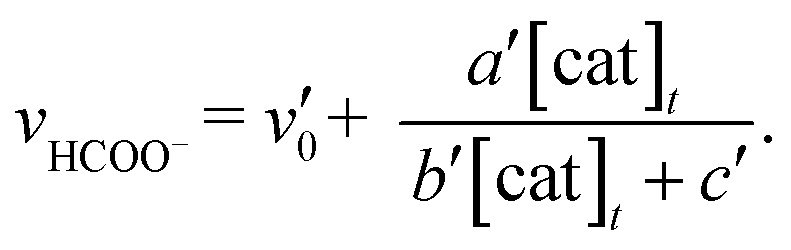 | (5) |
By assuming v0′ = 4.2 × 10−7 M s−1, the double-reciprocal plots of the rate of formate production versus the concentration of trans(Cl)–Ru(bpy)(CO)2Cl2 give a′/c′ = 0.10 s−1 and b′/c′ = 1.8 × 104 M−1 (Fig. 6). These simulation curves based on eqn (3) and (5) agree well with the experimental plots in Fig. 3 (top), and also reproduce the results for the selectivity in Fig. 3 (bottom).
 | ||
| Fig. 6 Double-reciprocal plots of the rate of the formate production versus the concentration of trans(Cl)–Ru(bpy)(CO)2Cl2. | ||
In eqn (3)–(5), the following relationships should be satisfied; b/c = b′/c′ and a/c = a′/b′. From eqn (3) and (5), b/c and b′/c′ are estimated to be 1.3 × 104 M−1 and 1.8 × 104 M−1, and a/c and a′/b′ to be 1.2 × 10−5 M s−1 and 0.6 × 10−5 M s−1, respectively. The results of the curve fittings satisfy the theoretical requirements. Furthermore, a/c and a′/b′ are expressed as eqn (6) and the value can be estimated using the values kq = 2.6 × 108 M−1 s−1, kr+nr = 1.2 × 106 s−1 and αIex = 3.3 × 10−5 M s−1, which are obtained from the Stern–Volmer plot, the emission lifetime of the excited [Ru(bpy)3]2+ and the simulation curve of the decrease of BNAH, respectively.47
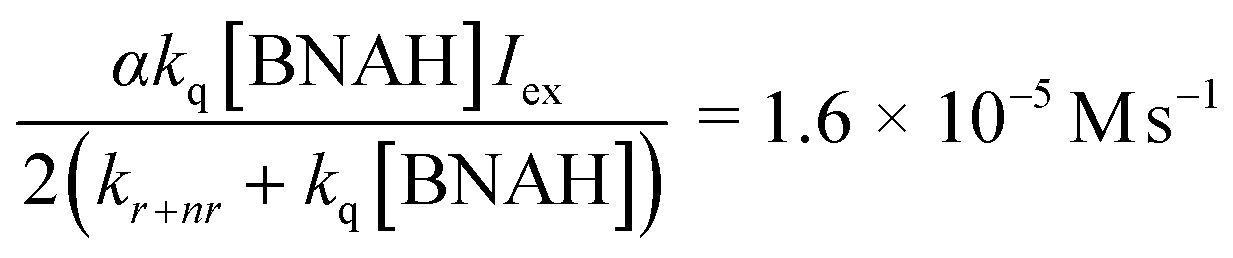 | (6) |
This value is consistent with those of a/c and a′/b′ estimated from eqn (3) and (5), indicating that the kinetic analyses strongly support the proposed mechanism.
Photochemical CO2 reduction using [Ru(4dmbpy)3]2+ as photosensitizer
As the photosensitizer in the photochemical CO2 reduction, we used [Ru(bpy)3]2+, whose first reduction potential is −1.68 V vs. Ag/Ag+ in DMA/water (9:1 v/v).47 In order to investigate the effect of the photosensitizer on the product selectivity, we used [Ru(4dmbpy)3]2+ (4dmbpy = 4,4′-dimethyl-2,2′-bipyridine) instead of [Ru(bpy)3]2+. [Ru(4dmbpy)3]2+ has a more negative reduction potential of −1.77 V vs. Ag/Ag+ (Fig. S4†) than that of [Ru(bpy)3]2+, indicating that [Ru(4dmbpy)3]2+ works as a powerful reductant after the photo-induced electron transfer from BNAH is completed. However, the more negative potential of [Ru(4dmbpy)3]2+ makes the quenching rate constant by BNAH more inefficient (kq ∼1.7 × 107 M−1 s−1)85 than that of [Ru(bpy)3]2+ (kq = 2.6 × 108 M−1 s−1)47 by one order of magnitude or more.
