
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSuccessive light-induced two electron transfers in a Ru–Fe supramolecular assembly: from Ru–Fe(II)–OH2 to Ru–Fe(IV)–oxo†
Christian
Herrero
a,
Annamaria
Quaranta
b,
Marie
Sircoglou
a,
Katell
Sénéchal-David
a,
Aurélie
Baron
b,
Irene Mustieles
Marín
a,
Charlotte
Buron
a,
Jean-Pierre
Baltaze
a,
Winfried
Leibl
*b,
Ally
Aukauloo
*ab and
Frédéric
Banse
*a
aInstitut de Chimie Moléculaire et des Matériaux d'Orsay, Université Paris Sud, CNRS, F-91405 Orsay, CEDEX, France. E-mail: ally.aukauloo@u-psud.fr; frederic.banse@u-psud.fr
bSB2SM, iBiTec-S, CEA, CNRS, UMR 8221, F-91191, Gif-sur-Yvette, France. E-mail: winfried.leibl@cea.fr
First published on 26th January 2015
Abstract
In the present work we describe the synthesis and study of a RuII–FeII chromophore–catalyst assembly designed to perform the light-induced activation of an iron bound water molecule and subsequent photo-driven oxidation of a substrate. Using a series of spectroscopic techniques, we demonstrate that excitation of the chromophore unit with 450 nm light, in the presence of a sacrificial electron acceptor, triggers a cascade of electron transfers leading to the formation of a high valent iron(IV)–oxo center from an iron(II)-bound water molecule. The activity of this catalytic center is illustrated by the oxidation of triphenyl phosphine.
Introduction
Despite the current efforts being deployed to reduce the impact of human activities on the planet and reach sustainability, polluting and energy consuming chemical processes remain to be adjusted. This is particularly true for oxidation reactions where stoichiometric amounts of often toxic oxidants are commonly used.1 In contrast, nature has developed several iron-based metalloenzymes that perform the oxidation of organic substrates via high valent iron–oxo species.2–4 To mimic these enzymes, a number of non-heme polypyridyl complexes have been studied extensively and employed in different oxidation reactions including C–H bond cleavage, epoxidations, and sulfoxidations, using chemical oxidants such as PhIO or peracids.5,6 In these cases, the role of the oxidant used is twofold; it activates the FeII precursor by the removal of two electrons to yield the active FeIV(O) intermediate, and acts as the source of the oxygen atom that will ultimately be inserted into the reaction products. This “shunt” mechanism, which has been favored in this research field over the naturally occurring reductive activation of O2 at iron centers, has proven to be an easier route to high valent iron–oxo centers but hinges on the use of the above mentioned oxygen containing oxidants.An alternative methodology to performing such reactions involves the use of both visible light as a source of energy, and water as a clean supply of electrons and O atoms in order to form energy rich reaction products. This approach, akin to the mechanism found in Photosystem II,7,8 utilizes visible light in order to activate a chromophore that in turn triggers the successive oxidation of a catalytic center which activates a bound water molecule in the form of a metal–oxo species through a series of proton coupled electron transfer reactions (PCET) (Fig. 1, eqn (1)–(6)).9,10 This strategy has inspired the development of a new area of research whose goal is to develop synthetic models capable of transforming light energy into chemical energy stored in chemical bonds.11–18
