
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEffect of phenolic glycolipids from Mycobacterium kansasii on proinflammatory cytokine release. A structure–activity relationship study†
Hassan R. H.
Elsaidi
and
Todd L.
Lowary
*
Alberta Glycomics Centre and Department of Chemistry, University of Alberta, Edmonton, AB, Canada T6G 2G2. E-mail: tlowary@ualberta.ca
First published on 26th March 2015
Abstract
The cell wall of pathogenic mycobacteria is abundant with virulence factors, among which phenolic glycolipids (PGLs) are prominent examples. Mycobacterium kansasii, an important opportunistic pathogen, produces seven PGLs and their effect on the release of important proinflammatory cytokines that mediate disease progression has not been investigated. We previously showed that proinflammatory cytokines are modulated by PGLs from M. tuberculosis, M. leprae and M. bovis. In this paper we describe the synthesis of a series of 17 analogs of M. kansasii PGLs containing a truncated aglycone. Subsequently, the effect of these compounds on the release of proinflammatory cytokines (TNF-α, IL-6, IL-1β, MCP-1) and nitric oxide (NO) was evaluated. These compounds exerted an immunoinhibitory effect on the release of the tested cytokines. The concentration-dependent inhibitory profile of the tested molecules was also found to be dependent on the methylation pattern of the molecule and was mediated via toll-like receptor (TLR)-2. This study led to the discovery of a glycolipid (18) that shows promising potent anti-inflammatory properties making it a potential candidate for further optimization of its anti-inflammatory profile.
Introduction
Emerging as an important non-tuberculous mycobacterium (NTM), Mycobacterium kansasii is the second most pathogenic NTM infecting humans after M. avium.1,2 Infections by M. kansasii cause chronic pulmonary illness that are indistinguishable from tuberculosis and have been suggested to have similar pathogenesis to M. tuberculosis.3–5 Phenolic glycolipids (PGLs) are important virulence factors expressed on the cell wall of M. kansasii. The structures of seven M. kansasii PGLs (1–7, Fig. 1) have been isolated and characterized to date.6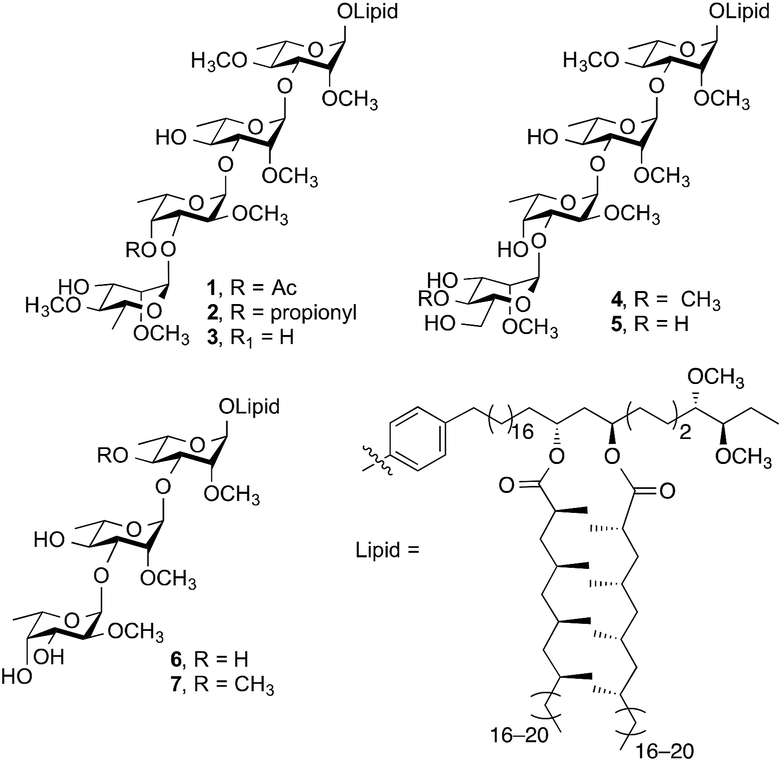 | ||
| Fig. 1 Structures of PGLs from M. kansasii. | ||
Despite the work done by Puzo7 and Minnikin8 on structural elucidation of M. kansasii PGLs, the impact of these molecules on proinflammatory cytokine release has not been studied. In a previous investigation,9 Barry and coworkers showed that disrupting PGL synthesis resulted in the loss of the hyperlethality of W-Beijing M. tuberculosis, which also correlated to increased release of proinflammatory cytokines tumor necrosis factor alpha (TNF-α), interleukin-6 (IL-6) and interleukin-12 (IL-12). Moreover, overproduction of PGLs by M. tuberculosis resulted in a dose-dependent inhibition of these proinflammatory cytokines.9 Recently, Cambier et al.10 showed that M. tuberculosis and M. marinum are capable of recruiting and infecting permissible macrophages and evading microbicidal ones through the use of phthiocerol dimycocerosate (PDIM) and structurally related PGL cell wall components. It was shown that PDIM evades the host immune response through masking the underlying pathogen-associated molecular pattern (PAMPs) thus altering the recruitment of microbicidal macrophages. On the other hand, PGLs increase infectivity through recruiting the permissible macrophages via chemokine receptor 2 (CCR2). Furthermore, Arbues et al.11 have recently reviewed the immunomodulatory properties of both PDIM and PGLs and proposed that PGLs might act as a ligand for carbohydrate-recognizing receptors on the surface of macrophages. It was also proposed that new strategies are needed for further assessment of the role of these biomolecules in virulence and immunomodulation.
Our previous work with truncated synthetic PGL analogs from M. leprae, M. bovis and M. tuberculosis12,13 established that these PGLs inhibit the release of nitric oxide (NO) as well as the proinflammatory cytokines TNF-α, IL-6, interleukin-1 beta (IL-1β) and chemokine monocyte chemotactic protein-1 (MCP-1) in a concentration-dependent manner through toll-like receptor 2 (TLR-2). Understanding the effect of PGLs from M. kansasii on the release of proinflammatory cytokines would lead to better understanding of their role in the progression of these infections. Moreover, the tested cytokines are also implicated in some inflammatory conditions14–20 such as rheumatoid arthritis (RA).2,21 Therefore, this study might lead to the discovery of compounds with potential use for treatment of these inflammatory conditions.
We report here the synthesis of a 17-member library of M. kansasii PGL analogs (8–24, Fig. 2). Seven of the targets (8, 9, 14, 15, 17, 20 and 23) are direct analogs of the native PGLs while the remaining ten have different methylation and substitution patterns to allow structure–activity relationships to be probed. Following the synthesis of the target compounds, their effect on proinflammatory cytokine release was assessed and the receptor that mediates the immunomodulatory activity of these compounds was determined. All of the targets were prepared with a simplified lipid core in which the complex phthiocerol dimycocerosate domain was replaced with a p-methoxyphenyl group. This modification simplifies the synthesis of the target compounds considerably and improves aqueous solubility, a key consideration in the use of these compounds in biological assays. Previous work with similar derivatives of M. leprae-derived PGLs suggested that this modification did not substantially alter the cytokine modulating activity compared to the parent compound present in nature.12 Hence these analogs are effective surrogates for the more complex natural products.