Fig. 7 shows the relationship between the concentration of the catalyst and the initial rates for CO and formate production using [Ru(4dmbpy)3]2+. The simulation curves based on eqn (3) and (4) well reproduce the experimental plots, where formate shows an induction region for its production at very low concentration, and the plots for CO show a downward convex shape at high concentration. From the curve fittings in Fig. 7 and the blank experiment involving [Ru(4dmbpy)3]2+ the parameters are given as: v0 = 1.5 × 10−6 M s−1, a/d = 2.5 s−1, b/d = 1.0 × 1010 M−2, c/d = 7.7 × 105 M−1, v0′ = 2.2 × 10−7 M s−1, a′/d′ = 1.5 × 104 M−1 s−1, b′/d′ = 8.4 × 109 M−2, c′/d′ = 2.1 × 105 M−1. While the values of b/c and b′/c′, estimated to be 1.3 × 104 M−1 and 4.0 × 104 M−1, are similar to those using [Ru(bpy)3]2+ (b/c = 1.3 × 104 M−1 and b′/c′ = 1.8 × 104 M−1 in Fig. 3), the values of a/c and a′/b′, estimated to be 3.3 × 10−6 M s−1 and 1.8 × 10−6 M s−1, are smaller than those using [Ru(bpy)3]2+ (a/c = 1.2 × 10−5 M s−1 and a′/b′ = 0.6 × 10−5 M s−1 in Fig. 3). Assuming that the cage escape efficiency is the same as that for [Ru(bpy)3]2+ (αIex = 3.3 × 10−5 M s−1), the value of eqn (6) is estimated to be 9.4 × 10−6 M s−1 using the quenching rate constant of [Ru(4dmbpy)3]2+. Thus, the smaller values of a/c and a′/b′ estimated in Fig. 7 well reflect the smaller quenching rate constant (kq) of [Ru(4dmbpy)3]2+. In addition, the maximum selectivity attained for the products ratio is ca. 13 at the concentration of 3 μM of the catalyst, indicating that the selectivity of CO is higher than that observed in Fig. 3. The result implies that the reduction potential of Ru+ is more negative than that of Ru2+–CO in Scheme 4, and that the powerful reductant smoothly supplies the second electron to Ru+ in the low catalyst concentration region.
 | ||
| Fig. 7 (Top) Plots of the amounts of the reduction products after 30 min of photo-irradiation (400 W Hg lamp, λ > 400 nm) versus the concentration of trans(Cl)–Ru(bpy)(CO)2Cl2 in CO2-saturated DMA/water (9:1, v/v) in the presence of [Ru(4dmbpy)3](PF6)2 (0.50 mM) and BNAH (0.10 M): CO (○), HCOO− (■), H2 (Δ) and CO + HCOO− (+). (Bottom) Plots of the CO/HCOO− ratio versus the concentration of trans(Cl)–Ru(bpy)(CO)2Cl2. The curves represent the theoretical fittings based on the kinetic analyses (see eqn (3) and (4)). | ||
Selective CO formation in photochemical CO2 reduction using trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2
While we have succeeded in explaining the product selectivity with the kinetic analyses, we have not yet directly detected the dimer during the CO2 reduction reaction by means of spectroscopic methods such as UV-vis absorption and ESI-MS. This is because the absorption band of the dimer is overlapped with that of [Ru(bpy)3]2+ and the absorption coefficient of the dimer at 450 nm is around one-quarter smaller than that of [Ru(bpy)3]2+ (Fig. 4b).86 Even if all of the trans(Cl)–Ru(bpy)(CO)2Cl2 (0.1 mM) transforms into the Ru(I)–Ru(I) dimer during the photochemical reaction, the contribution of the dimer would be only 2.5% of the whole absorption. In addition, polymerization easily occurs even in CO2-saturated DMA/water when a high concentration (>0.2 mM) of trans(Cl)–Ru(bpy)(CO)2Cl2 is used. The attempt to detect the intermediate by ESI-MS has also failed so far due to the lower stability during the ionization process of the measurement. Thus, we changed the strategy to verify the proposed mechanism: we synthesized a novel ruthenium complex, trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2, which has mesityl groups at the 6,6′-positions of the bipyridine ligand to suppress dimer formation, and investigated the product selectivity in the photocatalytic CO2 reduction.The synthesis