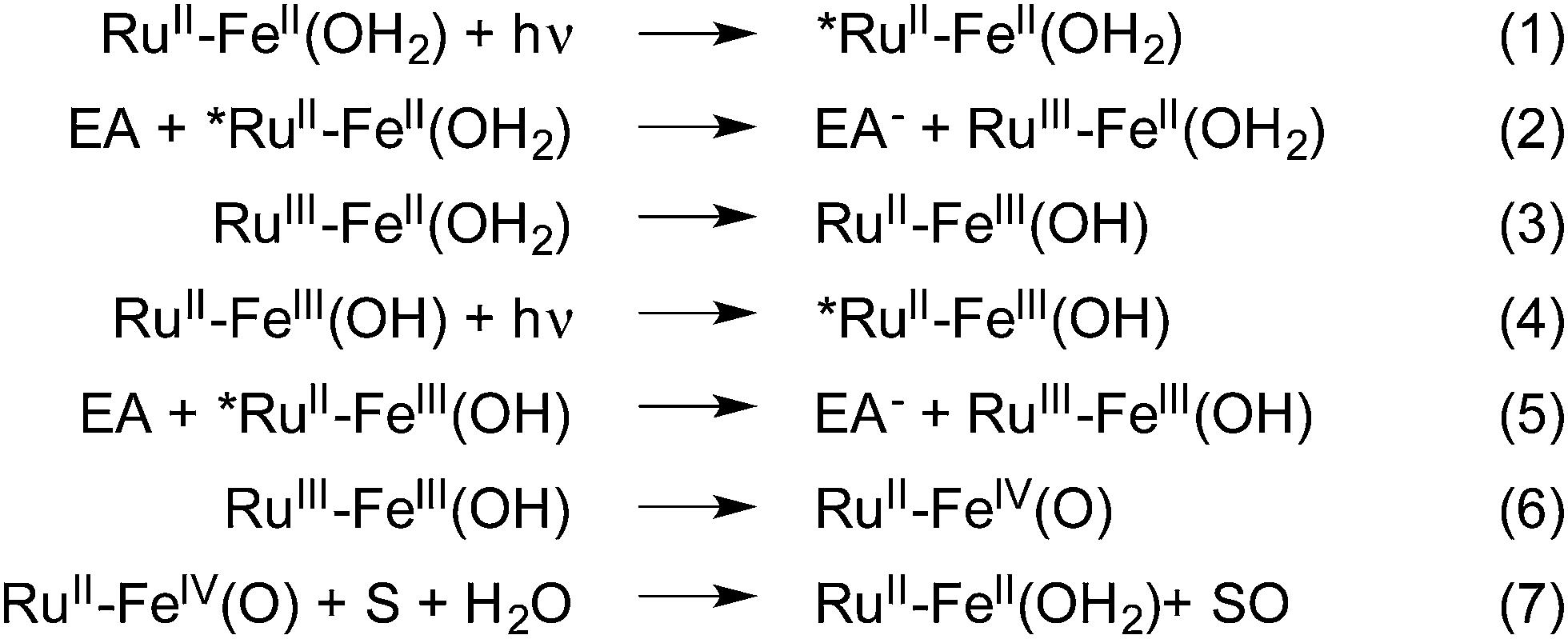 | ||
| Fig. 1 Overall mechanism to achieve oxygenation of a substrate by a RuII–FeII chromophore–catalyst complex upon light-induced activation of a water molecule. EA and S denote electron acceptor and substrate molecules, respectively. Protons released during steps 3 and 6 are omitted for clarity. | ||
In our laboratories we have focused our efforts on the study of covalently bound chromophore–catalyst complexes designed to perform the visible light activation of catalytic units.19,20 Photo-activation of such assemblies triggers a series of energy and electron transfer reactions which result in the formation of a charge-separated state where reductive equivalents are transferred onto a sacrificial electron acceptor while oxidative power is stored at the catalytic center.
In contrast to bimolecular chromophore/catalyst systems,14,21 these assemblies are versatile tools that not only allow the study of the complete cascade of reactions that follow activation by visible light but also offer the possibility to investigate the electronic communication between the chromophore and the catalyst. Furthermore, these types of assemblies are suitable candidates for incorporation in dye sensitized photo-electrochemical cells, where, as part of the photoanode22,23 they can furnish electrons and protons to a hydrogen- or fuel-forming cathode.
In this work, we report a full artificial photosynthetic system where light absorption, management of the chromophore's excited state, sequential electron transfers, accumulation of charges at the catalyst and catalysis are achieved and characterized. Using the chromophore–catalyst complex 1-OH2 depicted in Fig. 2, (for the detailed synthetic procedure see ESI†), visible light as the sole source of energy and water as the O atom source, we present the successive two-electron photo-oxidation of an iron-based catalyst. This process leads to the activation of a coordinated water molecule resulting in the formation of an active FeIV(O) species that performs subsequent oxygen atom transfer onto a substrate (Fig. 1). The two successive light-induced oxidations of a metal-bound water molecule have rarely been confirmed spectroscopically in the past.24
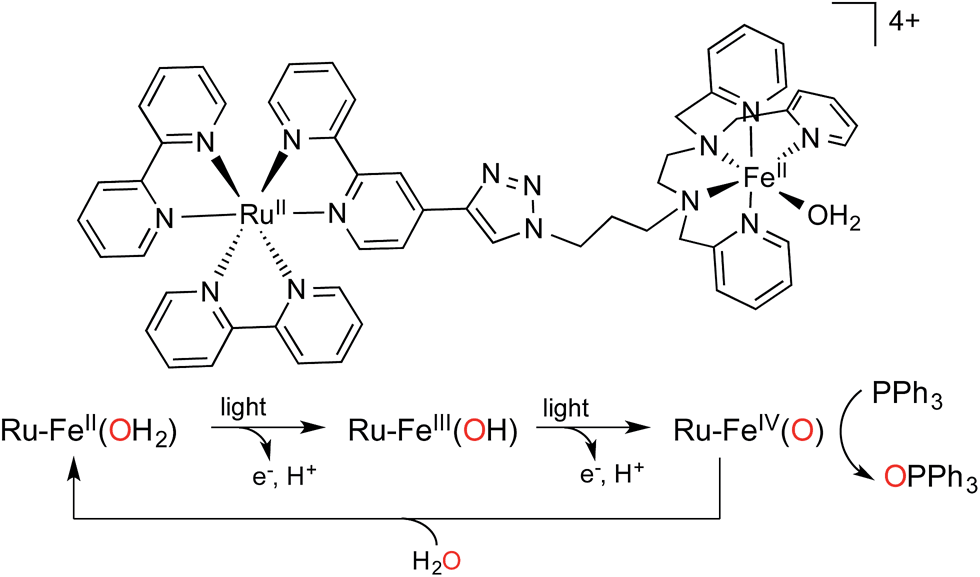 | ||
| Fig. 2 Structure of chromophore–catalyst complex 1-OH2 used in this study, and a simplified mechanism for the stepwise activation of the chromophore–catalyst complex prior to O atom transfer. | ||
Results and discussion