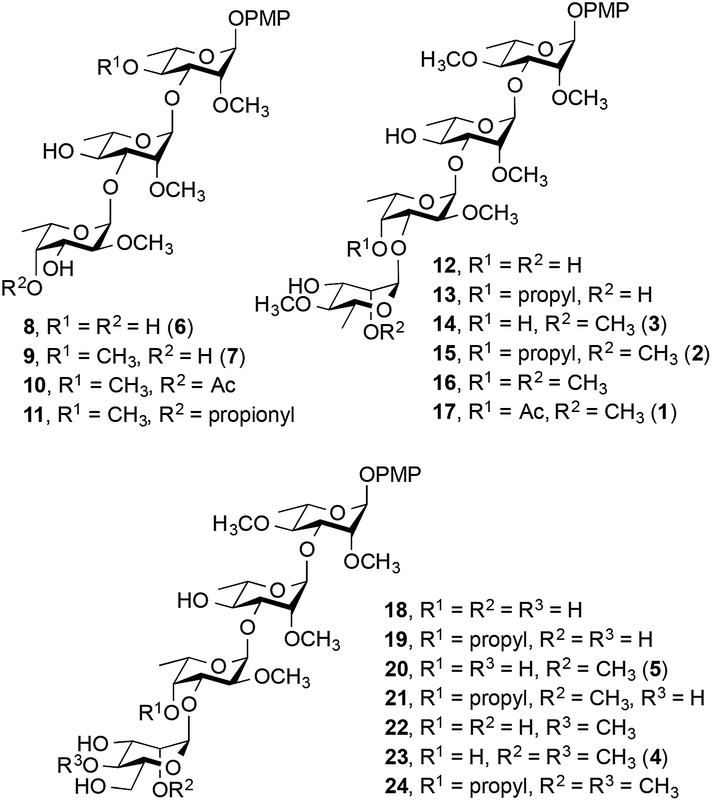 | ||
| Fig. 2 M. kansasii PGL analogs synthesized (8–24). The numbers in parentheses correspond to the natural compounds in Fig. 1. | ||
Results and discussion
Representative examples of how target compounds were synthesized are shown in Scheme 1, which illustrates the synthesis of 12–17. Details of the routes used for the synthesis of the other targets and all building blocks are provided in the ESI.† The synthesis commenced with an NIS–AgOTf-mediated glycosylation of disaccharide acceptor 26 with monosaccharide donor 25, followed by removal of the p-methoxybenzyl group furnishing trisaccharide 27 in 76% yield over two steps. Subsequent construction of tetrasaccharide 29 was achieved in 61% yield via glycosylation of trisaccharide 27 with the 6-deoxy-D-mannose thioglycoside 28. With 29 in hand, accessing targets 12 and 13 was possible via different deprotection sequences. Base-catalysed hydrolysis of C-3‴ benzoate followed by removal of allyl group using (Ph3P)4Pd (ref. 23) and finally hydrogenolysis were employed to obtain compound 12. On the other hand, base-catalysed hydrolysis of the C-3‴ benzoate followed by hydrogenolysis afforded compound 13. Accessing targets 14 to 17 required a methyl group to be added at O-2‴ of 29. This was achieved via deacylation and then treatment with methyl iodide and sodium hydride to give tetrasaccharide 30 in 81% yield. With 30 in hand, different deprotection protocols were employed to provide 14–17. Removal of the allyl group catalysed by (Ph3P)4Pd followed by hydrogenolysis gave compound 14, whereas direct hydrogenolysis afforded compound 15. Methylation of O-4″ after removal of allyl group using (Ph3P)4Pd and finally removal of all benzyl groups via hydrogenolysis gave compound 16. Finally, compound 17 was obtained after acylation of O-4″ following the removal of allyl group and then debenzylation using Pd–C under hydrogen atmosphere. The stereochemistry of all the newly synthesized glycosidic linkages were α, as confirmed by measuring the 1JC-1,H-1 for each monosaccharide residue using a coupled HSQC NMR experiment. The values, 170–171.5 Hz, agree with values typical of an α linkage.24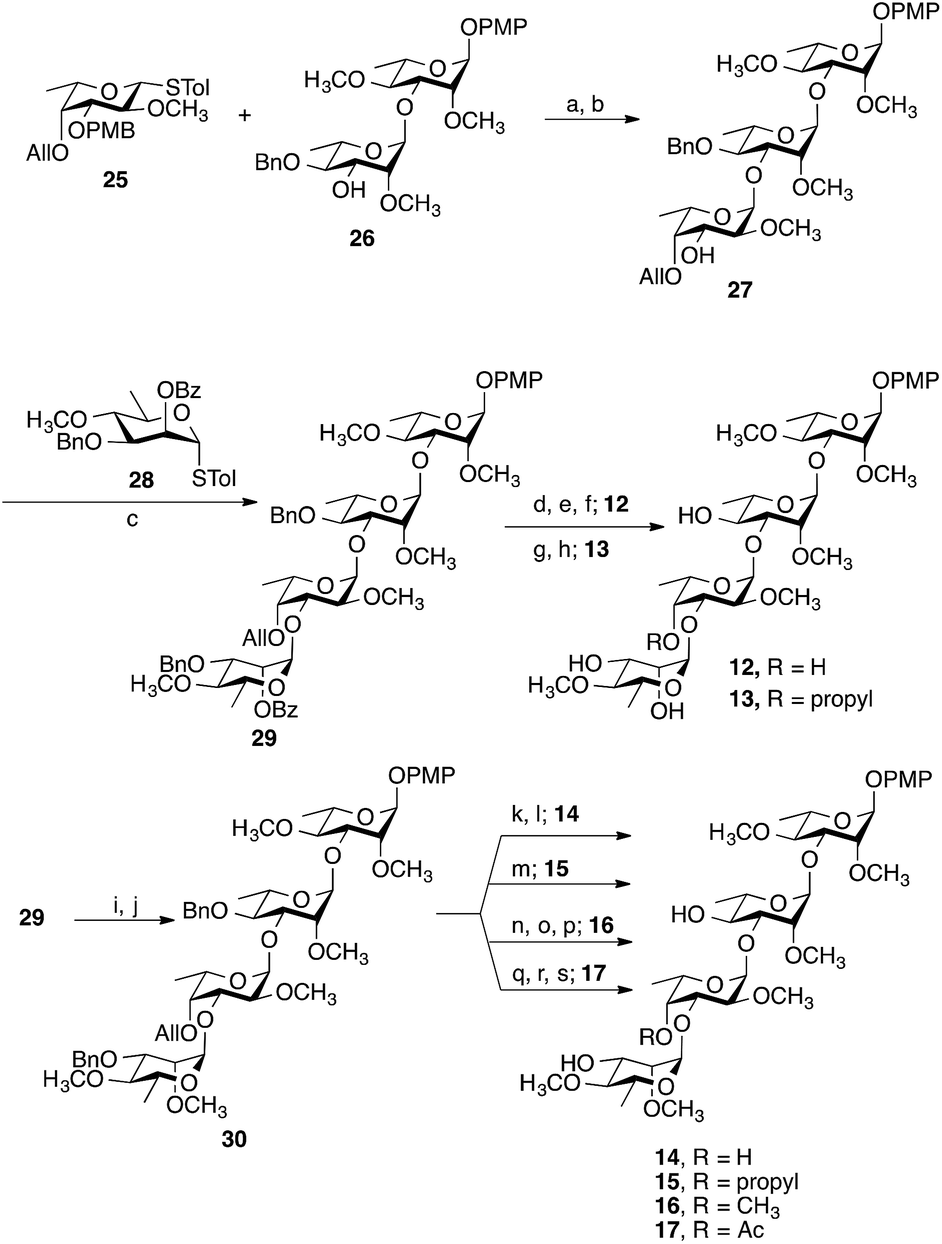 | ||
| Scheme 1 Synthesis of 12–17. Reagents and conditions: (a) NIS, AgOTf, −40 °C, 30 min; (b) 5% TFA, CH2Cl2, 0 °C, 20 min, 76% over two steps from 26; (c) NIS, AgOTf, CH2Cl2, −40 °C, 30 min, 61%; (d) NaOCH3, CH3OH, CH2Cl2, 3 h; (e) (Ph3P)4Pd, HOAc, overnight; (f) Pd–C, H2, CH3OH, CH2Cl2, overnight, 68% over three steps from 29; (g) NaOCH3, CH3OH/CH2Cl2, 3 h; (h) Pd–C, H2, CH3OH, CH2Cl2, overnight, 87% over two steps from 29; (i) NaOCH3, CH3OH, CH2Cl2, 5 h; (j) CH3I, NaH, DMF, 1 h, 81% over two steps from 29; (k) (Ph3P)4Pd, HOAc, overnight; (l) Pd–C, H2, CH3OH, CH2Cl2, overnight, 79% over two steps from 30; (m) Pd–C, H2, CH3OH, CH2Cl2, overnight, 78%; (n) (Ph3P)4Pd, HOAc, overnight; (o) CH3I, NaH, DMF, 1 h; (p) Pd–C, H2, CH3OH, CH2Cl2, overnight, 71% over three steps from 30; (q) (Ph3P)4Pd, HOAc, overnight; (r) Ac2O, pyridine, 2 h; (s) Pd–C, H2, CH3OH, CH2Cl2, overnight, 73% over three steps from 30. | ||
After accessing 8–24, each was tested for their effect on the release of NO and proinflammatory cytokines that have been shown to play an important role in the progression of mycobacterial infections: TNF-α, IL-6, IL-1β and MCP-1.9,25 To this end, the activated THP-1 (human acute leukemic monocyte/macrophage, A-THP-1) cell line was employed as it was widely used as model for macrophages.26 Upon challenge of A-THP-1 with the test compounds, the levels of the cytokines produced were comparable to the negative control (Fig. 3A), thus demonstrating the inability of these analogs to stimulate the release of TNF-α, IL-6, IL-1β and MCP-1 and NO.
 | ||
| Fig. 3 (A) Cytokine stimulation assay. THP-1 cells were differentiated (activated) into mature macrophages (A-THP-1) via treatment with 5 ng mL−1 of phorbol myristate acetate (PMA) for 18 h, treated with each synthetic compound at a concentration of 50 µg mL−1 (dissolved in 0.1% DMSO in RPMI), and then incubated for 24 h. Culture supernatants were then collected and tested for cytokine levels using ELISA. (B) TNF-α inhibition assay using Pam3CSK4, a TLR2 agonist, as the stimulant for compounds 8–11. Test compounds were added at concentrations of 10 or 50 µg mL−1 to A-THP-1 cells in presence of the stimulant. After 24 h incubation, supernatants were collected and analyzed for TNF-α using ELISA (see Experimental section for additional details). Each experiment was repeated twice and the concentration from each experiment was determined as triplicate using ELISA. The concentrations represented are the average of the six readings (±S.D.). (C) TNF-α inhibition assay of compounds 12–17. (D) TNF-α inhibition assay of compounds 18–24. *P > 0.05, **P < 0.05, ***P < 0.01, ****P < 0.0001. | ||
The potential of 8–24 to inhibit the release of these species was then evaluated by challenging A-THP-1 cells with the test compounds in the presence of a stimulant and measuring the concentrations of cytokines produced compared to a positive control. Trisaccharides 8–11 were evaluated first, using Pam3CSK4, a synthetic toll-like receptor 2 (TLR2) agonist27 as the stimulant. Fig. 3B shows the data obtained for TNF-α.
