
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePlasmonic giant quantum dots: hybrid nanostructures for truly simultaneous optical imaging, photothermal effect and thermometry†
Niladri S.
Karan
a,
Aaron M.
Keller
a,
Siddharth
Sampat
b,
Oleksiy
Roslyak
c,
Ayesha
Arefin
d,
Christina J.
Hanson
a,
Joanna L.
Casson
e,
Anil
Desireddy
a,
Yagnaseni
Ghosh
a,
Andrei
Piryatinski
f,
Rashi
Iyer
d,
Han
Htoon
a,
Anton V.
Malko
b and
Jennifer A.
Hollingsworth
*a
aMaterials Physics & Applications Division: Center for Integrated Nanotechnologies, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA. E-mail: jenn@lanl.gov
bDepartment of Physics, University of Texas at Dallas, Richardson, Texas 75080, USA
cDepartment of Physics, Fordham University, Bronx, New York 10458, USA
dDefense Systems and Analysis Division: Systems Analysis and Surveillance, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
eChemistry Division: Physical Chemistry & Applied Spectroscopy, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
fTheoretical Division: Physics of Condensed Matter & Complex Systems, Los Alamos National Laboratory, Los Alamos, New Mexico 87545, USA
First published on 9th February 2015
Abstract
Hybrid semiconductor–metal nanoscale constructs are of both fundamental and practical interest. Semiconductor nanocrystals are active emitters of photons when stimulated optically, while the interaction of light with nanosized metal objects results in scattering and ohmic damping due to absorption. In a combined structure, the properties of both components can be realized together. At the same time, metal–semiconductor coupling may intervene to modify absorption and/or emission processes taking place in the semiconductor, resulting in a range of effects from photoluminescence quenching to enhancement. We show here that photostable ‘giant’ quantum dots when placed at the center of an ultrathin gold shell retain their key optical property of bright and blinking-free photoluminescence, while the metal shell imparts efficient photothermal transduction. The latter is despite the highly compact total particle size (40–60 nm “inorganic” diameter and <100 nm hydrodynamic diameter) and the very thin nature of the optically transparent Au shell. Importantly, the sensitivity of the quantum dot emission to local temperature provides a novel internal thermometer for recording temperature during infrared irradiation-induced photothermal heating.
Introduction
Giant quantum dots (gQDs) are a unique functional class of colloidal semiconductor nanocrystals. They are core/shell QDs for which the shell has been grown to be especially thick (generally, >10 monolayers). While conventional core/shell QDs exhibit fluorescence intermittency or blinking when excited with a continuous light source, blinking is virtually eliminated in the case of gQDs.1–4 Furthermore, the thick shell limits oxidative photobleaching5 and metal-quenching effects.6,7 Significantly, while excitonic emission (emissive electron–hole recombination across the semiconductor bandgap) remains efficient, as for standard core/shell QDs, other photoexcitation/emission pathways are opened. Namely, in contrast with standard QDs, bi-excitons and multiple-excitons (2 or several electron–hole pairs, respectively, occupying a single QD) are long-lived and recombine via radiative emission even at room-temperature.8 Similarly, gQDs, as demonstrated for CdSe/CdS core/thick-shell systems, are further capable of emitting light when ionized, where the emission derives from recombination of a trion (charged electron–hole pair) to yield a reasonably bright “gray” state.9,10 In this way, gQDs have been shown to be practical bright and stable photoemitters for applications encompassing solid-state light-emitting devices11,12 and solution-phase optical probes in live-cell single-particle tracking.13Metal nanostructures afford scattering-based imaging and absorption-enabled photothermal effects, as well as nanoscale platforms for adhering cell-targeting agents, e.g., for selective targeting and thermal ablation of cancer cells in a milieu of healthy cells. Rod-shaped Au nanoparticles, or nanorods, as well as the more synthetically engineered Au nanoshell (silica/Au core/shell)14 and Au nanomatryoshka (Au/silica/Au core/shell/shell)15 have surface plasmon resonance bands tunable to the near-infrared (near-IR), an important spectral window where the penetration of light through biological tissue is the greatest. Infrared light absorbed by these Au nanostructures is converted to heat. Therefore, in a biological setting, once successfully located to a cellular target, such nanoparticles can be excited by infrared light and used to raise the temperature of targeted cells to initiate processes leading to cell death or even outright cell ablation. Thus, Au nanorods and nanoshells are inherently “multifunctional” nanomaterials in that the Au metal shell can be employed for both dark field imaging and photothermal cancer therapy.
Compared to nanorods, Au nanoshells afford enhanced extinction coefficients16 and the opportunity to precisely tune the surface plasmon resonance peak position from the visible to the near-IR as a function of the nanoshell geometry (dielectric core radius and Au shell thickness).17 Additional refinement of this material system has been achieved by combining nanoshells with magnetic nanoparticles and/or molecular fluorophores for including magnetic resonance and optical fluorescence imaging modalities, respectively. In these cases the magnetic or fluorescent components have been added to the outside of the nanoshell, with total particle size in the 150–180 nm range.18 Significantly, for the application of cancer tumor targeting and ablation, the optimal nanoparticle size for efficacious tissue penetration and subsequent elimination is 10–100 nm.19
Here, we explore the possibility of combining metal-shell nanostructures and semiconductor QDs in a single yet compact (<100 nm) construct with full retention of the distinct component properties. We do so by placing a gQD inside a Au shell. QDs have previously been enveloped within a metal shell,20 but despite theoretical modeling that demonstrates the possibility of realizing enhanced emission for an emitter inside a metal shell21 and even examples of enhanced fluorescence from ion emitters inside such a shell,22 the experimentally realized QD/Au core/shell constructs have yielded significant fluorescence quenching (45–75%).20 In our approach, we take advantage of the unique stability of the gQD emitter, as well as an improved “spacer” between the QD and the Au shell, to achieve a plasmonic gQD (pl-gQD) that combines the large extinction and photothermal effects of a Au shell with the brightness and stability of the gQD emitter with as little as 0% quenching. Furthermore, the thickness of the spacer used here—a silica layer—is tunable over a wide range. We show experimentally and theoretically that the interplay of silica spacer thickness with Au shell thickness in the ranges employed (∼10–17.5 nm and ∼2–5 nm, respectively) allows us to tune the surface-plasmon resonance band from the visible to the near-infrared (∼650–1000 nm). Furthermore, employing numerical simulations we demonstrate that the pl-gQD extinction is dominated by absorption rather than scattering and that the absorption cross-section is similar to larger and thicker-Au-shell structures previously reported, rendering pl-gQDs surprisingly efficient red/infrared photothermal transducers. Taken together, we demonstrate truly simultaneous optical imaging (QD emission) and efficient optically stimulated heating (Au-shell absorption and ohmic loss) in a compact, sub-100 nm structure. Furthermore, we effectively monitor for the first time the temperature of the nano-heater using the linear dependence of the QD fluorescence signal on temperature as an internal thermometer.
Results and discussion
Plasmonic-gQD chemistry and structural characterization
A schematic of the pl-gQD is shown in Fig. 1a. The center of this structure is a CdSe/CdS core/shell gQD, comprising a 4 nm CdSe core and a 16 monolayer-thick CdS shell (1 monolayer = 0.3375 nm). Light emission at 635 nm derives from the CdSe core, while absorption takes place principally in the CdS shell, which acts as a light antenna, harvesting but not consuming excitation photons.4 As prepared,3 CdSe/CdS gQDs are hydrophobic and form stable suspensions in standard nonpolar solvents. To transfer gQDs to the aqueous phase, we applied the reverse microemulsion technique to add a surface coating of negatively charged silica (see Methods).23–25 In addition to effecting the required phase transfer, the silica layer serves as our tunable spacer layer in the pl-gQD construct. It separates the emissive semiconductor core from the metallic Au shell and provides a semi-porous barrier during Au shell growth that limits penetration of Au3+ precursor ions to the QD surface (see below). In both capacities, the silica layer reduces fluorescence quenching that can result from either plasmonic nonradiative damping or metal-ion quenching.26,27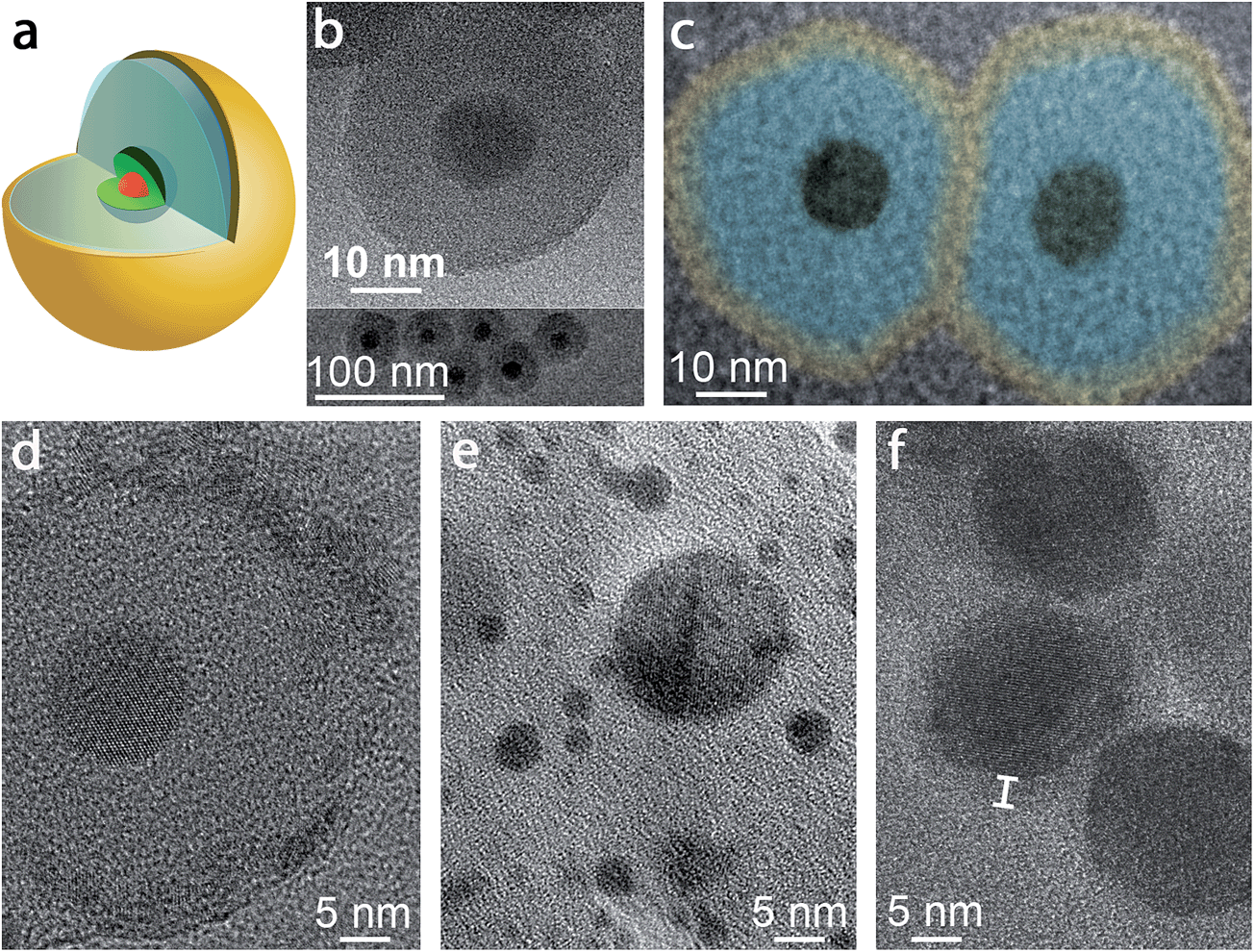 | ||