is described in the experimental section. The reduction potential of trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2 (−1.56 V vs. Ag/Ag+ in DMA/water (9:1 v/v), Fig. S7 in ESI†) is similar to that of trans(Cl)–Ru(bpy)(CO)2Cl2, indicating that the mesityl groups do not strongly affect the electronic structure. This also suggests that the electron transfer reaction from the reduced photosensitizer to trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2 occurs similarly to that of trans(Cl)–Ru(bpy)(CO)2Cl2. Before performing the photocatalytic CO2 reduction, we checked that trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2 does not form a dimer by monitoring the absorption spectra of the solutions containing the ruthenium complex, [Ru(bpy)3]2+ and BNAH during photo-irradiation under an Ar atmosphere. In contrast to trans(Cl)–Ru(bpy)(CO)2Cl2, as shown in Fig. 4a, no absorption band corresponding to the polymeric complex is observed, as shown in Fig. 8, indicating that the bulky substituents at the 6,6′-positions of the bipyridyl ligand suppress the formation of the Ru–Ru bond. The differential absorption spectra show small changes to the spectra with a broad shoulder between 550 and 600 nm (see the inset in Fig. 8). They are comparable with the changes to the spectra in electrolysis (Fig. S8 in the ESI†), because in the spectra measured during the photo-reaction (Fig. 8), the absorptions of the photosensitizer and the electron donor overlap. The changes in the spectra during the photo-reaction would be due to formation of the one-electron reduced but non-dimerised species of the ruthenium complex.
 | ||
| Fig. 8 Absorption spectra of the DMA/water (9:1, v/v) solution containing trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2 (0.20 mM), [Ru(bpy)3]2+ (0.50 mM) and BNAH (0.10 M) during the photo-irradiation with λ > 400 nm light with an intensity of 7.5 × 10−7 einstein s−1 under an Ar atmosphere. The inset shows the differential absorption spectra (optical path length: 10 mm). | ||
The photochemical CO2 reduction was carried out using trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2 as the catalyst in a DMA/water (9:1 v/v) solution containing [Ru(bpy)3]2+ (0.50 mM) as the photosensitizer and BNAH (0.10 M) as the electron donor. The relationships between the concentration of the catalyst and the amounts of CO and formate are shown in Fig. 9. In contrast with Fig. 3, Fig. 9 shows that CO mainly forms, accompanied by a small amount of formate. The formate production is independent of the catalyst concentration, indicating that the formate comes from the blank reaction by [Ru(bpy)3]2+. The plots of the CO production versus the catalyst concentration are well fitted by the kinetic analysis based on the mechanism which does not include dimer formation. When the dimer formation is negligible in Scheme 3 and 4, the production rate for CO is expressed as eqn (7) (ESI†):
 | (7) |
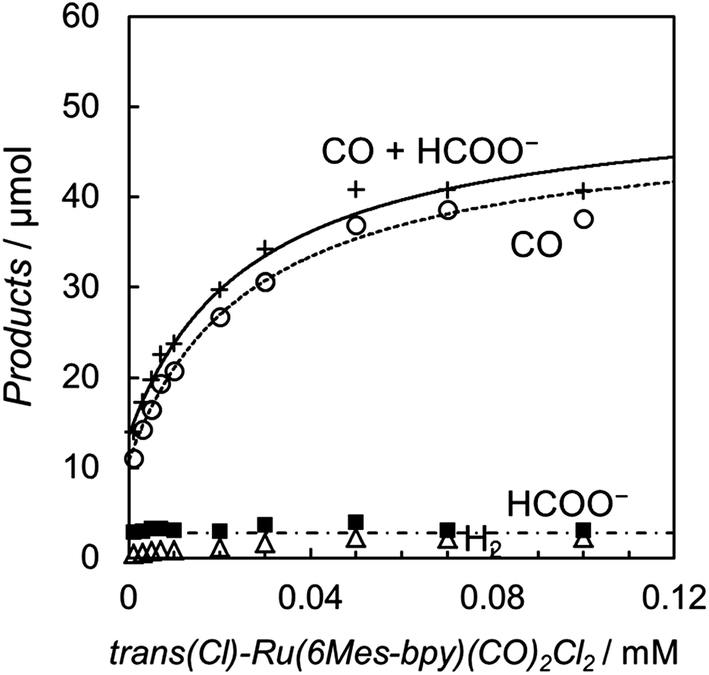 | ||
| Fig. 9 Plots of the amounts of the reduction products after 15 min of photo-irradiation (400 W Hg lamp, λ > 400 nm) versus the concentration of trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2 in CO2-saturated DMA/water (9:1, v/v) in the presence of [Ru(bpy)3](PF6)2 (0.50 mM) and BNAH (0.10 M): CO (○), HCOO− (■), H2 (Δ) and CO + HCOO− (+). The curve for CO represents the theoretical fittings based on the kinetic analysis (see eqn (7)). | ||