Compound 9 corresponding directly to the natural product 6 showed a low level of inhibitory activity on the release of TNF-α when tested at 50 µg mL−1, p < 0.01. Trisaccharide 8, which, compared to 9, lacks the methyl group at O-4 showed very weak inhibition of TNF-α production, p < 0.05. On the other hand, trisaccharide 9 showed a weak to moderate inhibition of TNF-α release. This result underscores the relative importance of the O-4 methyl group on the activity. Compounds 10 and 11 differ from 9 by the presence of an ester group at O-4″. The relative lack of activity of these compounds could be due to the size of the acyl group, which prohibits the binding of the molecule to TLR2. Similar trends were observed for 8–11 with regard to the other cytokines (data provided in the ESI, Fig. S1, S4, S7 and S10†).
Tetrasaccharides 12–17 were then evaluated using the same assay (Fig. 3C). Compounds 14 and 17 are analogs of the naturally occurring compounds 3 and 1, respectively. Unnatural analogs 13, 15 (contain a propyl group at O-4″) and 12, 16 (have different methylation pattern at both O-4″ and O-2‴) were also evaluated. These tetrasaccharides could be conveniently synthesized and were prepared to evaluate the importance of different substituents at these positions. As illustrated in Fig. 3C, a concentration-dependent, structure-dependent inhibitory pattern was obtained. Tetrasaccharide 16 showed the highest activity. At a concentration of 50 µg mL−1, 16 inhibited the release of TNF-α from the stimulated A-THP-1 cells to a level equivalent to the negative control. This molecule has all hydroxyl groups methylated except O-4′ and O-3‴. Removal of the O-4″ methyl group in 16 (compound 14), resulted in a significant loss of inhibitory activity. Replacing the methyl group at this position with a propyl group (compound 15) also led to a loss of activity. This latter result suggests that the observed differences between 14 and 16 are structure-dependent and not a non-specific effect resulting from the enhanced hydrophobicity of 16 compared to 14. Analogs 13 and 17, which differ in the substituents at O-4″ and O-2‴, exhibited similar inhibitory potency. Compound 13 has a propyl group at O-4 and analog 17 has an acetyl group compared to methyl group in compound 16. The lower inhibitory activity of both 13 and 17 illustrate the importance of methyl group at O-4 for binding to TLR2 and that any change in this group to a bigger alkyl group or an ester group greatly reduces the immunomodulatory activity. Finally, compound 12, which, compared to 16 lacks methyl groups at O-4″ and O-2‴, has the lowest level of activity amongst this series of compounds indicating the importance of these two hydrophobic groups on the inhibitory activity and underscoring the structure-dependent nature of the effect. As was observed for 8–11, the trends observed in TNF-α inhibition of 12–17 were also seen with the other cytokines evaluated, as well as NO (ESI, Fig. S2, S5, S8 and S11†).
Finally, tetrasaccharides 18–24 were evaluated (Fig. 3D). These compounds differ from 12–17 in that the 6-deoxy-α-D-mannopyranose capping residue is replaced with an α-D-mannopyranose motif. Evaluation of these compounds using Pam3CSK4 stimulation revealed that 23 showed the highest level of inhibition of TNF-α production. This compound lacks the O-4″ methyl group, but is methylated at both O-2‴ and O-4‴. Replacing the methyl group at O-4″ with a propyl group (compound 24), resulted in a significant loss of activity. Compound 18, which is not methylated at O-4″, O-2‴ or O-4‴ showed only weak inhibition activity. Both 19 and 20 have a propyl group at O-4″ and they showed weak activity, which agrees with the results obtained previously with compounds 13 and 15. Moreover, the absence of O-4″ methyl and O-2‴ or O-4‴ methyl groups resulted in the same effect of lowering the activity as shown for compounds 21 and 22. Finally, the weak-to-moderate activity of this series indicates that dimethylated 6-deoxy-α-D-mannopyranose residue leads to enhanced cytokine inhibition compared to those possessing a dimethylated mannose (12–17). Analogous patterns were observed for other tested cytokines, (IL-6, IL-1β, MCP-1 and NO; ESI, Fig. S3, S6, S9 and S12†).
Previous reports have shown that mycobacterial components such as lipomannan and lipoarabinomannan mediate their macrophage activation effects via TLR2.28 Previously, we showed that the immunoinhibitory activity of PGLs from M. tuberculosis, M. leprae and bovis is mediated via TLR2.12,13 To confirm that 8–24 exert their effect on cytokine and NO release through a similar pathway, they were tested using ultra-pure E. coli LPS, a TLR4 agonist,29 as the stimulant. As detailed in the ESI (Fig. S13–S24†), none of the compounds possessed any inhibitory activity thus confirming TLR2 mediates the immunomodulatory activity of 8–24.
When evaluated in the assay described above, the lipid core of native PGLs – phenolphthiocerol dimycocerosate (PDIM) – is a weak inhibitor of cytokine/NO release.12 However, M. leprae PGL-1 (31, Scheme 2), which contains PDIM glycosylated with a highly methylated trisaccharide, is a potent inhibitor.10 In previous work, we demonstrated that adding a simple lipid core to the glycan structure of PGL-1 resulted in a compound (32) with significantly more inhibitor activity than the carbohydrate domain (33) alone.12 We therefore pursued an analogous approach for the M. kansasii PGL structures and selected 34, which consists of the glycan portion of 16 conjugated to alkylphenol 35. Compound 16 was selected for this study as it possessed the most potent activity in the assays described above.
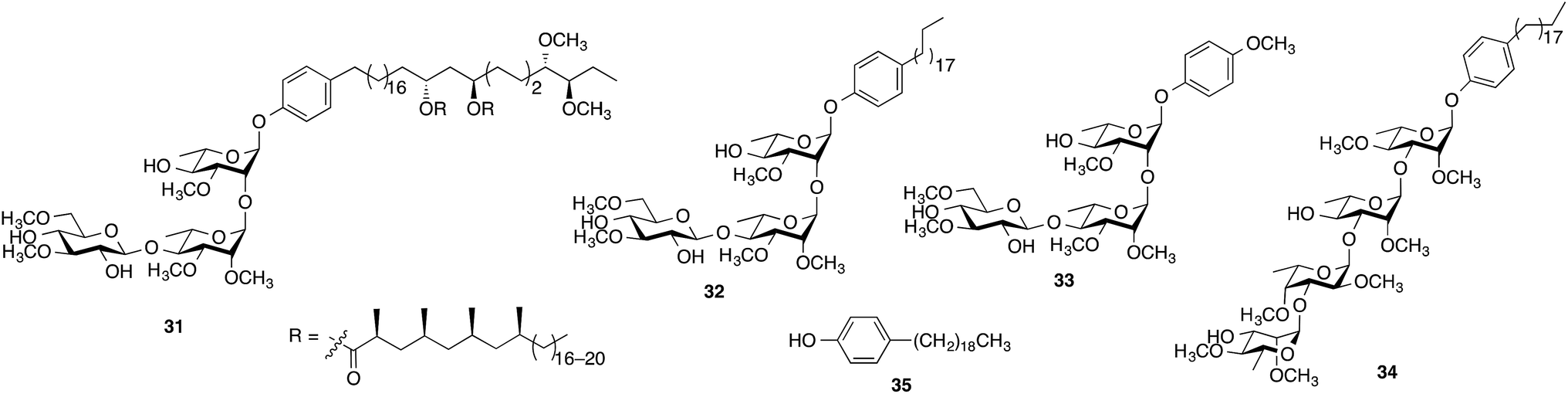 | ||
| Scheme 2 Structures of compounds 31–35. | ||
The synthesis of 34 is described in the ESI.† Its immunoinhibitory activity was tested at three concentrations, 10, 25 and 50 µg mL−1, using Pam3CSK4 as the agonist and the results were compared to glycan 16. As illustrated in Fig. 4, both 16 and 34 showed a concentration-dependent inhibitory pattern on the release of TNF-α and similar patterns were also obtained for the other cytokines and NO (ESI, Fig. S25–S28†). As we previously established for an M. leprae PGL analog,12 addition of a simple lipid domain (35) to carbohydrate analogs resulted in an increase of the inhibitory activity. Glycolipid 34 showed higher activity as well compared to 16. Moreover, glycolipid 34 was shown to be a potent inhibitor of the release of cytokines, causing inhibition of ∼67% of the induced TNF-α at a concentration of 10 µg mL−1.