| Fig. 1 (a) Schematic depiction of the pl-gQD structure: CdSe/CdS gQD center (red/green) is surrounded by a SiO2 spacer layer (pale blue) that is enveloped in a thin Au shell (yellow). (b) Transmission electron microscopy (TEM) images of gQD/SiO2 nanoparticles comprising ∼12 nm-thick SiO2 layers. (c) Color-enhanced TEM image of two representative pl-gQDs synthesized from the gQD/SiO2 nanoparticles shown in (b). The original image is provided in ESI Fig. S3,† but was magnified and colored here to facilitate direct size-comparison with the starting gQD/SiO2 nanoparticles and to indicate the approximate extent of the Au shell layer (“yellow” on “blue” SiO2) based on the observed change in total particle size (from 36.7 ± 2.8 to 47.0 ± 1.7 nm on average). (d) High-resolution TEM image of a pl-gQD [also representing the sample shown in (b) and (c)] reveals the characteristic lattice spacing of Au(111) (d = 2.3 Å) in the polycrystalline outermost shell and of CdS(002) (d = 3.3 Å) in the gQD center. The intermediate region is clearly amorphous and constitutes the SiO2 spacer layer. (e) TEM image of Au nanoparticles that form as a result of electron-beam damage to the pl-gQD Au shell. (f) TEM image of pl-gQDs for which the Au shell was grown directly onto the gQD, without an intervening SiO2 spacer layer. Marker delineates the approximate thickness of the Au shell that is observed in the low-resolution image as an electron-beam-stable and lower-contrast halo surrounding the CdSe/CdS gQD. | ||
As described first by Jin and Gao20,28,29 an ultrathin Au shell (<5 nm) can be grown onto nanocrystals by using the positively charged peptide poly-L-histidine hydrochloride (PLH) as a template for nucleation and growth of the metal. Compared to simple primary amines, the peptide promotes immobilization (through electrostatic interactions) of higher densities of Au3+ ions on the nanocrystal surface.30 The resulting high concentration of Au3+ ions is sufficient to promote shell nucleation and avoids the need for pre-synthesized Au nanoclusters as structural templates. Significantly, the nanocluster approach (employed in the synthesis of well-known Au nanoshells14 and the more recently described Au nanomatryoshkas15) yields relatively thick Au shells (5–20 nm and, most typically, 15–20 nm). For this reason, we adopted the peptide-template approach to ensure access to Au shells sufficiently thin to permit both excitation and readout of photoemission from the internalized gQD.
In this approach, the nanocrystal substrates (here, gQD/SiO2 functionalized with PLH) are incubated with an aqueous solution of HAuCl4 and NaOH. A mild reducing agent, NH2OH, is then added to initiate controlled reduction of Au3+ to Au0 metal. Importantly, while the gold incubation solution is maintained at pH 6–7, it is prepared from a HAuCl4 solution that is initially brought to a pH of 9–10 by addition of NaOH (Methods). The chemistry that underlies this selection of a basic pH for the starting HAuCl4 solution and the subsequent lower pH for the diluted incubation solution is based on consideration of the hydrolysis products of HAuCl4 as well as the acid/base chemistry of the PLH amine groups (ESI†). Briefly, At pH 9–10, Au(OH)4− is the dominant species31 and the hardest to reduce compared to less hydrolysed complexes,32 favoring a more controlled reduction process. But, why return to a lower pH in the incubation solution? The mildly acidic conditions ensure that both amine groups of the PLH binding layer are fully protonated and positively charged (imidazole side chain pKa: 6.6 and α-amino pKa: 9.3) for effective electrostatic binding to Au(OH)4− at high ion:surface densities. Subsequent addition of the mild reducing agent then affords controlled, heterogeneous nucleation and growth of uniform and thin gold shells, while avoiding separate, homogeneous nucleation of gold metal. In contrast, we observe that a basic incubation solution results in uncontrolled deposition of thick Au deposits (ESI Fig. S1†), possibly arising from amine-initiated Au3+ → Au0 reduction,33–35 while using even more basic conditions for the Au3+ reduction (pH > 10) results in Au0 deposition at the CdS surface, suggesting that the Au(OH)4− can penetrate the mesoporous silica spacer layer (Fig. S2 and ESI†).36
Nevertheless, the initial diversion of the concentrated HAuCl4 solution to a basic pH of 9–10 is a necessary step. If the entire process is instead conducted at pH <7, a very fast and uncontrolled reduction is observed with nucleation of free Au nanoparticles. Thus, the gold-reduction process for ultra-thin Au-shell growth from a HAuCl4 precursor must be considered as two steps: (1) hydrolysis of the gold chloride species to a less easily reduced form of Au3+ and (2) protonation of nanocrystal surface-amine groups for promotion of ideal electrostatic interactions with a negatively charged gold complex and minimization of amine-initiated gold reduction. The significance of the former step has been more31,32 or less20,28,29 appreciated in Au-reduction literature. We emphasize this point here to aid in future efforts directed at cluster-free synthesis of ultrathin-Au shells.
Finally, by controlling the Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :particle ratio (5–7.5 × 105) it is possible to tune the Au shell thickness from ∼2 to 5 nm. Immediately following conversion to the gold-terminated nanostructure (within a few minutes), the new gQD core/SiO2-shell/Au-shell nanocrystals are colloidally stabilized using a range of thiol-terminated ligands, where the thiol forms a strong bond with the gold surface. For biological applications that necessitate minimal “non-specific” binding to cells, a polyethyleneglycol (PEG) moiety is traditionally incorporated into the thiol ligand.37 The PEG group also contributes to the stability of the pl-gQD, such that optimal stability is only achieved using ligands containing long PEG-chains (average molecular weight, MW = 6000 Da) that mitigate Van der Waals interactions between the Au nanoshells avoiding irreversible aggregation. For coupling of the pl-gQDs to cancer-targeting antibodies, we employ a 4:1 molar ratio mixture of the long-chain PEG to maintain colloidal stability and short-chain carboxy-terminated PEG (MW = 635 Da) to support subsequent bioconjugation via carbodiimide chemistry.37 In all cases, hydrodynamic diameter is assessed and used to confirm non-aggregated status of the new nanocrystals directly following Au-shell growth as well as over time after storage (Methods).
:particle ratio (5–7.5 × 105) it is possible to tune the Au shell thickness from ∼2 to 5 nm. Immediately following conversion to the gold-terminated nanostructure (within a few minutes), the new gQD core/SiO2-shell/Au-shell nanocrystals are colloidally stabilized using a range of thiol-terminated ligands, where the thiol forms a strong bond with the gold surface. For biological applications that necessitate minimal “non-specific” binding to cells, a polyethyleneglycol (PEG) moiety is traditionally incorporated into the thiol ligand.37 The PEG group also contributes to the stability of the pl-gQD, such that optimal stability is only achieved using ligands containing long PEG-chains (average molecular weight, MW = 6000 Da) that mitigate Van der Waals interactions between the Au nanoshells avoiding irreversible aggregation. For coupling of the pl-gQDs to cancer-targeting antibodies, we employ a 4:1 molar ratio mixture of the long-chain PEG to maintain colloidal stability and short-chain carboxy-terminated PEG (MW = 635 Da) to support subsequent bioconjugation via carbodiimide chemistry.37 In all cases, hydrodynamic diameter is assessed and used to confirm non-aggregated status of the new nanocrystals directly following Au-shell growth as well as over time after storage (Methods).
Structural characterization of ultra-thin Au-shells by electron microscopy is challenged by the tendency of the Au to break apart and coalesce into small Au nanoparticles when exposed to an electron beam.28,35,38 At low resolution, it is possible to avoid damaging the Au shell; nonetheless, in this case the shell is not apparent in the image and must be distinguished by comparing the starting gQD/silica size with the particle size after Au shell growth (Fig. 1b and c). Here, before Au shell addition, the total particle size is 36.7 ± 2.8 nm, implying a ∼12 nm SiO2 spacer layer (12.2 ± 1.1 nm measured from TEM images of multiple gQD/SiO2 particles), while after Au shelling, the total particle size has increased to 47.0 ± 1.7 nm. Thus, on average ∼5 nm of Au has been added to the starting gQD/SiO2 nanoparticle in this example. Significantly, if the image is poorly focused, the Fresnel effect can cause the appearance of a shell, but this imaging artifact should not be confused with an actual shell, as an equivalent result is obtained for gQD/SiO2 nanostructures under the same defocusing conditions (ESI and Fig. S3†). Working quickly (several seconds), it is possible to capture an unambiguous image of the Au shell at high resolution (Fig. 1d). We observe a polycrystalline shell with the characteristic lattice spacing of Au(111) (d = 2.3 Å) that soon degenerates into Au nanoparticles under the beam (Fig. 1e). Interestingly, it is easier to resolve a Au shell grown directly on a gQD, which appears as a lower-contrast coating (Fig. 1f). Beyond electron microscopy imaging, evidence for formation of the Au shell is also found in the distinctive optical properties of the pl-gQDs.