Deronzier and Ziessel et al. reported that electrochemical CO2 reduction using ruthenium polymers such as [Ru(bpy)(CO)2]n affords CO selectively.63–68 These reports seem to be inconsistent with our results. It should be noted that the electrochemical reactions are different from the photochemical catalyses. In the electrochemical reaction, electrons can be efficiently supplied from the electrodes to the catalysts, resulting in the valences of the most ruthenium complexes being reduced to 0 or lower, to −1.68 On the contrary, in the photochemical catalyses discussed in this report, reduction of the catalysts occurs via the reaction between the reduced species of the photosensitizer and the catalyst. In the catalyses, there are various possible intermediates and reaction paths. The major reaction pathway is strongly dependent on the reaction (e.g., photochemical or electrochemical reaction) and the conditions (e.g., solvents, pH). The experimental results in this report suggest that the Ru(I) species of the dimer plays an important role in the reaction mechanism of formate production. It still remains unknown how the Ru(I) dimer can selectively produce formate from CO2. Further computational studies of the process are now under way.
Conclusion
We have carried out photochemical CO2 reduction catalyzed by trans(Cl)–Ru(bpy)(CO)2Cl2, and have unexpectedly found that the product ratio of CO to formate depends on the concentration of the catalyst. In order to explain the behavior of the CO/HCOO− selectivity, we have proposed a new reaction mechanism containing the formation of a catalyst dimer which selectively produces formate. The mechanism has strongly been supported by the kinetic analyses, the catalyses by [Ru(bpy)(CO)2Cl]2 and the light intensity dependence of the CO/HCOO− selectivity. The mechanism of the photocatalytic CO2 reduction consists of the electron relay cycle and the catalytic CO2 reduction cycle. The former is the process in which the reduced photosensitizer (PS−) supplies electrons to the catalyst, and the latter is the steps where the ruthenium complexes catalytically reduce CO2 by using the supplied electrons. At high catalyst concentration, the electron relay system would be rate-determining because the catalysis becomes faster than the electron supply. Under this condition, the ruthenium catalyst cannot be supplied with sufficient electrons; the one-electron reduced species of the catalyst is not able to receive more electrons, and it forms the dimer, which produces HCOO−. Therefore as the catalyst concentration increases, the product selectivity (CO/HCOO−) decreases. As the light intensity is reduced, the concentration of PS− also decreases, resulting in the same effect as a high concentration of the catalyst. The mechanism also explains the photo-irradiation time dependence of the CO2 reduction: the CO production reaches saturation. At longer reaction times, the electron donor BNAH is consumed to decrease the concentration of PS−. In this situation, the one-electron reduced species of the catalyst is not able to receive more electrons and forms the catalyst dimer, making the CO production decrease. However, the HCOO− formation continues until the electron donor is exhausted. We have further designed and synthesized a novel ruthenium complex, trans(Cl)–Ru(6Mes-bpy)(CO)2Cl2, which has a bulky ligand to eliminate the contribution of the dimer. By suppressing the dimer formation, the photochemical CO2 reduction produces CO selectively. Among photocatalytic systems for the CO2 reduction by ruthenium complexes, to the best of our knowledge, this system is the first case producing CO selectively. This finding not only elucidates the reaction mechanisms for the photocatalytic CO2 reduction but also leads us to design more effective metal complexes for the catalyses.Acknowledgements
This work was supported by the PRESTO Program of JST. We thank Mr Masaya Kamiya and Mr Kousuke Matsuura for experimental supports.References and Notes
- G. A. Olah, G. K. S. Prakash and A. Goeppert, J. Am. Chem. Soc., 2011, 133, 12881–12898 CrossRef CAS PubMed.