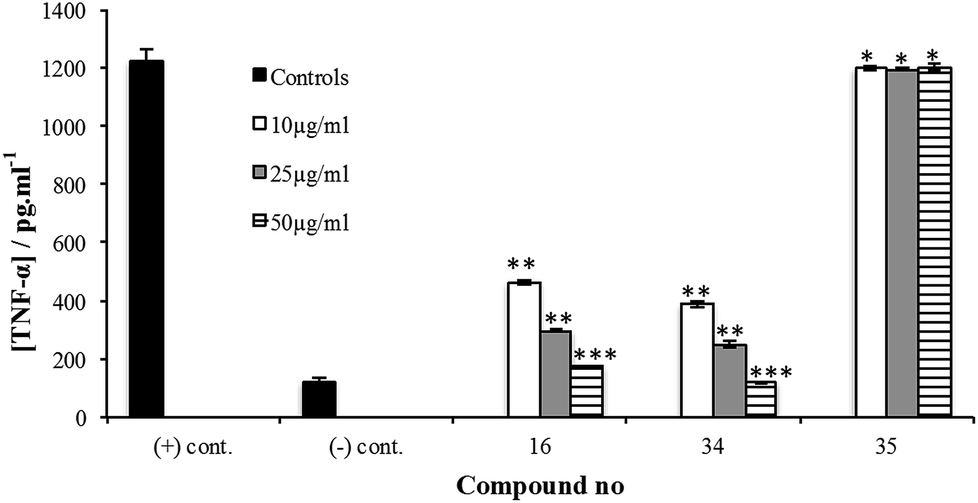 | ||
| Fig. 4 Cytokine inhibition assay of compound 16, glycolipid 34 and phenol 35 using Pam3CSK4, a TLR2 agonist, as stimulant. Test compounds were added at concentrations of 10, 25 or 50 µg mL−1 to the A-THP-1 in presence of the stimulant. After 24 h incubation, supernatants were collected and analyzed for TNF-α using ELISA. Each experiment was repeated twice and the concentration from each experiment was determined as triplicate using ELISA. The concentrations represented are the average of the six readings (±S.D.). *P > 0.05, **P < 0.001, ***P < 0.0001. | ||
Conclusion
We report here the synthesis of a panel of 17 synthetic analogs of PGLs from M. kansasii. This work represents the first synthetic investigations of M. kansasii PGLs. With these compounds in hand, we probed structure–activity relationships on the ability of these compounds to modulate cytokine and NO release. The investigations lead to the discovery of an active anti-inflammatory compound, 34, which targets proinflammatory cytokines implicated in many inflammatory and autoimmune conditions. The cytokine inhibition assays revealed that 8–24 inhibited the release of TNF-α, IL-6, MCP-1, IL-1β and NO to varying degrees in a concentration-dependent pattern. The methylation pattern of the carbohydrate domain was found to be a crucial structural feature that determines potency. Furthermore, the methyl group was found to be the only tolerated substituent; ester and larger alkyl groups resulted in significant loss of activity. In addition, the 6-deoxy-2,4-di-O-methyl-mannopyranoside capping motif was found to be crucial for the highest activity.The current study, as well as our previous studies,12,13 suggest that PGLs from M. tuberculosis, M. leprae, M. bovis, and M. kansasii attenuate the host immunity by inhibiting the release of important proinflammatory cytokines. The glycone part of PGLs was found to be the structural determinant for this activity and different methylation patterns of the glucan determines the potency of PGL. TLR2 was also shown to be the receptor mediating this activity and adding a simple lipid core to the most active glycans resulted in significant enhancement of the potency. Fig. 5 summarizes all the findings from our studies and provides a general pharmacophore for this class of compounds.
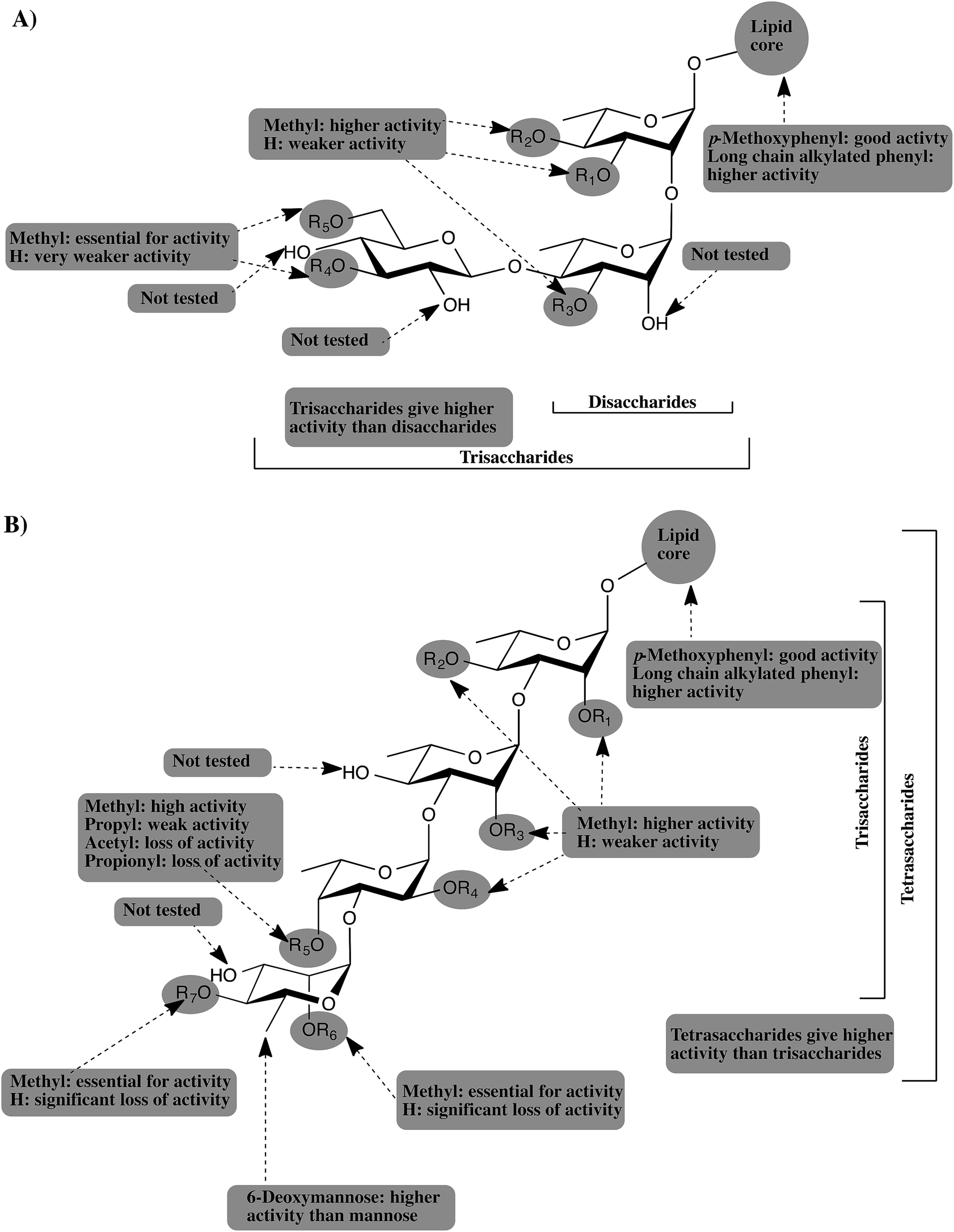 | ||
| Fig. 5 Schematic representation of the generic pharmacophore for PGLs from: (A) M. leprae, and M. bovis. (B) M. tuberculosis and M. kansasii. | ||
As depicted in Fig. 5A, PGLs from M. leprae and M. bovis are based upon a core structure of α-(1 → 2)-linked-L-rhamnopyranose (Rhap) residues. Trisaccharide PGLs showed higher cytokine inhibition activity than the disaccharide PGLs. The 3,6-di-O-methylated glucopyranose capping motif present in M. leprae PGLs, possess the greatest inhibitory potential. M. bovis produces three PGLs with a single Rhap residue, but those possess essentially no ability to inhibit cytokine release.12 On the other hand, PGLs from M. tuberculosis and M. kansasii (Fig. 5B) consist of an α-Rhap-(1 → 3)-α-Rhap-(1 → 3)-α-Rhap structure, which in the case of some M. kansasii is further functionalized by either a D-mannopyranose or 6-deoxy-D-mannopyranose residue. Comparison of the trisaccharides and tetrasaccharides from M. tuberculosis and M. kansasii PGLs revealed that the highest activity is obtained from the tetrasaccharides, particularly when a 6-deoxy-D-mannopyranose residue is present as a capping motif. Regardless of the size of the PGL, methylation of the glycan structure was found to be an important determinant for the activity, with higher levels of methylation generally leading to more potent compounds. However, the activity is dependent upon a distinct methylation pattern and, moreover, appears not to be a generally hydrophobic effect. For example, compounds containing a propyl group in place of a methyl group have significantly lower levels of activity. Finally, addition of a simple lipid structure such as long chain alkylated phenyl group significantly enhanced the immunoinhibitory activity.