Optical properties of the pl-gQD's thin Au shell: theory and experiment
Spherical Au nanocrystals are characterized by surface plasmon resonance (SPR) bands in the green/yellow visible part of the electromagnetic spectrum (modestly tunable from ∼515–550 nm for spherical nanoparticles ranging in size from 20–80 nm (ref. 39)). In contrast, traditional nanoshells afford greater tunability, allowing SPR bands to be located anywhere from ∼680–1000 nm depending upon the core/shell dimensions.17 Using the boundary element method (see Methods), we observe that the SPR maxima in the case of the ultra-thin Au shells grown here on silica-coated gQDs are similarly tunable from 680 to >1000 nm as a function of both silica spacer and Au shell thicknesses (modeled for: 10, 12.5, 15, 17.5 nm SiO2 and 2, 3, 4, 5 nm Au) (Fig. 2a). Clearly, plasmonic QDs built from a 15 nm gQD core can easily reach the “therapeutic window” (700–1000 nm) for optimal light penetration through biological tissue. However, the ability to reach near-IR wavelengths in these constructs depends on the Au shell being very thin. Where the total particle size is only 40–60 nm, as is the case here, thicker Au shells (>5 nm) would transition the SPR maxima into the visible. As shown in Fig. 2a, even a 4 nm thick Au shell yields an SPR band below 700 nm when the SiO2 spacer layer is only 10 nm and the total particle size is ∼40 nm. Experimental absorption spectra are shown in Fig. 2c and d for a range of Au-shell thicknesses constructed on either 12 nm or 15 nm SiO2 spacer layers, representing total particle sizes in the range from ∼45–60 nm and Au-shell thickness from ∼3–5 nm.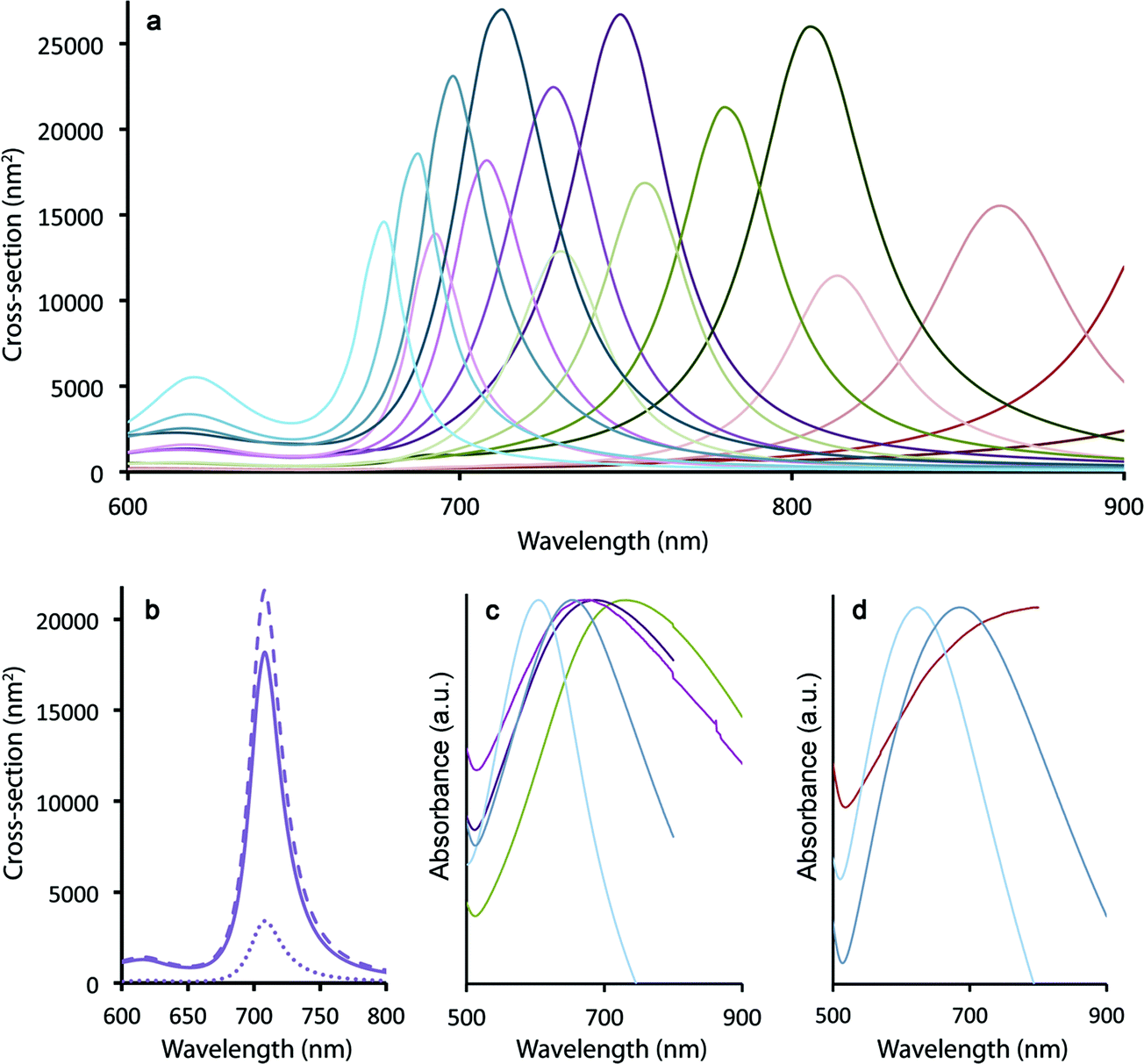 | ||
| Fig. 2 (a) Calculated total extinction cross-section for pl-gQDs characterized by four different Au shell thicknesses: 5 nm (blue series), 4 nm (purple series), 3 nm (green series) and 2 nm (red series); paired with four different SiO2 thicknesses: 10, 12.5, 15, and 17.5 nm (left-to-right in each Au-shell series). SPR is tuned from the visible to the near-IR as a function of both layer thicknesses. (b) Calculated total extinction cross-section for a pl-gQD comprising a 12.5 nm SiO2 spacer layer and a 4 nm Au shell (dashed line). Contributions to the cross-section from absorption (solid line) and scattering (spotted line) are distinguished. (c) and (d) Experimental absorption spectra for pl-gQDs having 12 and 15 nm thick SiO2 spacer layers, respectively. Spectral tuning in each case results from variations in Au shell thickness [roughly indicated by color-matching to (a)]. | ||
In contrast, traditional Au nanoshells and the newer nanomatryoshkas are much larger – “inorganic” diameters typically >150 and >85 nm, respectively. For these larger total particle sizes, thick Au shells (10–20 nm) still yield optimal near-IR SPR bands. Significantly, by starting with smaller-diameter particles, we gain an advantage for therapeutic applications. Namely, as we show below, even after surface functionalization for stability and cell targeting, pl-gQD hydrodynamic diameters remain <100 nm. Again, nanoparticles within the 10–100 nm size range are predicted to afford both optimal penetration into and movement within disseminated tumors, while at the same time allowing for sufficient circulation time followed by access to the liver, minimizing toxicity.19
Perhaps as significant as the spectral location of the plasmon resonance, the relative contribution to total extinction cross-section from absorption or scattering also significantly impacts the efficiency of Au-shell particles as phototransducers. Absorbed light contributes to particle heating, while scattered light does not (unless the photon that is scattered is ultimately absorbed by a particle prior to loss from the system40). We show in Fig. 2b that calculated total extinction for a pl-gQD is dominated by absorption rather than scattering. The example given is for a pl-gQD prepared using a 12.5 nm SiO2 spacer and a 4 nm Au shell, which approximates the pl-gQD shown in Fig. 1c and d. Significantly, the experimental absorption spectrum (green trace in Fig. 2c) corresponds well to the theoretical extinction spectrum (Fig. 2b). The inhomogeneous peak broadening in the experimental data results from distributions in particle size deriving from both variations in SiO2 spacer thickness (12.2 ± 1.1 nm) and Au shell thickness (∼4.5–5.5 nm, with the high-resolution data suggesting the possibility of even greater variation, though the apparent shell-thinning in certain regions may also have resulted from damage and shell reorganization under the electron-beam).41
For the ideal structure possessing a 12 nm SiO2 spacer and a ∼4 nm Au shell, the total extinction is ∼2.0 × 10−14 m2 compared to a typical nanoshell's almost 15 × 10−14 m2; however, absorption comprises almost 85% of the optical cross-section in the case of the pl-gQD. In contrast, the conventional nanoshell is dominated by scattering,15 so that absorption cross-sections are actually similar for the two types of structures: ∼1.7 × 10−14 m2 compared to the nanoshell's 2 × 10−14 m2 in the biologically relevant near-IR. In this way, despite a relatively small total particle size (∼50 nm) and an ultrathin Au shell (necessitated by the need for optical access to the gQD emitter core), the pl-gQD is predicted to be an efficient phototransducer, comparable to known nanoshell structures. As we show below, the effect is indeed sufficient to afford significant aqueous-phase heating, as well as ablation of biological cells.