- T. R. Cook, D. K. Dogutan, S. Y. Reece, Y. Surendranath, T. S. Teets and D. G. Nocera, Chem. Rev., 2010, 110, 6474–6502 CrossRef CAS PubMed.
- J. H. Alstrum-Acevedo, M. K. Brennaman and T. J. Meyer, Inorg. Chem., 2005, 44, 6802–6827 CrossRef CAS PubMed.
- H. Arakawa, M. Aresta, J. N. Armor, M. A. Barteau, E. J. Beckman, A. T. Bell, J. E. Bercaw, C. Creutz, E. Dinjus, D. A. Dixon, K. Domen, D. L. DuBois, J. Eckert, E. Fujita, D. H. Gibson, W. A. Goddard, D. W. Goodman, J. Keller, G. J. Kubas, H. H. Kung, J. E. Lyons, L. E. Manzer, T. J. Marks, K. Morokuma, K. M. Nicholas, R. Periana, L. Que, J. Rostrup-Nielson, W. M. H. Sachtler, L. D. Schmidt, A. Sen, G. A. Somorjai, P. C. Stair, B. R. Stults and W. Tumas, Chem. Rev., 2001, 101, 953–996 CrossRef CAS PubMed.
- K. Kobayashi and K. Tanaka, Phys. Chem. Chem. Phys., 2014, 16, 2240–2250 RSC.
- D. L. DuBois, Inorg. Chem., 2014, 53, 3955–3960 CrossRef PubMed.
- A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. DuBois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- C. D. Windle and R. N. Perutz, Coord. Chem. Rev., 2012, 256, 2562–2570 CrossRef CAS.
- T. Yui, Y. Tamaki, K. Sekizawa and O. Ishitani, Top. Curr. Chem., 2011, 303, 151–184 CrossRef CAS PubMed.
- H. Takeda and O. Ishitani, Coord. Chem. Rev., 2010, 254, 346–354 CrossRef CAS.
- K. Tanaka, Chem. Rec., 2009, 9, 169–186 CrossRef CAS PubMed.
- A. J. Morris, G. J. Meyer and E. Fujita, Acc. Chem. Res., 2009, 42, 1983–1994 CrossRef CAS PubMed.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- J.-M. Savéant, Chem. Rev., 2008, 108, 2348–2378 CrossRef PubMed.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- M. D. Sampson, A. D. Nguyen, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, J. Am. Chem. Soc., 2014, 136, 5460–5471 CrossRef CAS PubMed.
- H. Takeda, H. Koizumi, K. Okamoto and O. Ishitani, Chem. Commun., 2014, 50, 1491–1493 RSC.
- M. Bourrez, F. Molton, S. Chardon-Noblat and A. Deronzier, Angew. Chem., Int. Ed., 2011, 50, 9903–9906 CrossRef CAS PubMed.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- J. Grodkowski, D. Behar, P. Neta and P. Hambright, J. Phys. Chem. A, 1997, 101, 248–254 CrossRef CAS.