The chromophore employed is a [RuII(bpy)3]-like complex whose excited state can be oxidatively quenched by a number of electron acceptors, yielding a RuIII state which has 1.30 V vs. SCE oxidative power (Fig. 1, eqn (2) and (5)).25 Depending on the experiments performed, the electron acceptors used were either methyl viologen or [CoIII(NH3)5Cl]2+ salts which, unlike 4-nitrobenzene diazonium and sodium peroxosulfate that were also tried, were the only acceptors able to oxidatively quench the excited state of the ruthenium chromophore without performing oxidation of the FeII catalyst unit in the absence of light.26 The catalyst is an FeII ion wrapped by a pentadentate amine/pyridine ligand (noted as L52 hereafter), known to perform the catalytic oxidation of organic substrates in the presence of oxygen containing oxidants and for which the spectroscopic signature of the FeIV(O) intermediate has been reported.27–29The RuII–FeII complex was first isolated with a chloro exogenous ligand bound to FeII (1-Cl, see ESI†). When dissolved in 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 H2O:CH3CN (WAN), this complex exhibits two strong absorptions at 450 nm (ε = 17000 M−1 cm−1) due to a RuII to ligand charge transfer band (MLCT) and at 280 nm arising from bipyridine π–π* transitions (Fig. SI 6†). Additionally, the absorption spectrum exhibits a shoulder at 400 nm (ε = 5000 M−1 cm−1) which coincides with the absorption maximum observed for the [(L52)FeII(OH2)]2+ reference compound 2-OH2 (Fig. SI 5 and SI 6†) arising from a FeII to π* pyridine charge transfer band. In a 4:1 WAN mixture the chloro ligand is thus substituted by water leading to the Ru–FeII(OH2) precursor (1-OH2).30,31
:1 H2O:CH3CN (WAN), this complex exhibits two strong absorptions at 450 nm (ε = 17000 M−1 cm−1) due to a RuII to ligand charge transfer band (MLCT) and at 280 nm arising from bipyridine π–π* transitions (Fig. SI 6†). Additionally, the absorption spectrum exhibits a shoulder at 400 nm (ε = 5000 M−1 cm−1) which coincides with the absorption maximum observed for the [(L52)FeII(OH2)]2+ reference compound 2-OH2 (Fig. SI 5 and SI 6†) arising from a FeII to π* pyridine charge transfer band. In a 4:1 WAN mixture the chloro ligand is thus substituted by water leading to the Ru–FeII(OH2) precursor (1-OH2).30,31
This ligand substitution was further confirmed by electrochemical studies. For the synthetically prepared reference compound 2-OH2 the FeIII(OH2)/FeII(OH2) redox signal is observed at 0.51 V vs. SCE (ΔE = 140 mV) in acetone. This value matches the one obtained for 2-Cl when studied in a 4:1 WAN solvent mixture (E1/2 = 0.46 V) (Fig. SI 7†). Complexes 1-Cl, 1-CH3CN, and 1-OH2 were also prepared and studied by cyclic voltammetry (CV). In each case two different reversible processes are observed at positive potentials. A first, ligand-independent process corresponding to the RuIII/RuII couple is observed at E1/2 = 1.26 V vs. SCE (ΔE = 68 mV). A second wave, the potential of which varies as a function of the exogenous ligand bound to Fe, is attributed to the FeIII/II couple and is observed at either 0.97 V, 0.60 V or 0.50 V vs. SCE for 1-CH3CN, 1-Cl or 1-OH2, respectively (Fig. SI 8†). In any case, our electrochemical data for complexes 1 indicate that (i) the FeIII-X/FeII-X potential is almost identical to that of model complex 2-X (X = CH3CN, Cl or H2O) and; (ii) a photogenerated RuIII center should have enough driving force to oxidize the covalently linked [(L52)FeII] unit regardless of the nature of the exogenous ligand (Table SI 1†).