Compounds such as 34 represent a potential novel class of anti-inflammatory agents that could be used to treat diseases such as rheumatoid arthritis (RA), a chronic inflammatory autoimmune disease.21,22 A growing body of evidence has shown that proinflammatory cytokines, including those evaluated here (TNF-α, IL-1β IL-6, MCP-1), are directly implicated in RA pathogenesis.14–20 Accordingly, species that inhibit the release of these cytokines are potential RA therapeutics. To date, five anti-TNF-α, one anti-IL-6 and two anti-IL-1β drugs are in development for the treatment of RA, all of which are antibodies or soluble proteins.14,16,19 Only a single orally active small molecule inhibitor of IL-12/IL-23 is being evaluated in phase 2 clinical trials in patients with Crohn's disease and RA.30 Further investigations on the anti-inflammatory potential of this class of compounds are on going.
Experimental
General methods
Solvents used in reactions were purified by successive passage through columns of alumina and copper under an argon atmosphere. All reagents were purchased from commercial sources and were used without further purification unless noted otherwise. All reactions were carried out under a positive pressure of argon atmosphere and monitored by TLC on Silica Gel G-25 UV254 (0.25 mm) plates unless stated otherwise. Spots were detected under UV light and/or by charring with a solution of anisaldehyde in ethanol, acetic acid, and H2SO4. Column chromatography was performed on silica gel 60 (40–60 mm; silica gel–sample, 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1–20:1, w/w). Organic solutions were concentrated under vacuum below 50 °C. 1H NMR spectra were recorded at 400 or 500 MHz, and 1H NMR chemical shifts are referenced to TMS (0.0, CDCl3). 13C NMR spectra were recorded at 125.7 MHz, and chemical shifts are referenced to CDCl3 (77.23, CDCl3). 1H NMR data are reported as though they are first order, and the peak assignments were made on the basis of 2D NMR (1H, 1H COSY and HMQC) experiments. In reporting 13C chemical shifts, when two signals were so closely spaced that rounding to one decimal place would have led to identical values, the second decimal place is included in parentheses, e.g., 78.9(1) and 78.8(8). Optical rotations were measured at (21 ± 2) °C at the sodium D line (589 nm), and are in deg mL (dm g)−1. ESI-MS spectra were carried out on samples suspended in THF or CH3OH and added NaCl.
:1–20:1, w/w). Organic solutions were concentrated under vacuum below 50 °C. 1H NMR spectra were recorded at 400 or 500 MHz, and 1H NMR chemical shifts are referenced to TMS (0.0, CDCl3). 13C NMR spectra were recorded at 125.7 MHz, and chemical shifts are referenced to CDCl3 (77.23, CDCl3). 1H NMR data are reported as though they are first order, and the peak assignments were made on the basis of 2D NMR (1H, 1H COSY and HMQC) experiments. In reporting 13C chemical shifts, when two signals were so closely spaced that rounding to one decimal place would have led to identical values, the second decimal place is included in parentheses, e.g., 78.9(1) and 78.8(8). Optical rotations were measured at (21 ± 2) °C at the sodium D line (589 nm), and are in deg mL (dm g)−1. ESI-MS spectra were carried out on samples suspended in THF or CH3OH and added NaCl.
p-Methoxyphenyl 6-deoxy-4-O-methyl-α-d-mannopyranosyl-(1 → 3)-2-O-methyl-α-l-fucopyranosyl-(1 → 3)-2-O-methyl-α-l-rhamnopyranosyl-(1 → 3)-2,4-di-O-methyl-α-l-rhamnopyranoside (12)
To a solution of 29 (20 mg, 0.02 mmol) in 1:1 CH3OH–CH2Cl2 (10 mL), 1 M NaOCH3 (0.1 mL) was added and the reaction mixture was stirred for 3 h at rt. The reaction mixture was then neutralized by the addition of Amberlite IR-120 H+ resin, filtered and concentrated. The resulting residue was dissolved in AcOH (3 mL) and then (Ph3P)4Pd (4 mg) was added. The reaction mixture was stirred overnight and then filtered and water (5 mL) was added. The solution was diluted with CH2Cl2 (10 mL), washed with water (2 × 8 mL) and brine (10 mL). The organic layer was separated, dried (NaSO4), concentrated and the resulting residue was purified by chromatography (2:1 hexane–EtOAc) to give a colorless oil. To the solution of the oil in 1:1 CH3OH–CH2Cl2 (15 mL), Pd–C (5 mg) was added and the reaction mixture was stirred overnight under a hydrogen atmosphere before it was filtered and concentrated. The resulting crude product was purified by chromatography (20:1 CH2Cl2–CH3OH) to give 12 (9.6 mg, 62%) as a colorless oil: Rf 0.30 (20:1 CH2Cl2–CH3OH); [α]D −36.4 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3, δH) 6.99–6.96 (m, 2H, Ar-2,6), 6.83–6.80 (m, 2H, Ar-3,5), 5.38 (d, 1H, J1,2 = 1.6 Hz, H-1), 5.17 (br s, 1H, H-1′), 5.08 (d, 1H, J1″,2″ = 3.3 Hz, H-1″), 5.05 (d, 1H, J1‴,2‴ = 1.6 Hz, H-1‴), 4.19 (dq, 1H, J4″,5″ = 3.0 Hz, J5″,6″ = 6.3 Hz, H-5″), 4.08 (dd, 1H, J2′,3′ = 3.1 Hz, J3′,4′ = 9.4 Hz, H-3′), 4.05 (dd, 1H, J2,3 = 3.3 Hz, J3,4 = 9.5 Hz, H-3) 3.99–3.98 (m, 1H, H-3″), 3.87–3.84 (m, 1H, H-5′, H-5‴), 3.79–3.78 (m, 1H, H-5), 3.79 (s, 3H, OCH3), 3.73–3.65 (m, 3H, H-2, H-2′, H-2‴), 3.66–3.62 (m, 2H, H-2″, H-3‴), 3.61–3.57 (m, 2H, H-4′, H-4″), 3.57 (s, 3H, OCH3), 3.54 (s, 3H, OCH3), 3.51 (s, 3H, OCH3), 3.49 (s, 3H, OCH3), 3.46 (s, 3H, OCH3), 3.22 (app t, 1H, J3,4 = J4,5 = 9.5 Hz, H-4), 3.11 (app t, 1H, J3‴,4‴ = J4‴,5‴ = 9.6 Hz, H-4‴), 2.90 (br s, 1H, OH), 2.30 (br s, 1H, OH), 1.77 (br s, 2H, OH × 2), 1.37–1.32 (m, 6H, H-6′, H-6″), 1.27–1.24 (m, 6H, H-6, H-6‴); 13C NMR (125.7 MHz, CDCl3, δC) 154.9 (Ar), 150.4 (Ar), 117.5 (Ar × 2), 114.6 (Ar × 2), 101.2, 100.3, 98.9, 95.6, 83.1(0), 83.0(4), 82.3, 80.5, 80.3, 79.1, 78.6, 77.5, 72.0, 71.4, 71.3, 71.1, 69.0, 68.7, 68.4, 66.2, 61.1 (OCH3), 61.0 (OCH3), 59.6 (OCH3), 59.0 (OCH3), 58.4 (OCH3), 55.7 (OCH3), 18.0, 17.9, 17.8, 16.3. HRMS (ESI) calcd for (M + Na)+ C36H58NaO18: 801.3515. Found 801.3505.