- M. Hammouche, D. Lexa, M. Momenteau and J.-M. Savéant, J. Am. Chem. Soc., 1991, 113, 8455–8466 CrossRef CAS.
- R. Ziessel, J. Hawecker and J.-M. Lehn, Helv. Chim. Acta, 1986, 69, 1065–1084 CrossRef CAS.
- S. Matsuoka, K. Yamamoto, T. Ogata, M. Kusaba, N. Nakashima, E. Fujita and S. Yanagida, J. Am. Chem. Soc., 1993, 115, 601–609 CrossRef CAS.
- A. H. P. Tinnemans, T. P. M. Koster, D. H. M. W. Thewissen and A. Mackor, Recl. Trav. Chim. Pays-Bas, 1984, 103, 288–295 CrossRef CAS.
- M. Rudolph, S. Dautz and E.-G. Jäger, J. Am. Chem. Soc., 2000, 122, 10821–10830 CrossRef CAS.
- M. A. Mndez, P. Voyame and H. H. Girault, Angew. Chem., Int. Ed., 2011, 50, 7391–7394 CrossRef PubMed.
- J. L. Grant, K. Goswami, L. O. Spreer, J. W. Otvos and M. Calvin, J. Chem. Soc., Dalton Trans., 1996, 2581–2583 Search PubMed.
- M. L. Clark, K. A. Grice, C. E. Moore, A. L. Rheingold and C. P. Kubiak, Chem. Sci., 2014, 5, 1894–1900 RSC.
- S. C. Rasmussen, M. M. Richter, E. Yi, H. Place and K. J. Brewer, Inorg. Chem., 1990, 29, 3926–3932 CrossRef CAS.
- C. M. Bolinger, N. Story, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1988, 27, 4582–4587 CrossRef CAS.
- P. R. Bernatis, A. Miedaner, R. C. Haltiwanger and D. L. DuBois, Organometallics, 1994, 13, 4835–4843 CrossRef CAS.
- D. L. DuBois, A. Miedaner and R. C. Haltiwanger, J. Am. Chem. Soc., 1991, 113, 8153–8164 CrossRef.
- D. L. DuBois and A. Miedaner, J. Am. Chem. Soc., 1987, 109, 113–117 CrossRef CAS.
- Y. Kou, Y. Nabetani, D. Masui, T. Shimada, S. Takagi, H. Tachibana and H. Inoue, J. Am. Chem. Soc., 2014, 136, 6021–6030 CrossRef CAS PubMed.
- T. Morimoto, T. Nakajima, S. Sawa, R. Nakanishi, D. Imori and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 16825–16828 CrossRef CAS PubMed.
- T. Morimoto, C. Nishiura, M. Tanaka, J. Rohacova, Y. Nakagawa, Y. Funada, K. Koike, Y. Yamamoto, S. Shishido, T. Kojima, T. Saeki, T. Ozeki and O. Ishitani, J. Am. Chem. Soc., 2013, 135, 13266–13269 CrossRef CAS PubMed.
- Y. Tamaki, K. Koike, T. Morimoto, Y. Yamazaki and O. Ishitani, Inorg. Chem., 2013, 52, 11902–11909 CrossRef CAS PubMed.
- Y. Tamaki, K. Koike, T. Morimoto and O. Ishitani, J. Catal., 2013, 304, 22–28 CrossRef CAS.
- H. Takeda, K. Koike, H. Inoue and O. Ishitani, J. Am. Chem. Soc., 2008, 130, 2023–2031 CrossRef CAS PubMed.
- B. Gholamkhass, H. Mametsuka, K. Koike, T. Tanabe, M. Furue and O. Ishitani, Inorg. Chem., 2005, 44, 2326–2336 CrossRef CAS PubMed.
- Y. Hayashi, S. Kita, B. S. Brunschwig and E. Fujita, J. Am. Chem. Soc., 2003, 125, 11976–11987 CrossRef CAS PubMed.
- J. Hawecker, J.-M. Lehn and R. Ziessel, Helv. Chim. Acta, 1986, 69, 1990–2012 CrossRef CAS.