Fluorescent properties of pl-gQDs
As just discussed, the silica shell clearly serves as an adequate spacer in allowing for a range of tunable SPR bands by way of modulating the gQD core/Au-shell geometry. That said, its role as a barrier to metal-quenching effects must also be established. To this end, we assessed gQD optical performance before and after SiO2 and Au-shell additions using both ensemble solution-phase and single-dot solid-state methods. We note that Au-induced photoluminescence (PL) quenching can result from Au3+ ion-QD interactions and/or plasmonic quenching of QD emission, where the former is evident during Au-shell synthesis as quenching that occurs after Au3+ precursor addition but prior to Au3+ reduction to Au0. Au3+ ion-related quenching was consistently observed for gQD/SiO2 structures comprising thinner SiO2 spacer layers (10–12 nm: ∼25% quenched vs. the starting gQD/SiO2 nanoparticle), but was not a significant factor when the SiO2 layer was thick (15–17 nm). Thus, the SiO2 layer can serve as an adequate barrier to prevent Au3+ ion penetration to the gQD surface if it is sufficiently thick.Following Au-shell formation, additional PL quenching was observed in the case of pl-gQD constructs prepared using the thinner SiO2 spacer layers. As shown in Fig. 3a, PL intensity for a 12 nm-SiO2pl-gQD is approximately half that of the starting gQD/SiO2 nanoparticle. (PL spectra shown are representative of >10 syntheses performed using 10 or 12 nm SiO2 spacers.) However, the extent of Au-shell related quenching was observed to vary. In one case, using fluorescence correlation spectroscopy (FCS) to assess ‘brightness-per-particle’ (BPP), we found that Au-shell quenching for pl-gQDs having a 12 nm thick SiO2 spacer layer could be as little as 10% (ESI Fig. S4†). Notably, we observed that PL intensity for pl-gQDs prepared using the thicker SiO2 spacer layers ranged from ∼15% quenched to ∼15% enhanced compared to the starting gQD/SiO2 nanoparticles over >10 syntheses. The solution-phase PL spectra shown in Fig. 3d are representative of a (modestly) PL-enhanced reaction product. Overall, reduced PL intensity for constructs employing thinner spacer layers may be expected due to increased overlap between gQD emission (centered at 635 nm) and the Au shell SPR band. As shown in Fig. 2a, the Au shell SPR peak shifts to higher energy (lower wavelengths) for thinner SiO2 layers compared to thicker ones. Increased overlap may enhance absorption in the far-field of the emitted gQD PL in the case of the solution-phase ensembles, or may impact near-field non-radiative energy transfer to the metal shell.21,26
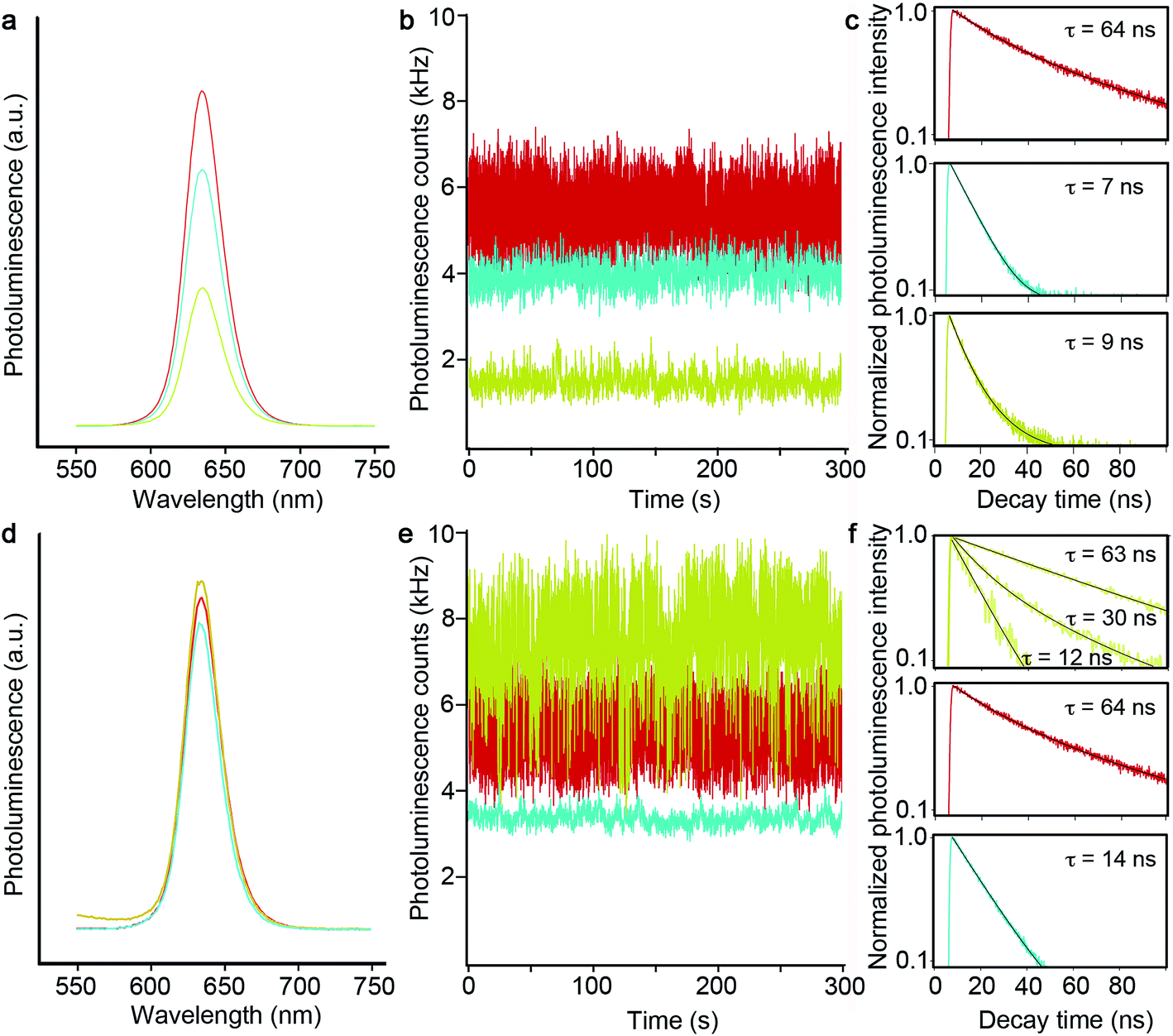 | ||
| Fig. 3 (a) Solution-phase emission spectra for gQDs in hexane (red), a gQD/12 nm-SiO2 sample in water (blue) and the resulting pl-gQD after Au-shell addition (green). (b) Single-dot photoluminescence time traces for the same series as shown in (a). (c) Correlated photoluminescence decay curves and measured emission lifetimes. (d) Solution-phase emission spectra for gQDs in hexane (red), a gQD/17 nm-SiO2 sample in water (blue) and the resulting pl-gQD after Au-shell addition (green). (e) Single-dot photoluminescence time traces for the same series as shown in (d). (f) Correlated photoluminescence decay curves and measured emission lifetimes. Here, the pl-gQD sample exhibits three distinct emission lifetimes, where the longest (63 ns: top of the photoluminescence intensity distribution) is similar to that observed for as-prepared g-QDs and, presumably, reflective of ideal excitonic emission, and the two shorter lifetimes likely derive from different charged states (30 ns and 12 ns: middle and bottom of the photoluminescence intensity distribution, respectively). | ||
To further probe 12 nm-SiO2pl-gQDs PL quenching, we investigated emitter properties at the level of a single dot via immobilization onto a glass substrate followed by intensity and fluorescence lifetime analysis using confocal microscopy. We found that the characteristic ‘gQD properties’ of suppressed blinking and photobleaching are retained (Fig. 3b). However, the PL decay time is rapid compared to the starting gQD emitter (Fig. 3c: 9 ns vs. 64 ns), and PL intensity is significantly reduced (from an average gQD value of 7500 ± 1600 Hz to an average 12 nm-SiO2pl-gQD value of 1300 ± 300 Hz). Interestingly, the reduction in per-dot PL intensity is not solely attributable to Au-shell effects. PL intensity was also quenched for gQD/SiO2 nanoparticles (without Au) – equally for both 12 and 17 nm SiO2 spacer layers (from the average gQD value of 7500 ± 1600 Hz to average gQD/12 nm-SiO2 and gQD/17 nm-SiO2 values of 4100 ± 1800 and 4200 ± 1600 Hz, respectively; Fig. 3b and e). Thus, in the solid-state (nanoparticles dispersed on a glass slide), the SiO2 shell itself can have a significant and deleterious impact on gQD emission intensity, while the effect is less in the solution environment. Furthermore, PL decay times for the gQD/12 nm-SiO2 and gQD/17 nm-SiO2 nanoparticles were fast compared to the starting gQD emitter and comparable to or even faster than the Au-shell products. Reductions in PL lifetime are expected if either plasmonic quenching or enhancement processes are active;26 however, in the absence of further experiments (underway), we are not currently able to ascribe changes in PL decay rates solely or even principally to plasmonic processes. The observation that the SiO2 shell itself causes significant changes in the PL lifetime suggests that other processes are active and contributing to gQD optical properties in these structures.
Toward understanding the effect of the SiO2 shell itself, we consider the possibility that the SiO2 layer may contribute to gQD charging, which could lead to both reduced PL intensity and PL lifetimes. To do so, we first note two features of the gQD/SiO2 nanoparticle: (1) gQDs are unique among well-studied QD emitters in that they are capable of emitting efficiently from “charged” states,9,10,42e.g., the emissive state comprises an exciton (electron–hole pair) and an extra electron, where such a ‘charged exciton’ is called a ‘trion,’ and (2) the SiO2 barrier layer is mesoporous rather than solid or crystalline, which is likely caused by the incomplete hydrolysis or polymerization of the starting tetraethyl orthosilicate (TEOS) precursor. We suggest that lone pair electrons on the oxygen atoms of the sol–gel siloxane bonds may serve as electron-donating traps for photogenerated charge carriers in the gQD core (in particular, hole carriers). Such an environment may lead to negative charging of the gQD, which would result in the observed retention of blinking-suppression combined with diminished emission intensity and faster radiative decay times (trion emission is less bright and more rapid compared to exciton emission). Furthermore, the reduced or absent quenching that is observed for gQD/SiO2 nanoparticles in the aqueous phase compared to the “dry” single-dot experiments may derive from the ability of water molecules to hydrogen bond with the ‘SiO2’ oxygen atoms, effectively passivating the otherwise numerous network of traps. We note that this is a tentative explanation, as it is not clear that sufficient “drying” of the individual gQD/SiO2 nanoparticles would take place to have the proposed impact on environmentally induced gQD charging.
The existence of charged-state emission in these constructs is also supported by the unusual single-dot behavior observed for the pl-gQDs made from 17 nm SiO2 spacer layers. Namely, 17 nm-SiO2pl-gQDs afforded single-dot PL intensities equal to or even somewhat enhanced compared to the starting gQDs (from the average gQD value of 7500 ± 1600 Hz to an average of 8200 ± 2700 Hz). More interestingly, however, the single PL-intensity time traces comprised contributions from multiple emissive states (2–7) characterized by different intensities and PL decay times (Fig. 3e and f and ESI Fig. S5†). In all cases, the higher-intensity states are the longer-lived ones, consistent with the less-intense emissions deriving from a range of charged states. Furthermore, decay times are overall faster for the gQD/17 nm SiO2 construct compared to its Au-shell counterpart, suggesting that charge-effects are somehow reduced by the inclusion of the outer Au layer. The mechanism responsible for the apparent ability of the Au shell to at least partially reverse gQD charging in this system is under further investigation.