The excited state properties of the photosensitizer (*RuII) in 1 were studied using steady state and time-resolved fluorescence spectroscopy techniques. The emission of 1-OH2 in 4:1 WAN is strongly quenched compared to the reference compound [Ru(bpy)3]2+ (λmax = 605 nm, Φ = 0.059)25 and shows a maximum at 619 nm with a quantum yield of 0.0091. Emission decay, as is often observed for dinuclear complexes based on [Ru(bpy)3]2+ chromophores,32,33 is biphasic with a main component of ∼80 ns (65%) and a minor one of ∼600 ns (35%). The excited state quenching is tentatively assigned to an energy transfer mechanism between the chromophore's excited state and low lying metal centered states of the catalyst unit since an electron transfer mechanism from the catalyst to the excited state of the chromophore can be ruled out by ligand exchange experiments (Table SI 1† and accompanying text) and a resonance energy transfer process is not consistent with the absorption/emission properties of the donor (Ru) and acceptor (Fe). Furthermore, the spin state of the FeII complex was found not to influence this quenching as the same phenomenon was observed in the case of the low spin (S = 0) cyano derivative [(L52)FeII(CN)]+.
The first photoinduced electron transfer (Fig. 1 and eqn (3)) was studied by laser flash photolysis. The differential absorption traces after excitation of 1-OH2 in the presence of methyl viologen (MV2+) as the reversible electron acceptor in a 4:1 WAN mixture, showed an increase of absorption at 605 nm together with a bleach at 450 nm (Fig. 3, top). These absorption changes are indications of the reduction of methyl viologen (MV2+ → MV˙+) and the concomitant formation of the oxidized state of the chromophore (RuII → RuIII) (Fig. 1, eqn (1) and (2)). Recovery of the chromophore's ground state is approximately 5 times faster than that of MV2+ implying an intramolecular electron transfer between the reduced form of the catalytic module and the oxidized chromophore (RuIII–FeII → RuII–FeIII) (Fig. 1, eqn (3)). In order to investigate the changes in electronic absorption features occurring during the electron transfer process, we replaced MV2+ by [RuIII(NH3)6]Cl3, which does not absorb in the visible region (Fig. 3, bottom). The strong initial bleaching of the RuII MLCT at 450 nm after excitation evolves in ∼60 μs into a depletion band with a maximum at 400 nm (Fig. 3, bottom) that is attributed to the formation of the FeIII state in the catalytic module. This loss of absorption at 400 nm, which originates from the loss of the FeII to π*pyridine MLCT band, was also observed upon chemical oxidation of 1-OH2, and more so with reference compound 2-OH2, when they were treated with 2 equivalents of H2O2 (Fig. SI 9†).