- J. Chauvin, F. Lafolet, S. Chardon-Noblat, A. Deronzier, M. Jakonen and M. Haukka, Chem.–Eur. J., 2011, 17, 4313–4322 CrossRef CAS PubMed.
- M. R. M. Bruce, E. Megehee, B. P. Sullivan, H. H. Thorp, T. R. O'TooIe, A. Downard, J. R. Pug and T. J. Meyer, Inorg. Chem., 1992, 31, 4864–4873 CrossRef CAS.
- S. Sato, T. Morikawa, T. Kajino and O. Ishitani, Angew. Chem., Int. Ed., 2013, 52, 988–992 CrossRef CAS PubMed.
- P. Kang, C. Cheng, Z. Chen, C. K. Schauer, T. J. Meyer and M. Brookhart, J. Am. Chem. Soc., 2012, 134, 5500–5503 CrossRef CAS PubMed.
- Y. Kuramochi, M. Kamiya and H. Ishida, Inorg. Chem., 2014, 53, 3326–3332 CrossRef CAS PubMed.
- P. Voyame, K. E. Toghill, M. A. Méndez and H. H. Girault, Inorg. Chem., 2013, 52, 10949–10957 CrossRef CAS PubMed.
- Y. Tamaki, T. Morimoto, K. Koike and O. Ishitani, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15673–15678 CrossRef CAS PubMed.
- J. R. Pugh, M. R. M. Bruce, B. P. Sullivan and T. J. Meyer, Inorg. Chem., 1991, 30, 86–91 CrossRef CAS.
- H. Ishida, T. Terada, K. Tanaka and T. Tanaka, Inorg. Chem., 1990, 29, 905–911 CrossRef CAS.
- H. Ishida, K. Tanaka and T. Tanaka, Chem. Lett., 1988, 339–342 CrossRef CAS.
- H. Ishida, K. Tanaka and T. Tanaka, Chem. Lett., 1987, 1035–1038 CrossRef CAS.
- H. Ishida, H. Tanaka, K. Tanaka and T. Tanaka, J. Chem. Soc., Chem. Commun., 1987, 131–132 RSC.
- H. Ishida, K. Tanaka and T. Tanaka, Organometallics, 1987, 6, 181–186 CrossRef CAS.
- H. Ishida, K. Tanaka and T. Tanaka, Chem. Lett., 1985, 405–406 CrossRef CAS.
- J. Hawecker, J.-M. Lehn and R. Ziessel, J. Chem. Soc., Chem. Commun., 1985, 56–58 RSC.
- K. Maeda, K. Sekizawa and O. Ishitani, Chem. Commun., 2013, 49, 10127–10129 RSC.
- T. M. Suzuki, H. Tanaka, T. Morikawa, M. Iwaki, S. Sato, S. Saeki, M. Inoue, T. Kajino and T. Motohiro, Chem. Commun., 2011, 47, 8673–8675 RSC.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS PubMed.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944–6946 RSC.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CrossRef CAS PubMed.
- (a) S. Chardon-Noblat, C. H. Cripps, A. Deronzier, J. S. Field, S. Gouws, R. J. Haines and F. Southway, Organometallics, 2001, 20, 1668–1675 CrossRef CAS; (b) S. Chardon-Noblat, C. P. Da, A. Deronzier, M. Haukka, T. A. Pakkanen and R. Ziessel, J. Electroanal. Chem., 2000, 490, 62–69 CrossRef CAS.
- S. Chardon-Noblat, A. Deronzier, R. Ziessel and D. Zsoldos, J. Electroanal. Chem., 1998, 444, 253–260 CrossRef CAS.
- S. Chardon-Noblat, A. Deronzier, R. Ziessel and D. Zsoldos, Inorg. Chem., 1997, 36, 5384–5389 CrossRef CAS.
- S. Chardon-Noblat, M. N. Collomb-Dunand-Sauthier, A. Deronzier, R. Ziessel and D. Zsoldos, Inorg. Chem., 1994, 33, 4410–4412 CrossRef CAS.