Photothermal effect and ‘taking the system's temperature’ locally and globally
In addition to the range of effects on gQD emission described above, the Au shell adds a distinct and new functionality to the gQD. Perhaps surprisingly, the ultrathin Au shell employed here is an efficient photo-transducer capable of converting infrared irradiation into heat. When suspended in water and stimulated with an infrared laser (800 nm at ∼1 W cm−2), the pl-gQDs cause an increase in the temperature of the surrounding water as a clear function of particle concentration (Fig. 4a). Significantly, this change in temperature is similar to the much larger and thicker Au-shell nanomatryoshkas and nanoshells when comparing similar experimental conditions (particle concentration, laser power, sample volume and distance of thermocouple from point of irradiation).14,15 Furthermore, we show that achievable temperature continues to increase with increasing particle concentration, without saturation. This trend implies that absorption rather than scattering processes dominate the pl-gQD's interactions with light,40 which is confirmed, as discussed above, by our theoretical calculations (Fig. 2b). In the absence of the Au shell, the gQD/silica structure does not produce a measurable temperature increase above that obtained simply by exposing water to the infrared laser. This result is consistent with the known heat generation rates for semiconductors compared to metals.43 We further show that by simply decreasing the heated volume from 1 mL to 400 μL we are able to double the amount of heating from ∼20 to >40 °C above room temperature (Fig. 4b) with no significant change in gQD BPP, fluorescence lifetime, or Au-shell absorption comparing emitter and metal characteristics before and after infrared irradiation (ESI Fig. S4, S6 and S7†).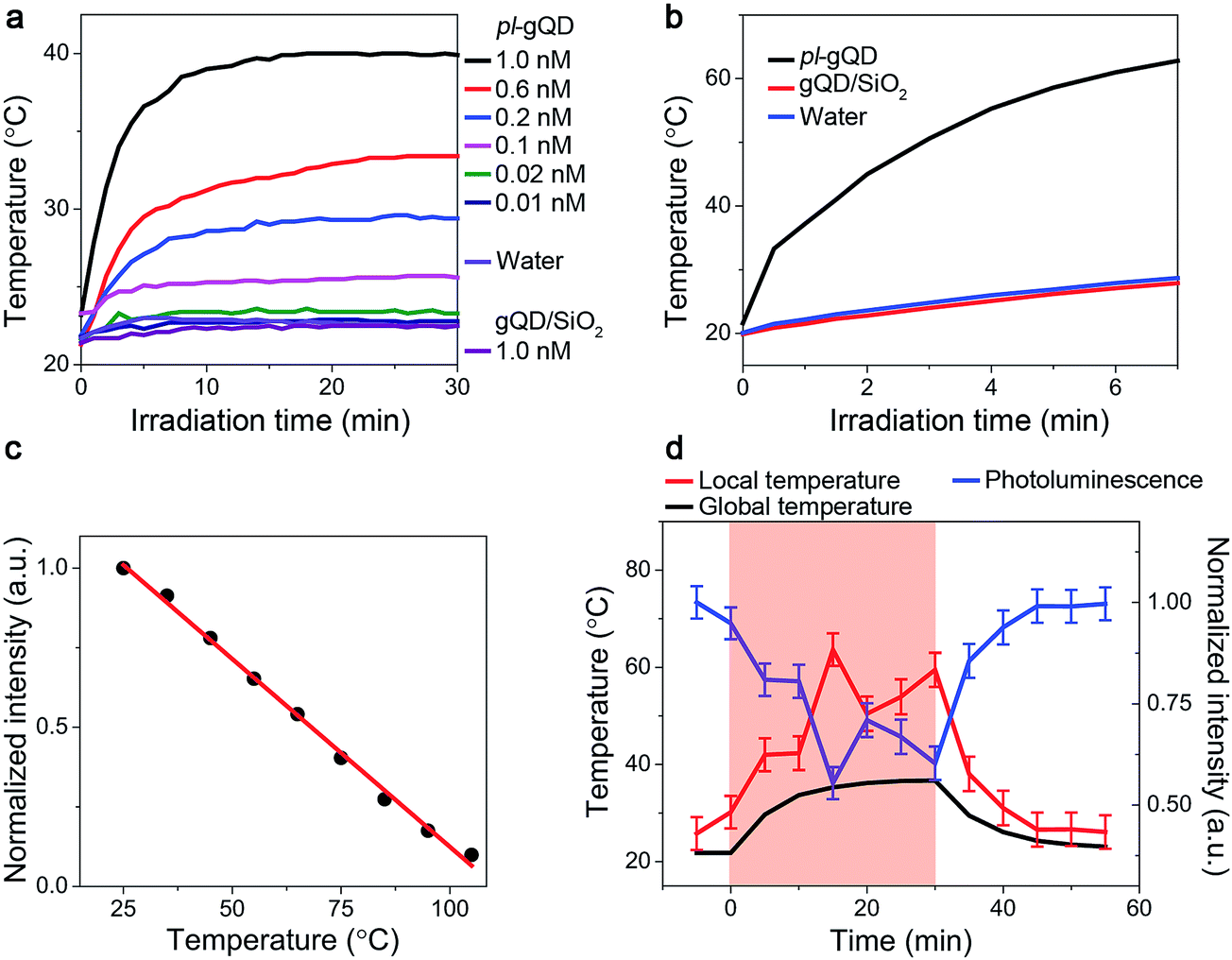 | ||
| Fig. 4 (a) Global solution temperature monitored using an inserted thermocouple probe as a function of duration of near-IR irradiation. pl-gQD solutions (1 mL sample volume) of different nanoparticle concentrations are compared with water and gQD/SiO2 solution controls. The concentration range shown in the figure inset represents from as many as ∼60 × 1010 nanoparticles per mL (1.0 nM) to as few as 6 × 109 nanoparticles per mL (0.01 nM), with a clear heating effect still evident when only 10 × 109 particles per mL (0.02 nM) are present in the solution. (b) Enhanced heating effect (an increase of ∼40 °C above room-temperature) observed for smaller sample volumes (0.4 mL of a ∼0.6 nM solution). (c) Temperature dependence of gQD/SiO2 photoluminescence used to approximate “local” temperature, i.e., nanoparticle temperature, during irradiation-induced heating. (d) Photothermal heating with temperature measured two ways: “globally” with inserted thermocouple probe and “locally” by correlating photoluminescence intensity quenching with nanoparticle temperature (2.0 mL of a ∼0.6 nM solution). Ultraviolet-light excitation of the QD emitter and near-IR excitation of the plasmonic shell are conducted simultaneously, as are optical readout of the QD signal and temperature readout from the thermocouple probe. Shaded region highlights time during which the near-IR laser source is on. For (a), (b), and (d): excitation wavelength is 800 nm; power density is 1.2 W cm−2. | ||
Further modification to our experimental setup (ESI Fig. S8†) allowed us to demonstrate for the first time the potential of this emitter-plasmonic hybrid nanostructure for simultaneous heating and temperature readout at the position of the heat source, i.e., the plasmonic nanoparticle. Specifically, by exposing the pl-gQD to both blue and infrared light sources, we are able to realize both gQD PL and Au-shell heating. Furthermore, as the gQD/SiO2 emission quenches as a linear function of temperature (Fig. 4c), we can use the change in gQD PL intensity as an internal thermometer for the plasmonic heating process. In Fig. 4d, we refer to the solution temperature measured by a thermocouple probe as the “global temperature” and that measured by gQD PL quenching as the “local temperature.” We find that the local temperature is always ∼15–20 °C higher than the global temperature, which is consistent with the observed retention in emitter and shell optical properties following irradiation. In other words, we do not observe particle “superheating” that was once thought to characterize the process of photothermal transduction and in line with more recent models.44
Breast cancer cell targeting and thermal ablation
pl-gQDs were conjugated to anti-HER2 monoclonal antibodies to promote selective targeting of SKBR3 mammary gland derived breast cancer cells (see Methods and Fig. 5a). Prior to cell exposure, colloidal stability of the conjugated pl-gQDs was confirmed by assessing hydrodynamic diameter in PBS buffer and cell culture media (Table 1 and ESI Fig. S9†). SKBR3 cells were initially exposed to both anti-HER2–pl-gQDs and PEGylated gQDs and imaged to determine the relative binding specificity afforded by the antibody-conjugated pl-gQDs compared to the non-conjugated PEG-pl-gQDs. In Fig. 5b we show that the conjugated nanoparticles specifically targeted the SKBR3 cell membranes, while the PEGylated nanoparticles did not bind to these cells.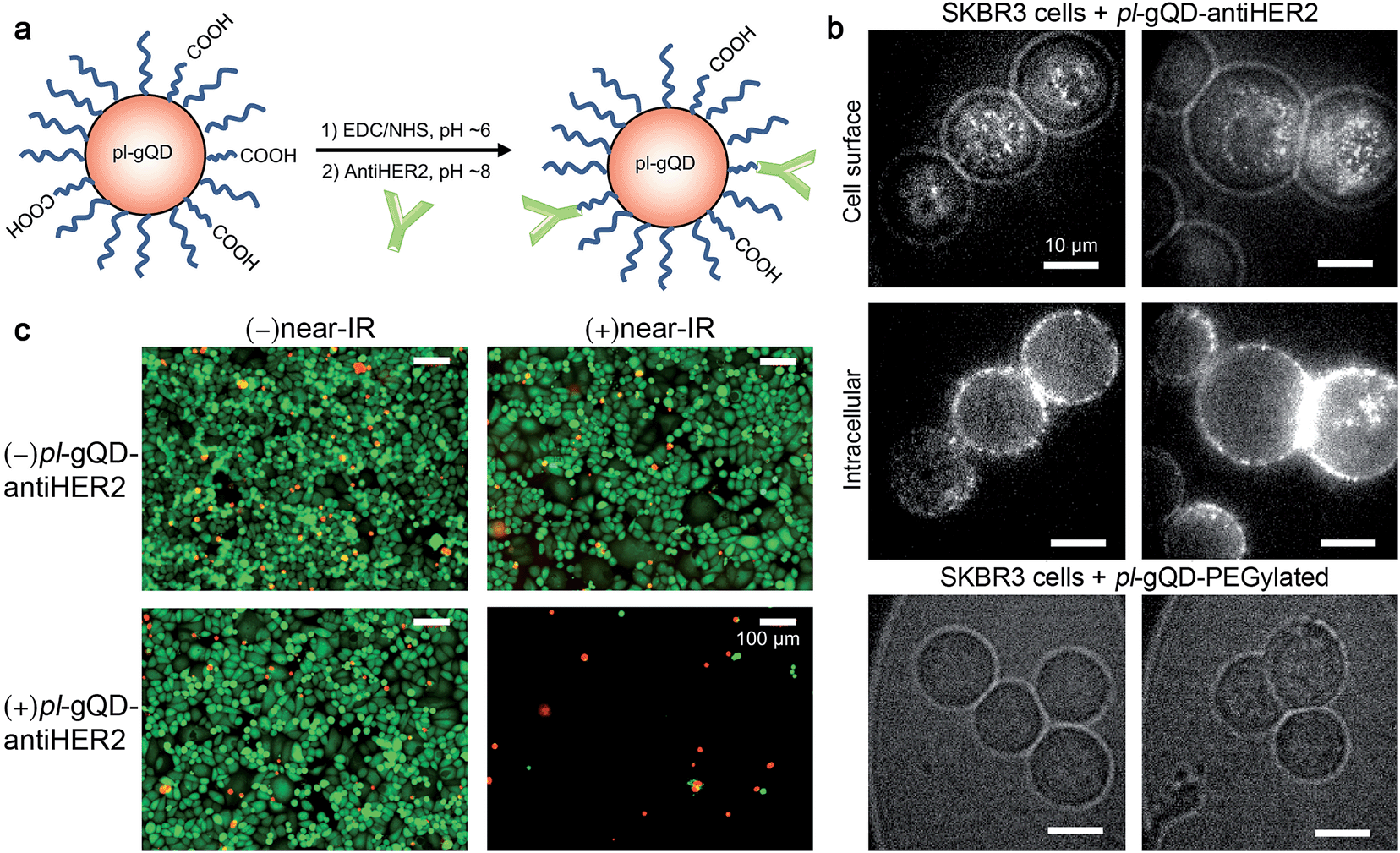 | ||
| Fig. 5 (a) Schematic diagram depicting conjugation of the antiHER2 antibody to the pl-gQD surface. (b) Confocal images of SKBR3 breast cancer cells incubated with either antiHER2-conjugated pl-gQDs or unconjugated pl-gQDs reveal cell outline (white light induced) and pl-gQD photoluminescence (excitation wavelength: 470 nm). The bright speckle in the centers of the cells shown in the top panel images, as well as the bright rings on the outside of the cells shown in the middle panel images, derive from pl-gQD emission. The difference between the top and middle panels arises from a shift in the focusing plane from the upper cell surface (top panel) to the middle of the cells (middle panel). Comparing the location of the pl-gQDs at the two different cellular positions suggests that the nanoparticles are located at the cell surfaces, rather than in their interior. No bright speckle or rings were observed in the case of cells incubated with non-conjugated pl-gQDs (bottom panel). (c) Results of live/dead cell assay following different exposure conditions: neither nanoparticle nor near-IR irradiation (top left), nanoparticle-free cells exposed to near-IR irradiation (top right), nanoparticle-exposed cells without near-IR irradiation (bottom left), and nanoparticle-exposed with near-IR irradiation (bottom right). Green emission from calcein AM staining indicates live cells while red emission from ethidium homodimer staining indicates dead cells. Only cells incubated with pl-gQD-antiHER2 and irradiated with near-IR showed evidence of cell death; here, loss of cells from cell chamber. | ||
The effectiveness of the pl-gQDs as agents for SKBR3 cell thermal ablation was subsequently tested. Cells were incubated with and without anti-HER2–pl-gQDs prior to treatment with either near-IR irradiation or no irradiation and then imaged using a standard live/dead cell assay to determine the impact of the exposure conditions on gross cell viability (see Methods and ESI Fig. S10†). Of these four treatments, only the combination of anti-HER2–pl-gQD and near-IR exposure resulted in evidence of cell death, which in this case was observed as catastrophic cell death (sloughing off of cells from the culture dish). Cell viability in samples treated with anti-HER2–pl-gQD incubation without near-IR irradiation and near-IR irradiation in the absence of anti-HER2–pl-gQD was similar to untreated control cells (indicated by “green” color in Fig. 5c images). The lack of dead cells (where dead cells would be “red” in color) is due to the rapid cell death induced by anti-HER2–pl-gQD/near-IR treatment causing enhanced sloughing-off of cells. Both results – retention of dead cells and sloughing-off of cells from the culture dish – have been reported in the literature following photothermal heating.45,46 The result here suggests that we have likely heated the cells beyond ∼45 °C, as less rapid cell death and, thereby, retention of dead cells on the culture dish is expected for relatively milder heating. The extent of photothermal heating can be easily controlled by tuning the nanoparticle concentration and/or the power of the near-IR laser used for Au-shell excitation.