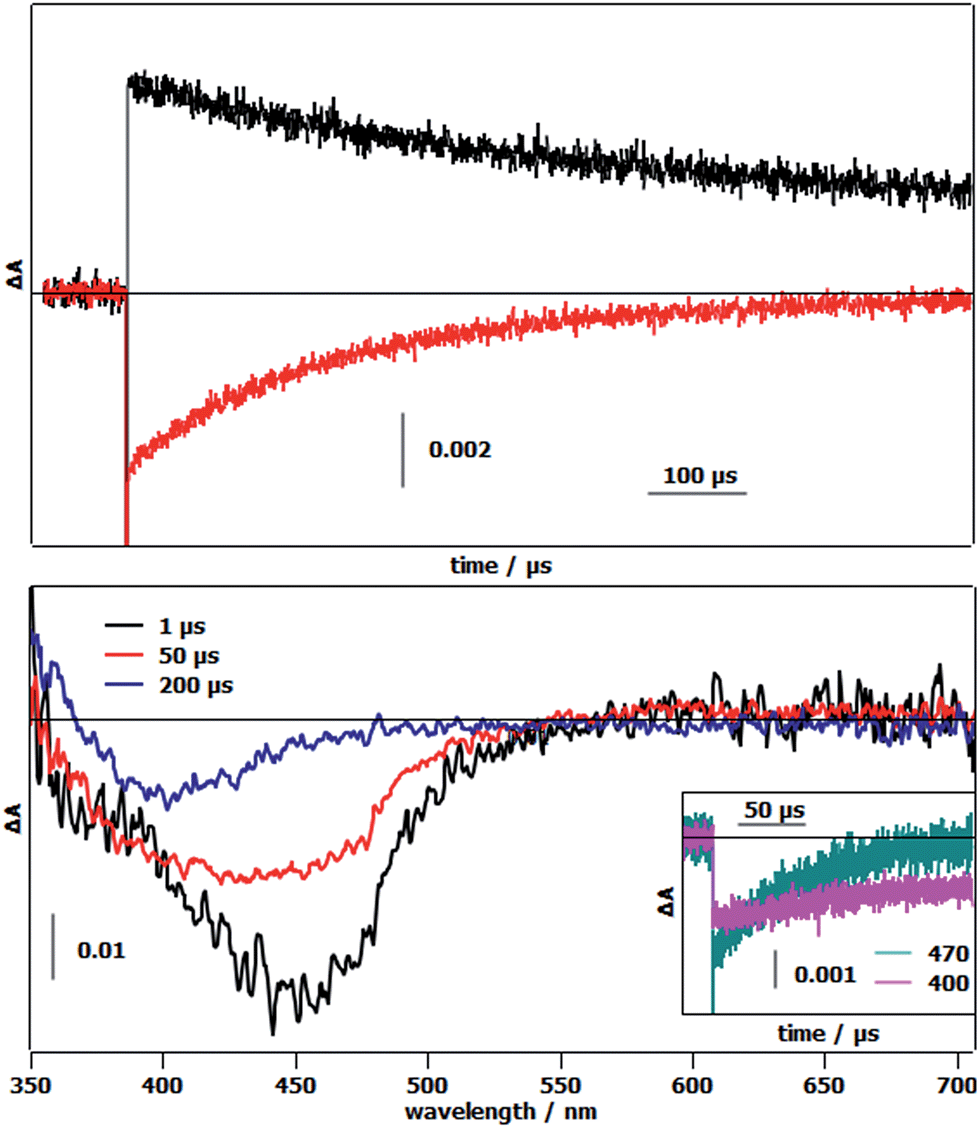 | ||
| Fig. 3 (Top) Transient absorption kinetics of 1-OH2 in the presence of 20 mM methyl viologen (MV2+) after 460 nm laser flash excitation. Black trace: decay of MV˙+ to MV2+ followed at 605 nm. Red trace: recovery of RuII from RuIII monitored at 450 nm. (Bottom) Transient absorption spectra of 1-OH2 in the presence of 20 mM [RuIII(NH3)6]Cl3 at 1, 50, and 200 μs after laser flash excitation. Inset: kinetic traces for the recovery of RuII (470 nm, green trace) and the disappearance of FeII (400 nm, pink trace). Laser energy: 10 mJ. | ||
To further study the change in oxidation state of the initial FeII ion, we performed X-band EPR measurements after continuous light excitation of a solution of 1-OH2 in 4:1 WAN in the presence of [CoIII(NH3)5Cl]2+ acting as an irreversible electron acceptor. The initial spectrum shows no observable signal due to the diamagnetic nature of the CoIII and RuII ions and the high spin FeIIS = 2 state (Fig. 4, left, blue trace). After irradiation of this sample with 450 nm light for 2 min, the EPR spectrum exhibits a resonance at g = 4.44 due to the formation of high spin CoII ions in solution and signals at g = 2.34, 2.15 and 1.91 which are characteristic of low spin FeIII(OH) (S = 1/2) in this family of complexes (Fig. 4, left, red trace).21,34–36 Furthermore, these resonances match those observed when 1-OH2 is chemically oxidized using 2 eq. H2O2 (Fig. 4, right). In parallel, the results of a controlled chemical oxidation performed on reference compound 2-OH2 are consistent with the above observations (Fig. SI 10†). Altogether, these results clearly evidence the first activation step of the Fe-bound water molecule leading to RuII–FeIII(OH).
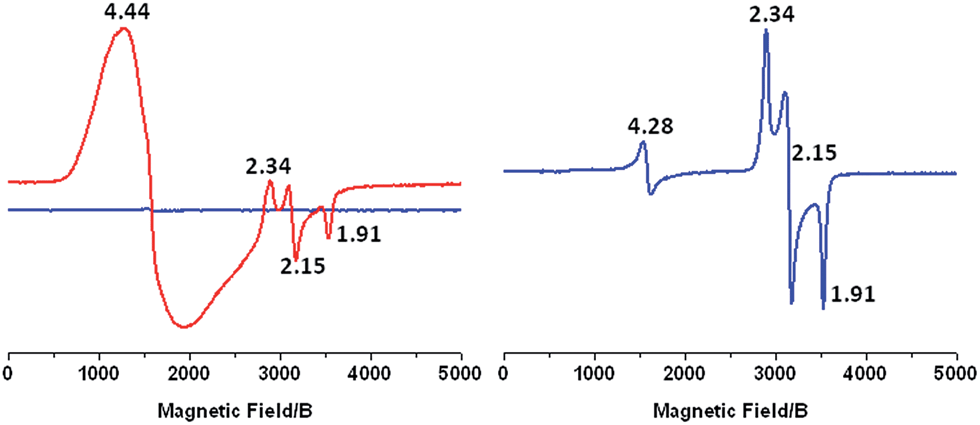 | ||
| Fig. 4 X-band EPR of: (left) 16 mM [CoIII(NH3)5Cl]2+ and 0.80 mM complex 1-OH2 in a 4:1 H2O:CH3CN solvent mixture before (blue) and after (red) 2 min illumination with 450 nm light. (Right) 1.65 mM solution of complex 1-OH2 after addition of 2 eq. H2O2. The weak resonance at g = 4.28 is due to the ubiquitous S = 5/2 FeIII species. | ||
The second light-induced electron transfer (Fig. 1, eqn (6)) needed to generate the targeted FeIV(O) species was evidenced by studying the evolution of the EPR signals at different time intervals during continuous illumination of a solution of 1-OH2 in 4:1 WAN in the presence of [CoIII(NH3)5Cl]2+. An increase in intensity of the EPR signals accounting for the formation of the low spin FeIII(OH) species was observed, followed by a decrease until the complete disappearance of the rhombic low-spin FeIII signal (Fig. SI 11†). Concomitantly, we observed a persistent