- M. N. Collomb-Dunand-Sauthier, A. Deronzier and R. Ziessel, Inorg. Chem., 1994, 33, 2961–2967 CrossRef CAS.
- M. N. Collomb-Dunand-Sauthier, A. Deronzier and R. Ziessel, J. Chem. Soc., Chem. Commun., 1994, 189–191 RSC.
- H. Ishida, K. Fujiki, T. Ohba, K. Ohkubo, K. Tanaka, T. Terada and T. Tanaka, J. Chem. Soc., Dalton Trans., 1990, 2155–2160 RSC.
- J.-M. Lehn and R. Ziessel, J. Organomet. Chem., 1990, 382, 157–173 CrossRef CAS.
- S. Chardon-Noblat, A. Deronzier, D. Zsoldos, R. Ziessel, M. Haukka, T. Pakkanen and T. Venalainen, J. Chem. Soc., Dalton Trans., 1996, 2581–2583 RSC.
- S. Myllynen and M. Wasberg, J. Electroanal. Chem., 2008, 623, 93–101 CrossRef CAS.
- M. Haukka, J. Kiviaho, M. Ahlgren and T. A. Pakkanen, Organometallics, 1995, 14, 825–833 CrossRef CAS.
- A. Paul, D. Connolly, M. Schulz, M. T. Pryce and J. G. Vos, Inorg. Chem., 2012, 51, 1977–1979 CrossRef CAS PubMed.
- P. A. Anderson, G. B. Deacon, K. H. Haarmann, F. R. Keene, T. J. Meyer, D. A. Reitsma, B. W. Skelton, G. F. Strouse, N. C. Thomas, J. A. Treadway and A. H. Whit, Inorg. Chem., 1995, 34, 6145–6157 CrossRef CAS.
- K. Suzuki, A. Kobayashi, S. Kaneko, K. Takehira, T. Yoshihara, H. Ishida, Y. Shiina, S. Oishi and S. Tobita, Phys. Chem. Chem. Phys., 2009, 11, 9850–9860 RSC.
- M. Schmittel, A. Ganz, W. A. Schenk and M. Z. Hagel, Z. Naturforsch., B: J. Chem. Sci., 1999, 54, 559–564 CAS.
- D. Mauzerall and F. H. Westheimer, J. Am. Chem. Soc., 1955, 77, 2261–2264 CrossRef CAS.
- M. N. Collomb-Dunand-Sauthier, A. Deronzier and R. Ziessel, J. Electroanal. Chem., 1991, 319, 347–353 CrossRef CAS.
- M. N. Collomb-Dunand-Sauthier, A. Deronzier and R. Ziessel, J. Electroanal. Chem., 1993, 350, 43–55 CrossRef CAS.
- G. J. Kavarnos, Fundamentals of Photoinduced Electron Transfer, Wiley-VCH, Weinheim, 1993 Search PubMed.
- M. Z. Hoffman, J. Phys. Chem., 1988, 92, 3458–3464 CrossRef CAS.
- E. Fujita, M. Chou and K. Tanaka, Appl. Organomet. Chem., 2000, 14, 844–846 CrossRef CAS.
- Y. Kuramochi, Y. Ito and H. Ishida, Eur. J. Inorg. Chem., 2012, 2012, 1167–1170 CrossRef CAS.
- The quenching rate constant was estimated by using the Stern–Volmer constant of KSV = 13 M−1 (Fig. S5†) and the emission lifetime of [Ru(4dmbpy)3]2, assumed to be 766 ns. See ref. 49.
- S. Myllynen, M. Wasberg and M. Haukka, J. Electroanal. Chem., 2006, 586, 217–224 CrossRef CAS.
- We have also examined the photochemical CO2 reduction catalysed by trans(Cl)–Ru(4dmbpy)(CO)2Cl2, whose first reduction potential is −1.58 V vs. Ag/Ag+ in DMA/water (9:1 v/v). The dependence of the product selectivity on the catalyst concentration is similar to that of trans(Cl)–Ru(bpy)(CO)2Cl2, eliminating the possibility that the simple electric effect of the substituents changes the product selectivity.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5sc00199d |
| This journal is © The Royal Society of Chemistry 2015 |