p-Methoxyphenyl 6-deoxy-4-O-methyl-α-d-mannopyranosyl-(1 → 3)-2-O-methyl-4-O-propyl-α-l-fucopyranosyl-(1 → 3)-2-O-methyl-α-L-rhamnopyranosyl-(1 → 3)-2,4-di-O-methyl-α-L-rhamnopyranoside (13)
To a solution of 29 (12 mg, 0.011 mmol) in 1:1 CH3OH–CH2Cl2 (10 mL), 1 M NaOCH3 (0.1 mL) was added and the reaction mixture was stirred for 3 h at rt. The reaction mixture was neutralized by the addition of Amberlite IR-120 H+ resin, filtered and concentrated. The resulting residue was purified by chromatography (1:1 hexane–EtOAc) to give oil. This oil was dissolved in 1:1 CH3OH–CH2Cl2 (15 mL), Pd–C (3 mg) was added and the reaction mixture was stirred overnight under hydrogen before it was filtered and concentrated. The resulting residue was purified by chromatography (20:1 CH2Cl2–CH3OH) to give 13 (7.8 mg, 87%) as a colorless oil: Rf 0.38 (20:1 CH2Cl2–CH3OH); [α]D −28.2 (c 0.4, CHCl3); 1H NMR (500 MHz, CDCl3, δH) 6.99–6.96 (m, 2H, Ar-2,6), 6.83–6.80 (m, 2H, Ar-3,5), 5.39 (br s, 1H, H-1), 5.16 (br s, 1H, H-1′), 5.07 (d, 1H, J1″,2″ = 3.5 Hz, H-1″), 5.03 (br s, 1H, H-1‴), 4.14 (dd, 1H, J2′,3′ = 3.2 Hz, J3′,4′ = 9.5 Hz, H-3′), 4.10 (dd, 1H, J2,3 = 3.1 Hz, J3,4 = 9.4 Hz, H-3), 4.07 (dq, 1H, J4″,5″ = 2.9 Hz, J5″,6″ = 6.5 Hz, H-5″), 4.00–3.98 (m, 1H, H-3″), 3.86 (dq, 1H, J4′,5′ = 9.4 Hz, J5′,6′ = 6.3 Hz, H-5′), 3.82–3.78 (m, 2H, H-5, H-5‴), 3.76 (s, 3H, OCH3), 3.70–3.65 (m, 5H, H-2, H-2′, H-2‴, CH2O × 2), 3.64–3.60 (m, 3H, H-2″, H-3‴, H-4′), 3.57 (s, 3H, OCH3), 3.53 (s, 3H, OCH3), 3.50 (s, 3H, OCH3), 3.49 (s, 3H, OCH3), 3.46 (s, 3H, OCH3), 3.40–3.37 (m, 1H, H-4‴), 3.21 (app t, 1H, J3,4 = J4,5 = 9.5 Hz, H-4), 3.10–2.98 (m, 1H, H-4″), 1.65–1.58 (m, 2H, CH2CH3), 1.33 (d, 3H, J5″,6″ = 6.5 Hz, H-6″), 1.27–1.24 (m, 6H, H-6, H-6′), 1.21 (d, 3H, J5‴,6‴ = 6.5 Hz, H-6‴), 0.93 (t, 3H, J = 7.5 Hz, CH2CH3); 13C NMR (125.7 MHz, CDCl3, δC) 154.9 (Ar), 150.4 (Ar), 117.5 (Ar × 2), 114.6 (Ar × 2), 101.2, 100.2, 99.3, 95.6, 83.4, 83.0, 82.2, 80.8, 80.4, 80.3, 80.0, 79.4, 76.4, 75.8 (CH2O), 71.4, 71.3(3), 71.3(0), 69.0, 68.7, 68.2, 67.5, 61.1 (OCH3), 60.9 (OCH3), 59.4 (OCH3), 58.9 (OCH3), 58.4 (OCH3), 55.7 (OCH3), 23.5 (CH2CH3), 18.0, 17.9, 17.8, 16.6, 10.8 (CH2CH3). HRMS (ESI) calcd for (M + Na)+ C39H64NaO18: 843.3985. Found 843.3978.
p-Methoxyphenyl 6-deoxy-2,4-di-O-methyl-α-d-mannopyranosyl-(1 → 3)-2-O-methyl-α-l-fucopyranosyl-(1 → 3)-2-O-methyl-α-L-rhamnopyranosyl-(1 → 3)-2,4-di-O-methyl-α-L-rhamnopyranoside (14)
To a solution of 30 (15 mg, 0.015 mmol) in HOAc (3 mL), (Ph3P)4Pd (3 mg) was added and the reaction mixture was stirred overnight at rt. The reaction mixture was then filtered, water (8 mL) was added and then it was diluted with CH2Cl2 (15 mL), washed with water (2 × 10 mL) and brine (10 mL). The organic layer was separated, dried (Na2SO4) and concentrated. The resulting residue was purified by chromatography (1:1 hexane–EtOAc) to give a colorless oil. The oil was dissolved in 1:1 CH2Cl2–CH3OH (20 mL) and Pd–C (4 mg) was added. The reaction mixture was stirred overnight under hydrogen before it was filtered, concentrated and the resulting residue was purified by chromatography (20:1 CH2Cl2–CH3OH) to give 14 (9.3 mg, 79%) as a thick syrup: Rf 0.44 (20:1 CH2Cl2–CH3OH); [α]D −23.5 (c 0.7, CHCl3); 1H NMR (500 MHz, CDCl3, δH) 7.00–6.97 (m, 2H, Ar-2,6), 6.83–6.80 (m, 2H, Ar-3,5), 5.39 (d, 1H, J1,2 = 1.8 Hz, H-1), 5.17–5.16 (m, 2H, H-1′, H-1‴), 5.07 (d, 1H, J1″,2″ = 3.1 Hz, H-1″), 4.20 (dq, 1H, J4″,5″ = 2.7 Hz, J5″,6″ = 6.5 Hz, H-5″), 4.11–4.06 (m, 2H, H-3, H-3′), 3.89–3.81 (m, 2H, H-3″, H-5′), 3.80–3.78 (m, 1H, H-5‴), 3.76 (s, 3H, OCH3), 3.71–3.67 (m, 4H, H-2, H-2′, H-2″, H-5), 3.66–3.63 (m, 3H, H-3‴, H-4′, H-4″), 3.60 (dd, 1H, J1‴,2‴ = 1.8 Hz, J2‴,3‴ = 3.3 Hz, H-2‴), 3.58 (s, 3H, OCH3), 3.54 (s, 3H, OCH3), 3.52 (s, 3H, OCH3), 3.50 (s, 3H, OCH3), 3.49 (s, 3H, OCH3), 3.47 (s, 3H, OCH3), 3.22 (app t, 1H, J3,4 = J4,5 = 9.5 Hz, H-4), 3.00 (app t, 1H, J3‴,4‴ = J4‴,5‴ = 9.7 Hz, H-4‴), 2.43 (d, 1H, J = 9.0 Hz, OH), 2.12 (br s, 1H, OH), 1.65 (br s, 1H, OH), 1.35 (d, 3H, J5′,6′ = 6.2 Hz, H-6′), 1.30 (d, 3H, J5,6 = 6.3 Hz, H-6), 1.27–1.25 (m, 6H, H-6″, H-6‴); 13C NMR (125.7 MHz, CDCl3, δC) 154.9 (Ar), 150.4 (Ar), 117.5 (Ar × 2), 114.6 (Ar × 2), 100.6, 99.4, 97.9, 95.6, 83.6, 83.3, 82.3, 80.7, 80.6, 80.3, 78.9(3), 78.8(7), 78.6, 71.6(3), 71.5(9), 71.2, 68.9, 68.7, 68.2, 66.3, 61.1 (OCH3), 61.0 (OCH3), 59.7 (OCH3), 59.0 (OCH3), 58.8 (OCH3), 58.6 (OCH3), 55.7 (OCH3), 17.9(6), 17.9(4), 17.9, 16.3. HRMS (ESI) calcd for (M + Na)+ C37H60NaO18: 815.3672. Found 815.3663.