increase of the signal related to the photo-reduced free CoII ions in solution. These observations are supportive of a second electron transfer process from the FeIII(OH) intermediate to photogenerated RuIII leading to the probable formation of a low spin S = 1 FeIV species. A conclusive kinetic study of this second electron transfer by laser flash photolysis of RuII–FeIII(OH) (prepared with H2O2) in the presence of MV2+ has not yet been possible due to competitive back electron transfer processes between the reduced reversible electron acceptor and the singly oxidized FeIII(OH) form of the catalyst (Fig. SI 12†). Definitive evidence for the photogeneration of an FeIV(O) species comes from the ESI-MS characterization of an irradiated solution of 1-OH2 under similar experimental conditions. The mass spectrum shows a signal at 688.1087 amu corresponding to {[RuII–FeIV(O)](PF6)(OTf)}2+ (Fig. 5, top). Isotopic labelling experiments with H218O were performed to confirm the origin of the oxygen atom in the metal–oxo species. A peak at 689.1026 was detected corresponding to {[RuII–FeIV(18O)](PF6)(OTf)}2+. This result evidences that the oxidized form FeIV(O) stems from the activation of a metal-bound water molecule. Additionally, the formation of the FeIV(O) species was revealed by its characteristic absorbance band at 750 nm (Fig. SI 13†) which compares well with the reported values of 756 nm for [(L52)FeIV(O)]2+.29
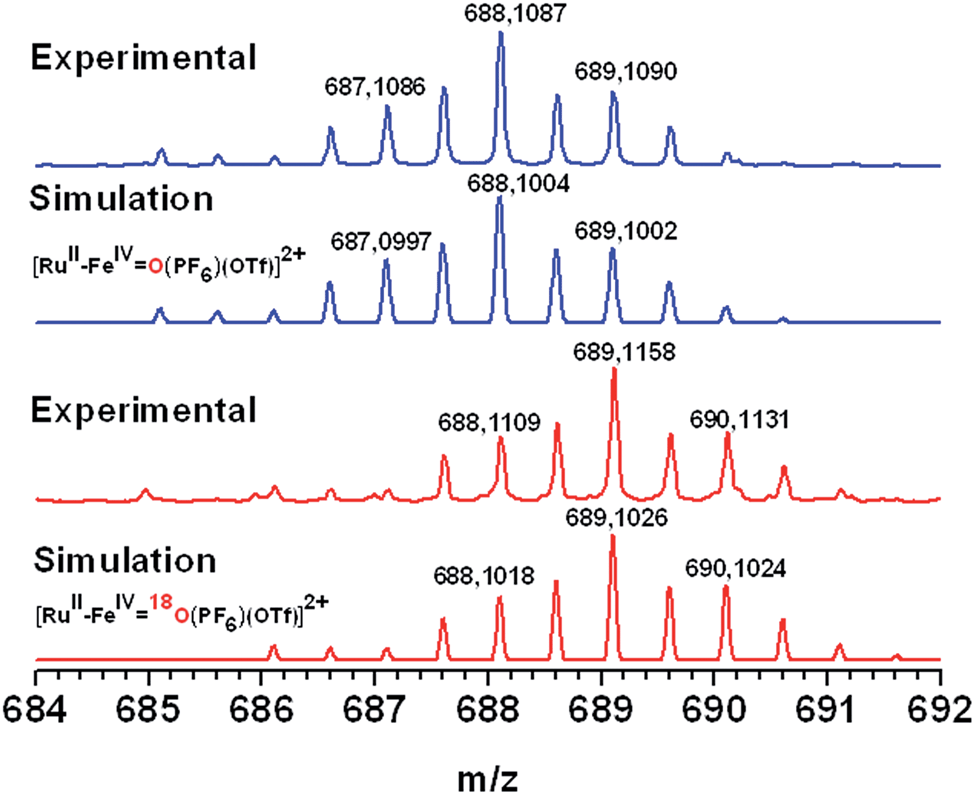 | ||
| Fig. 5 HR ESI-MS of RuII–FeIV(O) produced after illumination of complex 1-OH2 with 450 nm light in the presence of H2O (blue) or H218O (red). Simulation spectra are shown for comparison of isotopic patterns. | ||
A preliminary investigation of the catalytic activity of the photogenerated FeIV(O) species was performed using triphenyl phosphine as the substrate. Reaction mixtures containing complex 1-OH2, [CoIII(NH3)5Cl]2+, triphenyl phosphine and acetate buffer in a 4:1 WAN mixture were illuminated with 450 nm light. GC monitoring showed the formation of increasing amounts of triphenyl phosphine oxide during the first 10 minutes of illumination (Fig. SI 14†), reaching a turnover number (TON) of 3.2. This TON corresponds to a 20% efficiency with respect to the amount of [CoIII(NH3)5Cl]2+ used, which in our case, is the limiting reagent. Removal of any of the components in the reaction mixture, [CoIII(NH3)5Cl]2+, complex 1-OH2, substrate, light, as well as substitution of complex 1-OH2 by [Ru(bpy)3]2+, yielded no oxidation product during control runs. Proof of the insertion of oxygen from water in the substrate was obtained by use of H218O and the detection of isotopically labelled triphenyl phosphine oxide (Fig. SI 15†).