Truly simultaneous optical imaging, heating and temperature readout
To demonstrate truly simultaneous imaging and photothermal heating toward deep-tissue therapy applications, we embedded the pl-gQDs within a tissue-scaffold protein matrix and imaged the gQD emission via confocal scanning microscopy during irradiation with a near-infrared laser (see Methods). As shown in Fig. 6 (and ESI Movie 1†), the gQD emission within the region of the near infrared laser is quenched ∼50% over the course of 30 min irradiation. From our linear calibration curve of the gQD emission response as a function of temperature (Fig. 4c), this corresponds to a local temperature of >60 °C. Notably, in response to the local temperature increase, the protein tissue-scaffold also expands. The expansion of the matrix and quenching of the pl-gQDs extends beyond the region of the near-IR laser likely due to local dissipation/transduction of heat. Ten minutes following the near infrared irradiation, the gQD emission is fully restored and the tissue scaffold contracts to its original state. These results demonstrate the powerful multiplexed potential of pl-gQDs as agents for truly simultaneous fluorescence imaging, thermometry and photothermal treatment in vivo.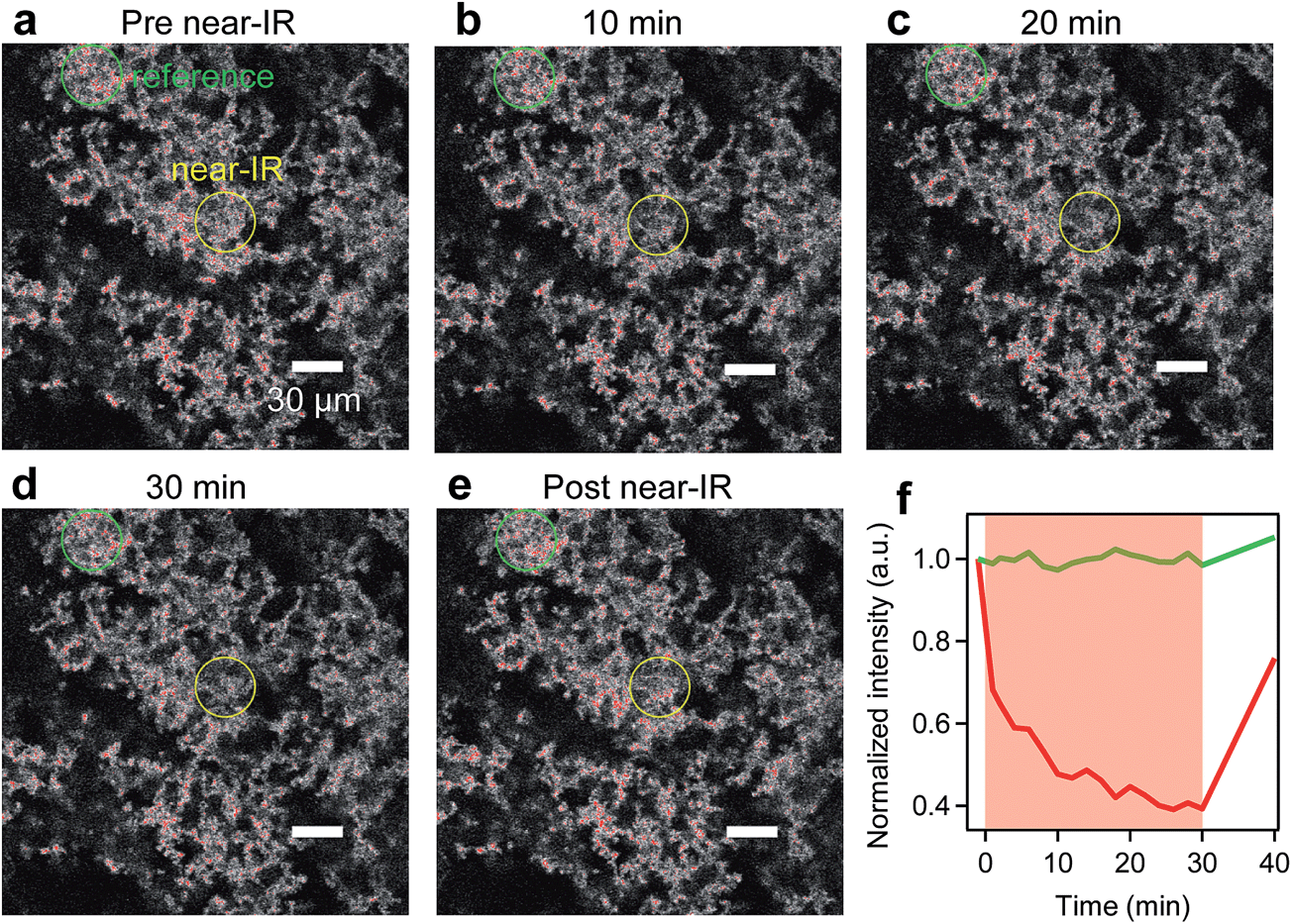 | ||
| Fig. 6 Simultaneous confocal imaging and near-IR irradiation-induced heating: imaging 45 μm into a suspension of pl-gQDs in a tissue-scaffold matrix (a) before, (b–d) during near-IR irradiation, and (e) 10 min after exposure. The region of pl-gQDs in the vicinity of the near-IR laser is marked by a yellow circle (∼30 μm in diameter), while a region of pl-gQDs outside of the near-IR irradiation (“reference”) is marked by a green circle. (f) Average pl-gQD intensity for each region versus time: near-IR exposed region (red curve; transparent red highlights time span of active near-IR exposure) shows significant quenching, while the non-exposed pl-gQDs (green curve) are not measurably quenched. | ||
Experimental methods
Materials
Tetraethyl orthosilicate (TEOS, 99.999%), Igepal CO-520, NH4OH solution (ACS reagent, 28–30%), gold chloride trihydrate (HAuCl4·3H2O, ≥99.99% trace metal basis), hydroxylamine hydrochloride (NH2OH·HCl, 99.995%, trace metal basis), poly-L-histidine hydrochloride (PLH, molecular weight ≥ 5000), and mPEG thiol (molecular weight ∼ 6 kDa) were purchased from Sigma-Aldrich. Cyclohexane (spectroscopy grade) was purchased from Acros Organics. Sodium hydroxide (certified ACS) was purchased from Fisher Scientific. Nanopure water (18 MΩ) was used for all aqueous-phase syntheses.Synthesis of pl-gQDs
000 rpm for 10 min. The precipitation and washing steps were repeated three times. Finally, the product was re-dispersed in 10 mL nanopure water (18 MΩ) with a final concentration ∼5 nM. Silica thicknesses were tuned by changing the amount of TEOS as well as the NH4OH and Igepal CO-520 ratio. Since bare silica has an isoelectric point of ∼2 at pH 7, the silica-coated gQDs are negatively charged due to the presence of Si–O− bonds on the surface. This negative charge was evident in the measured zeta potential (ζ) of ζ = −35 mV.