p-Methoxyphenyl 6-deoxy-2,4-di-O-methyl-α-d-mannopyranosyl-(1 → 3)-2-O-methyl-4-O-propyl-α-l-fucopyranosyl-(1 → 3)-2-O-methyl-α-L-rhamnopyranosyl-(1 → 3)-2,4-di-O-methyl-α-L-rhamnopyranoside (15)
To a solution of 30 (10 mg, 0.01 mmol) in 1:1 CH2Cl2–CH3OH (10 mL), Pd–C (4 mg) was added and the reaction mixture was stirred overnight under hydrogen at rt before it was filtered and concentrated. The resulting residue was purified by chromatography (20:1 CH2Cl2–CH3OH) to give 15 (6.4 mg, 78%) as a colorless oil: Rf 0.51 (20:1 CH2Cl2–CH3OH); [α]D −25.0 (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3, δH) 6.99–6.95 (m, 2H, Ar-2,6), 6.82–6.79 (m, 2H, Ar-3,5), 5.38 (d, 1H, J1,2 = 1.7 Hz, H-1), 5.16–5.14 (m, 2H, H-1′, H-1‴), 5.05 (d, 1H, J1″,2″ = 3.5 Hz, H-1″), 4.15–4.06 (m, 3H, H-3, H-3′, H-5″), 3.86 (dq, 1H, J4′,5′ = 9.6, J5′,6′ = 6.2 Hz, H-5′), 3.87–3.77 (m, 2H, H-5, H-5‴), 3.75 (s, 3H, OCH3), 3.73–3.59 (m, 8H, H-2, H-2′, H-2″, H-4′, H-3″, H-3‴, CH2O × 2), 3.57 (s, 3H, OCH3), 3.52 (s, 3H, OCH3), 3.52–3.51 (m, 1H, H-2‴), 3.50 (s, 3H, OCH3), 3.47 (s, 3H, OCH3), 3.46 (s, 3H, OCH3), 3.45 (s, 3H, OCH3), 3.38–3.36 (m, 1H, H-4), 3.20 (app t, 1H, J3‴,4‴ = J4‴,5‴ 9.5 Hz, H-4‴), 3.10–2.96 (m, 1H, H-4″), 1.63–1.56 (m, 2H, CH2CH3), 1.34 (d, 3H, J5′,6′ = 6.2 Hz, H-6′), 1.31 (d, 3H, J5,6 = 6.2 Hz, H-6), 1.26 (d, 3H, J5″,6″ = 6.5 Hz, H-6″), 1.20 (d, 3H, J5‴,6‴ = 6.3 Hz, H-6‴), 0.92 (t, 3H, J = 7.5 Hz, CH2CH3); 13C NMR (125.7 MHz, CDCl3, δC) 154.9 (Ar), 150.4 (Ar), 117.5 (Ar × 2), 114.6 (Ar × 2), 100.5, 99.5, 97.5, 95.6, 83.8, 83.2, 82.2, 80.8, 80.7, 80.5, 80.3, 80.3, 79.3, 76.2, 75.8 (CH2O), 71.5, 71.2, 69.0, 68.7, 68.0, 67.6, 61.1 (OCH3), 61.0 (OCH3), 59.3 (OCH3), 58.9 (OCH3), 58.7 (OCH3), 58.5 (OCH3), 55.7 (OCH3), 23.5 (CH2CH3), 18.0, 17.8(9), 17.8(4), 16.6, 10.80 (CH3CH2). HRMS (ESI) calcd for (M + Na)+ C40H66NaO18: 857.4141. Found 857.4134.
p-Methoxyphenyl 6-deoxy-2,4-di-O-methyl-α-d-mannopyranosyl-(1 → 3)-2,4-O-dimethyl-α-l-fucopyranosyl-(1 → 3)-2-O-methyl-α-L-rhamnopyranosyl-(1 → 3)-2,4-di-O-methyl-α-L-rhamnopyranoside (16)
To a solution of 30 (15 mg, 0.015 mmol) in AcOH (5 mL), (Ph3P)4Pd (4 mg, 20% w/w) was added and the reaction mixture was stirred overnight at rt. The reaction mixture was filtered and water (10 mL) was added. The solution was diluted with CH2Cl2 (15 mL), washed with water (2 × 10 mL) and brine (10 mL). The organic layer was separated, dried (NaSO4), concentrated and the resulting residue was dissolved in DMF (2 mL) and CH3I (0.1 mL) was added. To this solution, NaH (5 mg) was added at 0 °C and it was stirred for 1 h at rt and then chilled water (5 mL) was added before being diluted with CH2Cl2 (10 mL) and washed with water (2 × 10 mL). The organic layer was concentrated and the resulting residue was purified by chromatography (2:1 hexane–EtOAc) to give a colorless oil. To a solution of the oil in 1:1 CH3OH–CH2Cl2 (10 mL), Pd–C (4 mg, 20% w/w) was added and the reaction mixture was stirred overnight under hydrogen before it was filtered and concentrated. The resulting residue was purified by chromatography (20:1 CH2Cl2–CH3OH) to 16 (8.5 mg, 71%) as a thick syrup: Rf 0.56 (20:1 CH2Cl2–CH3OH); [α]D −41.9 (c 0.8, CHCl3); 1H NMR (500 MHz, CDCl3, δH) 6.98–6.97 (m, 2H, Ar-2,6), 6.82–6.80 (m, 2H, Ar-3,5), 5.38 (d, 1H, J1,2 = 1.8 Hz, H-1), 5.15–5.13 (m, 2H, H-1′, H-1″), 5.05 (d, 1H, J1‴,2‴ = 1.6 Hz, H-1‴), 4.19–4.10 (m, 3H, H-3, H-3′, H-5″), 3.93–3.84 (m, 3H, H-3″, H-5, H-5′), 3.76 (s, 3H, OCH3), 3.77–3.63 (m, 6H, H-2, H-2′, H-2″, H-3‴, H-4′, H-5‴), 3.58 (s, 3H, OCH3), 3.57 (s, 3H, OCH3), 3.53 (s, 3H, OCH3), 5.21–5.10 (m, 1H, H-2‴), 3.50 (s, 3H, OCH3), 3.49 (s, 3H, OCH3), 3.42 (s, 3H, OCH3), 3.46 (s, 3H, OCH3), 3.32–3.31 (m, 1H, H-4″), 3.21 (app t, 1H, J3,4 = J4,5 = 9.6 Hz, H-4), 2.98 (app t, 1H, J3‴,4‴ = J4‴,5‴ = 9.5 Hz, H-4‴), 1.37 (d, 3H, J6′,5′ = 6.2 Hz, H-6′), 1.35 (d, 3H, J6,5 = 6.2 Hz, H-6), 1.29 (d, 3H, J6‴,5‴ = 6.2 Hz, H-6‴), 1.25 (d, 3H, J6″,5″ = 6.5 Hz, H-6″); 13C NMR (125.7 MHz, CDCl3, δC) 154.9 (Ar), 150.4 (Ar), 117.5 (Ar × 2), 114.6 (Ar × 2), 100.5, 99.6, 97.7, 95.5, 83.8, 83.2, 82.3, 82.2, 80.8, 80.5, 80.3, 80.1, 79.2, 76.4, 71.5, 71.2, 69.0, 68.7, 68.0, 67.3, 62.0 (OCH3), 61.0(7) (OCH3), 61.0(6) (OCH3), 59.4 (OCH3), 58.9 (OCH3), 58.6 (OCH3), 58.6 (OCH3), 55.7 (OCH3), 18.0, 17.8(7), 17.8(4), 16.4. HRMS (ESI) calcd for (M + Na)+ C38H62NaO18: 829.3828. Found 829.3828.