Conclusion
In summary, we have evidenced the two sequential light-induced electron transfer reactions from a FeII(OH2) species to a covalently bound chromophore and subsequent oxygen atom transfer from the resulting FeIV(O) intermediate. After the first photon absorption, the one-electron oxidized intermediate RuII–FeIII(OH) was identified by X-band EPR and flash photolysis methods. Further illumination of this species leads to the RuII–FeIV(O) intermediate which has been characterized by ESI-MS and UV-visible spectroscopy. Oxygen atom transfer from the FeIV(O) moiety to triphenyl phosphine has been proven using 18O labelling experiments.While the present work confirms the validity of the concept, current efforts in our laboratories are directed towards the optimization of the system. These include improvement of the excited state properties of the chromophore–catalyst assembly in order to optimize the photo-driven electron transfer processes, implementation of a more potent oxidation catalyst, as well as synthetic modification for its incorporation in Graetzel-type semiconducting electrodes23 in order to avoid both the use of sacrificial electron acceptors and to recover the electrons obtained during light-driven oxidation reactions for their further use.
Acknowledgements
The authors thank the Labex CHARMMMAT for a postdoctoral fellowship to CH as well as the ANR for financial support (project ANR Blanc Cathymetoxy). This work was also supported by the French Infrastructure for Integrated Structural Biology (FRISBI) ANR-10-INSB-05-01, EU-COST Actions CM1003 and Perspect-H2O.Notes and references
- J. A. Labinger, J. Mol. Catal. A: Chem., 2004, 220, 27–35 CrossRef CAS
.
- P. C. A. Bruijnincx, G. van Koten and R. J. M. Klein Gebbink, Chem. Soc. Rev., 2008, 37, 2716 RSC
- M. Costas, M. P. Mehn, M. P. Jensen and L. Que, Chem. Rev., 2004, 104, 939–986 CrossRef CAS PubMed
- C. Krebs, D. Galonić Fujimori, C. T. Walsh and J. M. Bollinger, Acc. Chem. Res., 2007, 40, 484–492 CrossRef CAS PubMed
- W. Nam, Acc. Chem. Res., 2007, 40, 522–531 CrossRef CAS PubMed
- W. Nam, Y.-M. Lee and S. Fukuzumi, Acc. Chem. Res., 2014, 47, 1146–1154 CrossRef CAS PubMed
- J. Barber, Chem. Soc. Rev., 2009, 38, 185 RSC
- J. P. McEvoy and G. W. Brudvig, Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS PubMed
- T. J. Meyer, M. H. V. Huynh and H. H. Thorp, Angew. Chem., Int. Ed., 2007, 46, 5284–5304 CrossRef CAS PubMed
- M. H. V. Huynh and T. J. Meyer, Chem. Rev., 2007, 107, 5004–5064 CrossRef CAS PubMed
- M. E. Ener, Y.-T. Lee, J. R. Winkler, H. B. Gray and L. Cheruzel, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 18783–18786 CrossRef CAS PubMed
- S. Fukuzumi, T. kishi, H. Kotani, Y. M. Lee and W. Nam, Nat. Chem., 2011, 3, 38–41 CrossRef CAS PubMed
- G. Knör, Chem.–Eur. J., 2009, 15, 568–578 CrossRef PubMed
- H. Kotani, T. Suenobu, Y.-M. Lee, W. Nam and S. Fukuzumi, J. Am. Chem. Soc., 2011, 133, 3249–3251 CrossRef CAS PubMed
- O. Hamelin, P. Guillo, F. Loiseau, M.-F. Boissonnet and S. Ménage, Inorg. Chem., 2011, 50, 7952–7954 CrossRef CAS PubMed
- H. Inoue, T. Shimada, Y. Kou, Y. Nabetani, D. Masui, S. Takagi and H. Tachibana, ChemSusChem, 2011, 4, 173–179 CAS