Basic structural and optical characterization
Transmission electron microscopy (TEM) images were acquired using a JEOL JEM-2010 machine operated at 200 kV acceleration voltage. A FEI Tecnai F30 electron microscope was used for high-resolution imaging (HRTEM). All of the absorption spectra were recorded using a Cary 5000 spectrophotometer. Solution-phase photoluminescence was measured using Horiba Jobin Yvon Fluorolog spectrometer.Theory
The absorption/scattering simulations were performed by the boundary element method (BEM) including the retardation effects.48Single-dot spectroscopy measurements
Nanoparticles suspended in hexane were dispersed onto a quartz substrate with the density on the order of 0.01 per μm2. The sample was mounted on a translation stage of an optical microscope and excited at 405 nm with 50 ps pulses through a 100×, 1.45 NA oil-immersion objective lens that is also used to collect PL. The pulse-to-pulse separation (variable from 400 ns to 2000 ns) is set to be much longer than the PL decay times in order to ensure complete relaxation of excitons between sequential laser pulses. Collected PL is sent to a pair of Perkin-Elmer avalanche photodiodes (SPCM AQR-14) positioned at two arms of the standard Hanbury-Brown–Twiss (HBT) arrangement with 50/50 emission signal split. For PL blinking traces only one of the detectors is used. We maintained average number of excitations-per-pulse low, in the range of 0.2 < 〈N〉 < 0.5, in order to avoid higher order exciton effects. Before going into the HBT system, the PL emission is passed through long-pass excitation/emission filters that reject scattered laser light. Additionally, each of the detectors has a 680 nm short-pass filter to avoid avalanche photodiode (APD) after-pulsing light. Time-correlated single photon counting (TCSPC) is performed using PicoQuant TimeHarp electronics.Fluorescence correlation spectroscopy (FCS) measurements
FCS measurements were used to determine the average brightness per particle (BPP) of the gQD/SiO2 before and after Au shelling. On an Olympus IX71 microscope, each sample was excited using a pulsed (10 MHz) 485 nm laser at 5 μW (PicoQuant PDL 800-B) through an Olympus 60× NA 1.2 water-immersion objective. The diffraction-limited probe volume was focused 25 μm above the coverslip into the solution. After passing through a 488 nm long-pass dichroic (Di02-R488, Semrock), the emission was filtered with a 640 nm band pass (FF01-641/75, Semrock), split by a 50/50 beam splitter (BS016, ThorLabs), and collected by two fiber-coupled single photon counting avalanche photodiodes (SPCM-AQ4C, Excelitas). The reflected and transmitted channels were cross-correlated using ALV5000 hardware correlators. The correlation function was averaged from at least three 5 second measurements. The BPP was determined by multiplying the average count rate by the amplitude of the correlation function, which is inversely equal to the number of particles in the probe volume.Near-IR-induced heating and simultaneous photoluminescence measurements
A diagram of our experimental setup to simultaneously measure temperature and PL emission is shown in ESI Fig. S8.† A Titanium Sapphire laser (Mai Tai, Spectra Physics) tuned to 800 nm was used as a continuous wave near infrared (near-IR) source by disabling the mode-locking feature. The near-IR beam was expanded to a diameter of ∼1 cm using a telescope lens system. The pl-gQD samples were irradiated at 1.2 W cm−2 for 30 min in a 1 cm path length quartz cuvette containing 1 mL of sample solution at various concentrations. The “global” temperature was recorded by placing a thermocouple (Fisher Scientific) in the bottom corner of the cuvette just outside the irradiated region. Controls with water and gQD/SiO2 showed minimal residual heating from the laser at this position compared to the pl-gQD samples. The gQD PL spectrum was simultaneously recorded using a portable fiber-coupled spectrometer (CCS200, ThorLabs) with a 10 s integration time upon excitation with a UV lamp at 312 nm (SpectraLine Longlife Filter). To estimate the “local” temperature (temperature of the pl-gQD), the relative area of the emission peak at ∼635 nm was determined by Gaussian fitting, which was in turn used to estimate the percentage of quenching. Sample concentrations were estimated using the Nanosight Nanoparticle Tracking Analysis (NTA) software to track and count pl-gQDs in fluorescence mode. The determined particles per mL values (obtained for three replicates) were multiplied by a factor of 2 to account for an approximate bright/dark fraction of ∼50% (ESI Fig. S9†). The local temperature was calculated using a calibration curve of gQD PL quenching vs. temperature (see Fig. 4c). The calibration curve was generated using temperature-dependent PL spectra acquired on a Horiba Jobin Yvon Nanolog with an attached temperature-controlled sample holder. gQD/SiO2 nanoparticles in a solution of 60% water/40% glycol were used as the temperature standard. These were heated in 10 °C increments (5 min were allowed at each point to ensure that the solution had come to thermal equilibrium). The integrated area of each PL spectrum obtained at elevated temperatures was compared to that obtained at room temperature to assess the extent of PL quenching for each temperature point. (Note: Due to limited solubility of pl-gQDs in the water–glycol mixture, the calibration curve was obtained using gQD/SiO2 nanoparticles. For a smaller temperature range in pure water, we confirmed that pl-gQD PL responds similarly to heating: ESI Fig. S12†).Bio-conjugation for cell targeting using anti-HER2
For carbodiimide-mediated coupling of the pl-gQDs to anti-HER2, a 4:1 molar mixture of large methoxy-terminated thiol-PEG, CM(PEG)6000, and small carboxy-terminated thiol-PEG, CT(PEG)635, was added immediately after Au-shelling. Specifically, to 1.5 mL of 20 nM pl-gQDs, 5 μL of 333 mM CT(PEG)635 (MW 635 Da) (Pierce) stock dissolved in anhydrous DMSO was added resulting in a final CT(PEG)635 concentration of 1.1 mM. Subsequently, 40 mg of CM(PEG)6000 (MW ∼ 6 kDa, Sigma-Aldrich) was added with stirring resulting in a final CM(PEG)6000 concentration of 4.4 mM. To deprotonate the thiol ligand and facilitate binding to the Au surface, the pH was adjusted to ∼8 with NaOH and the solution was allowed to stir overnight. The excess ligands were removed via multiple centrifugation–dilution cycles with a 50 kDa molecular weight cutoff concentrator (EMD Millipore, Amicon Ultra-15), followed by sonication for ∼30 min to re-disperse the sample at a final volume of 0.5 mL. The hydrodynamic diameter was subsequently assessed to ensure the sample remained mono-dispersed ESI Fig. S9†). The carboxy-terminated ligands were converted to succinimidyl ester (NHS) groups by adding a solid mixture of 2 mg of 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (Pierce) and 4 mg sulfo-NHS (Pierce), followed by stirring at room temperature for ∼20 min at pH ∼6. To 0.5 mL of the NHS-activated pl-gQDs, 1 mL of 150 nM anti-HER2 (Pierce) in 100 mM NaHCO3 pH 8.3 buffer was added. The pH was adjusted to ∼8 if needed and the sample was stirred overnight at 4 °C. The hydrodynamic diameter, ∼85 nm, was checked following bio-conjugation of the pl-gQDs to anti-HER2 (ESI Fig. S9†).
Cell culture, targeting, treatment and imaging
Mammary gland derived breast cancer, SKBR3, cells were cultured using standard aseptic tissue culture techniques in McCoy's 5A media supplemented with 10% fetal bovine serum and 1% antibiotics. At passage number 6, the cells were seeded in 8 well glass bottom dishes at a density of 125000 cells cm−2. The cells were cultured for 3 days in a humidified cell culture incubator. The cells were then washed twice with PBS to remove the serum and incubated with either 0.01 nM anti-HER2–pl-gQD or 0.01 nM PEGylated pl-gQDs in PBS with calcium and magnesium for 1 h for: (a) imaging experiments to confirm successful cell targeting and (b) cell ablation experiments involving pl-gQD exposure (with and without near-IR irradiation). To confirm specific targeting of the SKBR3 cells by the anti-HER2–pl-gQD compared to the pl-gQD with just the PEG ligand for (a), the cells were imaged with an electron multiplying charge coupled device (EMCCD) camera (Photon Max, Princeton Instruments) using a combination of white light to observe the cell outline and wide-field 470 nm LED illumination (ThorLabs) to excite gQD PL. For (b), in addition to the nanoparticle-targeted cell samples, another set of cell samples was simply washed with PBS and left in the incubator for 1 h in PBS for use in cell ablation experiments entailing only near-IR irradiation, i.e., with pl-gQDs absent. For experiments using near-IR irradiation, the samples were exposed to 800 nm irradiation at 2.6 W cm−2 (Spectra Physics, Mai Tai Titanium Sapphire operated in continuous wave). After irradiation, the cells were washed with PBS and incubated in McCoy's 5A media for 4 h. For samples not exposed to near-IR irradiation, the cells were subjected to the same processing. Following final incubation, all samples were incubated with 2 mM of ethidium homodimer and 0.5 mM of calcein AM for 30 min. The samples were then imaged on a fluorescence microscope. Live cells were revealed by their green color, while dead cells imaged as red. Cell ablation was indicated as an absence of cells in the field of view, i.e., consistent with cell detachment.
Simultaneous near-IR irradiation and imaging within tissue-scaffold matrix
A mixture of 0.10 nM pl-gQDs and 4.5 mg mL−1 tissue-scaffold matrix (Matrigel Basement Membrane Matrix, BD Biosciences) was prepared at 4 °C within a multi-well chamber slide. The matrix was polymerized at ∼40 °C for 30 min. To perform simultaneous near-IR irradiation and pl-gQD imaging, near-IR laser was coupled into the back port of an Olympus IX81 Flouview Microscope (FV300) with a near-IR dichroic (FF720-SDi01, Semrock) and an Olympus UIS2 60× NA 0.70 air objective. The near-IR profile was imaged using a solution of up-converting nanoparticles (carboxylated Sunstone UCP 545, Sigma-Aldrich) drop cast onto a coverslip. Multiple images of the up-converting nanoparticles were taken using an Olympus DP71 camera, which were subsequently registered onto the image formed by the photomultiplier tube using a standard ruler. The circular near-IR profile was determined to have a full-width-half-max of ∼7.6 μm, corresponding to an irradiated area of ∼45 μm2. The pl-gQD emission was imaged 45 μm into the tissue-scaffold matrix by confocal microscopy (Olympus Fluoview FV300) using a 300 μm pinhole, filtered with a 640 nm band pass (FF01-641/75, Semrock) and a 488 nm laser as the excitation source (Coherent Sapphire). Confocal images of the gQD emission were acquired every 2 min during the course of a 30 min near-IR irradiation at a power of 30 mW and wavelength of 800 nm. The background-subtracted average gQD intensity in the region irradiated by near-IR laser was quantified using ImageJ and used to assess the relative gQD quenching.Conclusions
In summary, we have previously demonstrated the potential of photostable giant or thick-shell CdSe/CdS QDs for the range of applications from solid-state lighting to live-cell single-particle tracking.11–13 Here, we show that such novel emitters are ideal components for hybrid semiconductor–metal nanocomposites for which the ensemble and single-dot gQD optical properties can be retained and new functionality realized. Namely, a photothermal heating effect and an ‘on-board’ temperature sensing capability located precisely at the point of heating (which could be further improved by including advanced dual-emitting nanoscale temperature sensors49) are added. Furthermore, as theoretically and experimentally shown, photo-thermal transduction is comparable to that achieved by larger metal-shell constructs comprising substantially thicker Au shells. The compact size realized here (<100 nm hydrodynamic diameter) affords colloidal stability in a range of liquid media and, thereby, enhanced potential for in vivo applications as well as solution-based processing into optically transparent monoliths. Finally, although the key g-QD property of blinking-suppressed emission was retained in all cases, in developing the pl-gQDs we observed a range of effects with respect to introduction of new emissive states (beneficially faster but reduced in intensity) that are the subject of ongoing investigations.Acknowledgements
For this effort, J.A.H., H.H., J.L.C and C.J.H. were supported by a Single Investigator Small Group Research Grant (2009LANL1096), Division of Materials Science and Engineering (MSE), Office of Basic Energy Sciences (OBES), Office of Science (OS), U.S. Department of Energy (DOE). A.V.M. and S.S were supported by grant DE-SC0010697, OBES, OS, DOE. N.S.K., A.A., Y.G. and R.I. were supported by Los Alamos National Laboratory (LANL) Directed Research and Development (LDRD) funds. A.M.K. and O.R. were supported by LANL Center for Integrated Nanotechnologies (CINT) postdoctoral funding. A.D. was supported by the OBES, OS, DOE MSE Biomolecular Materials Program. This work was performed in large part at CINT, a DOE, OBES Nanoscale Science Research Center and User Facility. Los Alamos National Laboratory, an affirmative action equal opportunity employer, is operated by Los Alamos National Security, LLC, for the National Nuclear Security Administration of the U.S. Department of Energy under contract DE-AC52-06NA25396. We acknowledge the technical assistance from Piyush Bajaj in execution of the cell exposure experiments and analysis. We thank James Werner for instrumentation resources, helpful discussion regarding experimental design, and assistance in setting up the near-infrared irradiation with simultaneous photoluminescence imaging.References
- Y. Chen, J. Vela, H. Htoon, J. L. Casson, D. J. Werder, D. A. Bussian, V. I. Klimov and J. A. Hollingsworth, J. Am. Chem. Soc., 2008, 130, 5026–5027 CrossRef CAS PubMed
.