- D. W. Low, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 1996, 118, 117–120 CrossRef CAS
- F. Li, Y. Jiang, B. Zhang, F. Huang, Y. Gao and L. Sun, Angew. Chem., Int. Ed., 2012, 51, 2417–2420 CrossRef CAS PubMed
- C. Herrero, A. Quaranta, R.-A. Fallahpour, W. Leibl and A. Aukauloo, J. Phys. Chem. C, 2013, 117, 9605–9612 CAS
- C. Herrero, A. Quaranta, W. Leibl, A. W. Rutherford and A. Aukauloo, Energy Environ. Sci., 2011, 4, 2353–2365 CAS
- A. Company, G. Sabenya, M. González-Béjar, L. Gómez, M. Clémancey, G. Blondin, A. J. Jasniewski, M. Puri, W. R. Browne, J.-M. Latour, L. Que Jr, M. Costas, J. Pérez-Prieto and J. Lloret-Fillol, J. Am. Chem. Soc., 2014, 136, 4624–4633 CrossRef CAS PubMed
- A. Nayak, R. R. Knauf, K. Hanson, L. Alibabaei, J. J. Concepcion, D. L. Ashford, J. L. Dempsey and T. J. Meyer, Chem. Sci., 2014, 5, 3115–3119 RSC
- D. L. Ashford, W. Song, J. J. Concepcion, C. R. K. Glasson, M. K. Brennaman, M. R. Norris, Z. Fang, J. L. Templeton and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 19189–19198 CrossRef CAS PubMed
- J. Berglund, T. Pascher, J. R. Winkler and H. B. Gray, J. Am. Chem. Soc., 1997, 119, 2464–2469 CrossRef CAS
- A. Juris, V. Balzani, F. Barigelleti, S. Campagna, P. Belser and A. Von Zelewsky, Coord. Chem. Rev., 1988, 84, 85–277 CrossRef CAS
- A. R. Parent, R. H. Crabtree and G. W. Brudvig, Chem. Soc. Rev., 2013, 42, 2247 RSC
- N. Ségaud, J.-N. Rebilly, K. Sénéchal-David, R. Guillot, L. Billon, J.-P. Baltaze, J. Farjon, O. Reinaud and F. Banse, Inorg. Chem., 2013, 52, 691–700 CrossRef PubMed
- A. Thibon, J.-F. Bartoli, S. Bourcier and F. Banse, Dalton Trans., 2009, 9587 RSC
- V. Balland, M.-F. Charlot, F. Banse, J.-J. Girerd, T. A. Mattioli, E. Bill, J.-F. Bartoli, P. Battioni and D. Mansuy, Eur. J. Inorg. Chem., 2004, 2004, 301–308 CrossRef
- P. Comba, H. Wadepohl and A. Waleska, Aust. J. Chem., 2014, 67, 398–404 CrossRef CAS
- A. Draksharapu, Q. Li, H. Logtenberg, T. A. van den Berg, A. Meetsma, J. S. Killeen, B. L. Feringa, R. Hage, G. Roelfes and W. R. Browne, Inorg. Chem., 2012, 51, 900–913 CrossRef CAS PubMed
- M. L. A. Abrahamsson, H. B. Baudin, A. Tran, C. Philouze, K. E. Berg, M. K. Raymond-Johansson, L. Sun, B. Åkermark, S. Styring and L. Hammarström, Inorg. Chem., 2002, 41, 1534–1544 CrossRef CAS PubMed
- C. Herrero, L. Batchelor, A. Baron, S. El Ghachtouli, S. Sheth, R. Guillot, B. Vauzeilles, M. Sircoglou, T. Mallah, W. Leibl and A. Aukauloo, Eur. J. Inorg. Chem., 2013, 494–499 CrossRef CAS
- M. Martinho, P. Dorlet, E. Rivière, A. Thibon, C. Ribal, F. Banse and J.-J. Girerd, Chem.–Eur. J., 2008, 14, 3182–3188 CrossRef CAS PubMed
- G. Roelfes, M. Lubben, K. Chen, R. Y. N. Ho, A. Meetsma, S. Genseberger, R. M. Hermant, R. Hage, S. K. Mandal, V. G. Young, Y. Zang, H. Kooijman, A. L. Spek, L. Que and B. L. Feringa, Inorg. Chem., 1999, 38, 1929–1936 CrossRef CAS PubMed
- L. Duelund, R. Hazell, C. J. McKenzie, L. Preuss Nielsen and H. Toftlund, J. Chem. Soc., Dalton Trans., 2001, 152–156 RSC
Footnote |
| † Electronic supplementary information (ESI) available: Full description of synthetic methods as well as additional data including NMR analyses, cyclic voltammetry, UV/vis and experimental methods. See DOI: 10.1039/c5sc00024f |
| This journal is © The Royal Society of Chemistry 2015 |