- B. Mahler, P. Spinicelli, S. Buil, X. Quelin, J.-P. Hermier and B. Dubertret, Nat. Mater., 2008, 7, 659–664 CrossRef CAS PubMed
- Y. Ghosh, B. D. Mangum, J. L. Casson, D. J. Williams, H. Htoon and J. A. Hollingsworth, J. Am. Chem. Soc., 2012, 134, 9634–9643 CrossRef CAS PubMed
- A. M. Dennis, B. D. Mangum, A. Piryatinski, Y.-S. Park, D. C. Hannah, J. L. Casson, D. J. Williams, R. D. Schaller, H. Htoon and J. A. Hollingsworth, Nano Lett., 2012, 12, 5545–5551 CrossRef CAS PubMed
- J. Vela, H. Htoon, Y. Chen, Y.-S. Park, Y. Ghosh, P. M. Goodwin, J. H. Werner, P. N. Wells, J. L. Casson and J. A. Hollingsworth, J. Biophotonics, 2010, 3, 706–717 CrossRef CAS PubMed
- Y.-S. Park, Y. Ghosh, Y. Chen, A. Piryatinski, P. Xu, N. H. Mack, H.-L. Wang, V. I. Klimov, J. A. Hollingsworth and H. Htoon, Phys. Rev. Lett., 2013, 110, 117401 CrossRef
- Y.-S. Park, Y. Ghosh, P. Xu, N. H. Mack, H.-L. Wang, J. A. Hollingsworth and H. Htoon, J. Phys. Chem. Lett., 2013, 4, 1465–1470 CrossRef CAS
- H. Htoon, A. V. Malko, D. Bussian, J. Vela, Y. Chen, J. A. Hollingsworth and V. I. Klimov, Nano Lett., 2010, 10, 2401–2407 CrossRef CAS PubMed
- P. Spinicelli, S. Buil, X. Quelin, B. Mahler, B. Dubertret and J. P. Hermier, Phys. Rev. Lett., 2009, 102, 136801 CrossRef CAS
- C. Galland, Y. Ghosh, A. Steinbrueck, M. Sykora, J. A. Hollingsworth, V. I. Klimov and H. Htoon, Nature, 2011, 479, 203–207 CrossRef CAS PubMed
- B. N. Pal, Y. Ghosh, S. Brovelli, R. Laocharoensuk, V. I. Klimov, J. A. Hollingsworth and H. Htoon, Nano Lett., 2012, 12, 331–336 CrossRef CAS PubMed
- J. Kundu, Y. Ghosh, A. M. Dennis, H. Htoon and J. A. Hollingsworth, Nano Lett., 2012, 12, 3031–3037 CrossRef CAS PubMed
- A. M. Keller, Y. Ghosh, M. S. DeVore, M. E. Phipps, M. H. Stewart, B. S. Wilson, D. S. Lidke, J. A. Hollingsworth and J. H. Werner, Adv. Funct. Mater., 2014, 24, 4796–4803 CrossRef CAS
- B. E. Brinson, J. B. Lassiter, C. S. Levin, R. Bardhan, N. Mirin and N. J. Halas, Langmuir, 2008, 24, 14166–14171 CrossRef CAS PubMed
- C. Ayala-Orozco, C. Urban, M. W. Knight, A. S. Urban, O. Neumann, S. W. Bishnoi, S. Mukherjee, A. M. Goodman, H. Charron, T. Mitchell, M. Shea, R. Roy, S. Nanda, R. Schiff, N. J. Halas and A. Joshi, ACS Nano, 2014, 8, 6372–6381 CrossRef CAS PubMed
- R. Bardhan, N. K. Grady, J. R. Cole, A. Joshi and N. J. Halas, ACS Nano, 2009, 3, 744–752 CrossRef CAS PubMed
- S. J. Oldenburg, R. D. Averitt, S. L. Westcott and N. J. Halas, Chem. Phys. Lett., 1998, 288, 243–247 CrossRef CAS
- R. Bardhan, W. Chen, C. Perez-Torres, M. Bartels, R. M. Huschka, L. L. Zhao, E. Morosan, R. G. Pautler, A. Joshi and N. J. Halas, Adv. Funct. Mater., 2009, 19, 3901–3909 CrossRef CAS
- M. E. Davis, Z. Chen and D. M. Shin, Nat. Rev. Drug Discovery, 2008, 7, 771–782 CrossRef CAS PubMed
- Y. Jin and X. Gao, Nat. Nanotechnol., 2009, 4, 571–576 CrossRef CAS PubMed
- X. Miao, I. Brener and T. S. Luk, J. Opt. Soc. Am. B, 2010, 27, 1561–1570 CrossRef CAS
- J. Zhang, Y. Fu and J. R. Lakowicz, J. Phys. Chem. C, 2009, 113, 19404–19410 CAS
- H. L. Ding, Y. X. Zhang, S. Wang, J. M. Xu, S. C. Xu and G. H. Li, Chem. Mater., 2012, 24, 4572–4580 CrossRef CAS
- R. Koole, M. M. van Schooneveld, J. Hilhorst, C. d. M. Donega, D. C. t. Hart, A. van Blaaderen, D. Vanmaekelbergh and A. Meijerink, Chem. Mater., 2008, 20, 2503–2512 CrossRef CAS
- D. K. Yi, S. T. Selvan, S. S. Lee, G. C. Papaefthymiou, D. Kundaliya and J. Y. Ying, J. Am. Chem. Soc., 2005, 127, 4990–4991 CrossRef CAS PubMed
- J. R. Lakowicz, Anal. Biochem., 2001, 298, 1–24 CrossRef CAS PubMed
- J. A. Schuller, E. S. Barnard, W. Cai, Y. C. Jun, J. S. White and M. L. Brongersma, Nat. Mater., 2010, 9, 193–204 CrossRef CAS PubMed
- Y. Jin and X. Gao, J. Am. Chem. Soc., 2009, 131, 17774–17776 CrossRef CAS PubMed
- Y. Jin, C. Jia, S.-W. Huang, M. O'Donnell and X. Gao, Nat. Commun., 2010, 1, 41 Search PubMed
- R. Djalali, Y. F. Chen and H. Matsui, J. Am. Chem. Soc., 2003, 125, 5873–5879 CrossRef CAS PubMed
- C. K. Chang, Y. J. Chen and C. T. Yeh, Appl. Catal., A, 1998, 174, 13–23 CrossRef CAS
- J. C. Y. Kah, N. Phonthammachai, R. C. Y. Wan, J. Song, T. White, S. Mhaisalkar, I. Ahmad, C. Sheppard and M. Olivo, Gold Bull., 2008, 41, 23–36 CrossRef CAS
- J. D. S. Newman and G. J. Blanchard, Langmuir, 2006, 22, 5882–5887 CrossRef CAS PubMed
- X. Yang, M. Shi, R. Zhou, X. Chen and H. Chen, Nanoscale, 2011, 3, 2596–2601 RSC
- X. Lu, M. S. Yavuz, H.-Y. Tuan, B. A. Korgel and Y. Xia, J. Am. Chem. Soc., 2008, 130, 8900–8901 CrossRef CAS PubMed
- C.-M. Huang, S.-H. Cheng, U. S. Jeng, C.-S. Yang and L.-W. Lo, Nano Res., 2012, 5, 654–666 CrossRef CAS
- T. Liu and B. Thierry, Langmuir, 2012, 28, 15634–15642 CrossRef CAS PubMed
- M. Meyns, N. G. Bastus, Y. Cai, A. Kornowski, B. H. Juarez, H. Weller and C. Klinke, J. Mater. Chem., 2010, 20, 10602–10605 RSC
- P. K. Jain, J. Phys. Chem. Lett., 2014, 5, 3112–3119 CrossRef CAS
- N. J. Hogan, A. S. Urban, C. Ayala-Orozco, A. Pimpinelli, P. Nordlander and N. J. Halas, Nano Lett., 2014, 14, 4640–4645 CrossRef CAS PubMed
- S. L. Westcott, J. B. Jackson, C. Radloff and N. J. Halas, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 66, 155431 CrossRef
- C. Galland, Y. Ghosh, A. Steinbrueck, J. A. Hollingsworth, H. Htoon and V. I. Klimov, Nat. Commun., 2012, 3, 908 CrossRef PubMed
- A. O. Govorov and H. H. Richardson, Nano Today, 2007, 2, 30–38 CrossRef
- S. V. Boriskina, H. Ghasemi and G. Chen, Mater. Today, 2013, 16, 375–386 CrossRef CAS PubMed
- A. R. Lowery, A. M. Gobin, E. S. Day, N. J. Halas and J. L. West, Int. J. Nanomed., 2006, 1, 149–154 CrossRef CAS
- L. R. Hirsch, R. J. Stafford, J. A. Bankson, S. R. Sershen, B. Rivera, R. E. Price, D. J. Hazle, N. J. Halas and J. L. West, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13549–13554 CrossRef CAS PubMed
- D. Hanaor, M. Michelazzi, C. Leonelli and C. C. Sorrell, J. Eur. Ceram. Soc., 2012, 32, 235–244 CrossRef CAS PubMed
- U. Hohenester and A. Truegler, Comput. Phys. Commun., 2012, 183, 370–381 CrossRef CAS PubMed
- E. J. McLaurin, L. R. Bradshaw and D. R. Gamelin, Chem. Mater., 2013, 25, 1283–1292 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available: Further information on Au shelling chemistry and imaging of the Au shell by electron microscopy. Figures and Movie. See DOI: 10.1039/c5sc00020c |
| This journal is © The Royal Society of Chemistry 2015 |