p-Methoxyphenyl 6-deoxy-2,4-di-O-methyl-α-d-mannopyranosyl-(1 → 3)-4-O-acetyl-2-O-methyl-α-l-fucopyranosyl-(1 → 3)-2-O-methyl-α-L-rhamnopyranosyl-(1 → 3)-2,4-di-O-methyl-α-L-rhamnopyranoside (17)
To a solution of 30 (10 mg, 0.01 mmol) in AcOH (5 mL), (Ph3P)4Pd (2 mg, 10% w/w) was added and the reaction mixture was stirred overnight at rt before it was filtered water (10 mL) was added. The solution was diluted with CH2Cl2 (20 mL), washed with water (2 × 10 mL) and brine (10 mL). The organic layer was separated, dried (NaSO4) and concentrated. The resulting residue was dissolved in pyridine (2 mL), Ac2O (0.25 mL) was added and the reaction mixture was stirred for 2 h at rt. Water (5 mL) was added, the solution was diluted with CH2Cl2 (10 mL) and washed with water (2 × 10 mL). The organic layer was concentrated and the resulting residue was purified by chromatography (2:1 hexane–EtOAc) to give a syrup (39 mg). To the solution of the syrup in 1:1 CH3OH–CH2Cl2 (15 mL), Pd–C (3 mg) was added and the reaction mixture was stirred overnight under hydrogen before it was filtered, concentrated and the resulting residue was purified by chromatography (20:1 CH2Cl2–CH3OH) to 17 (6.5 mg, 79%) as a colorless oil: Rf 0.48 (20:1 CH2Cl2–CH3OH); [α]D −27.1 (c 0.5, CHCl3); 1H NMR (500 MHz, CDCl3, δH) 7.00–6.98 (m, 2H, Ar-2,6), 6.83–6.81 (m, 2H, Ar-3,5), 5.40 (br s, 1H, H-1), 5.22–5.21 (d, 1H, J1″,2″ = 3.1 Hz, H-1″), 5.18 (br s, 1H, H-1′), 5.12–5.09 (m, 2H, H-1‴, H-4″), 4.29 (dq, 1H, J4″,5″ = 2.9 Hz, J5″,6″ = 6.5H, H-5″), 4.18 (dd, 1H, J2′,3′ = 3.3 Hz, J3′,4′ = 9.6 Hz, H-3′), 4.10 (dd, 1H, J2,3 = 3.1 Hz, J3,4 = 9.5 Hz, H-3), 3.89–3.84 (m, 1H, H-3″), 3.77 (s, 3H, OCH3), 3.74–3.56 (m, 8H, H-2, H-2′, H-2″, H-3‴, H-4′, H-5, H-5′, H-5‴), 3.54 (s, 6H, OCH3 × 2), 3.52 (s, 6H, OCH3 × 2), 3.46–3.44 (m, 7H, OCH3 × 2, H-2‴), 3.22 (app t, 1H, J3,4 = J4,5 = 9.6 Hz, H-4), 2.97 (app t, 1H, J3‴,4‴ = J4‴,5‴ = 9.5 Hz, H-4‴), 1.80 (br s, 1H, OH), 1.66 (br s, 1H, OH), 2.14 (s, 3H, CH3CO), 1.34 (d, 3H, J5″,6″ = 6.5 Hz, H-6″), 1.30 (d, 3H, J5′,6′ = 6.2 Hz, H-6′), 1.27 (d, 3H, J5,6 = 6.6 Hz, H-6), 1.10 (d, 3H, J5‴,6‴ = 6.4 Hz, H-6‴); 13C NMR (125.7 MHz, CDCl3, δC) 170.3 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 154.9 (Ar), 150.4 (Ar), 117.5 (Ar × 2), 114.6 (Ar × 2), 100.5, 99.3, 98.1, 95.6, 83.4, 83.3, 82.3, 80.8, 80.5, 80.4, 79.7, 79.0, 74.2, 73.1, 71.6, 70.7, 68.9, 68.7, 67.9, 65.9, 61.0 (OCH3), 60.3 (OCH3), 59.7 (OCH3), 59.0 (OCH3), 58.7 (OCH3), 58.6 (OCH3), 55.7 (OCH3), 20.8 (CH3CO), 17.9 (C × 3), 16.3. HRMS (ESI) calcd for (M + Na)+ C39H62NaO19: 857.3783. Found 857.3782.
O), 154.9 (Ar), 150.4 (Ar), 117.5 (Ar × 2), 114.6 (Ar × 2), 100.5, 99.3, 98.1, 95.6, 83.4, 83.3, 82.3, 80.8, 80.5, 80.4, 79.7, 79.0, 74.2, 73.1, 71.6, 70.7, 68.9, 68.7, 67.9, 65.9, 61.0 (OCH3), 60.3 (OCH3), 59.7 (OCH3), 59.0 (OCH3), 58.7 (OCH3), 58.6 (OCH3), 55.7 (OCH3), 20.8 (CH3CO), 17.9 (C × 3), 16.3. HRMS (ESI) calcd for (M + Na)+ C39H62NaO19: 857.3783. Found 857.3782.
p-Methoxyphenyl 4-O-allyl-2-O-methyl-α-L-fucopyranosyl-(1 → 3)-4-O-benzyl-2-O-methyl-α-L-rhamnopyranosyl-(1 → 3)-2,4-di-O-methyl-α-L-rhamnopyranoside (27)
Two solutions were prepared. Solution A was prepared by dissolving donor 25 (0.29 g, 0.65 mmol) in CH2Cl2 (15 mL); containing crushed 4 Å molecular sieves (50 mg). Solution B was prepared by dissolving acceptor 26 (300 mg, 0.55 mmol) in CH2Cl2 (15 mL); containing crushed 4 Å molecular sieves (100 mg). Both solutions were then stirred for 30 min at rt before solution B solution was cooled to −40 °C, NIS (144 mg, 0.64 mmol) and AgOTf (39 mg, 0.15 mmol) were added. Solution A was then added to solution B dropwise over 5 min while stirring. Then, the reaction mixture was stirred for additional 30 min at −40 °C before it was neutralized by the addition of Et3N (1 mL). The solution was filtered, concentrated and the resulting residue was dissolved in CH2Cl2 (15 mL). To this solution, TFA (0.75 mL, 5% v/v) was added dropwise over 1 min at 0 °C and reaction mixture was then stirred for additional 30 min at 0 °C before it was neutralized by the addition of Et3N (2 mL). The solution was concentrated and the resulting crude product was purified by chromatography (1:1 hexanes–EtOAc) to give 27 (311 mg, 76%) as a colorless oil: Rf 0.54 (1:1 hexanes–EtOAc); [α]D +25.0 (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3, δH) 7.37–7.23 (m, 5H, Ar), 7.00–6.96 (m, 2H, Ar-2,6), 6.83–6.80 (m, 2H, Ar-3,5), 6.01–5.93 (m, 1H, CH2CH), 5.39 (d, 1H, J1,2 = 1.9 Hz, H-1), 5.31–5.27 (m, 1H, CH2CH), 5.23 (d, 1H, J1″,2″ = 3.4 Hz, H-1″), 5.20–5.18 (m, 1H, CH2CH), 5.16 (d, 1H, J1′,2′ = 1.7 Hz, H-1′) 5.13, 4.58 (ABq, 2H, J = 11.5 Hz, ArCH2), 4.35–4.31 (m, 1H, CH2O), 4.22–4.17 (m, 2H, CH2O, H-5″), 4.14–4.08 (m, 2H, H-4″, H-3″), 4.06 (dd, 1H, J2,3 = 3.3 Hz, J3,4 = 9.6 Hz, H-3), 4.02 (dd, 1H, J2′,3′ = 3.2 Hz, J3′,4′ = 9.5 Hz, H-3′) 3.95 (dq, 1H, J4,5 = 9.4 Hz, J5,6 = 6.2 Hz, H-5), 3.76 (s, 3H, OCH3), 3.74–3.72 (m, 2H, H-2, H-2′), 3.68 (dq, 1H, J4′,5′ = 9.5 Hz, J5′,6′ = 6.2 Hz, H-5′), 3.59–3.58 (m, 1H, H-2″), 3.54 (s, 3H, OCH3), 3.50 (s, 3H, OCH3), 3.48 (s, 3H, OCH3), 3.49–3.46 (m, 1H, H-4′), 3.29 (s, 3H, OCH3), 3.21 (app t, 1H, J3,4 = J4,5 = 9.6 Hz, H-4), 2.34 (d, 1H, J3″,OH-3″ = 2.5 Hz, OH-3″), 1.30 (d, 3H, J5″,6″ = 6.5 Hz, H-6″), 1.27 (d, 3H, J5′,6′ = 6.2 Hz, H-6′), 1.25 (d, 3H, J5,6 = 6.2 Hz, H-6); 13C NMR (125.7 MHz, CDCl3, δC) 154.9 (Ar), 150.5 (Ar), 139.1 (Ar), 135.0 (CH), 128.2 (Ar × 2), 127.4 (Ar × 2), 127.3 (Ar), 117.5 (Ar × 2), 117.4 (CH2CH), 114.6 (Ar × 2), 99.1, 98.6, 95.5, 81.9, 80.7, 80.2, 80.1, 79.5, 79.4, 78.8, 74.9, 74.8, 70.2, 68.7(0), 68.6(8), 66.4, 61.2, 58.8 (OCH3), 58.1 (OCH3), 57.7(1) (OCH3), 55.6(5) (OCH3 × 2), 18.3, 17.9, 16.9. HRMS (ESI) calcd for (M + Na)+ C39H56NaO14: 771.3562. Found 771.3560.
p-Methoxyphenyl 2-O-benzoyl-3-benzyl-6-deoxy-4-O-methyl-α-D-mannopyranosyl-(1 → 3)-4-O-allyl-2-O-methyl-α-L-fucopyranosyl-(1 → 3)-4-O-benzyl-2-O-methyl-α-L-rhamnopyranosyl-(1 → 3)-2,4-di-O-methyl-α-L-rhamnopyranoside (29)
To a solution of donor 28 (131 mg, 0.27 mmol) and acceptor 27 (120 mg, 0.16 mmol) in CH2Cl2 (20 mL), crushed 4 Å molecular sieves (200 mg) were added. After the mixture was stirred at rt for 30 min, it was cooled to −20 °C, NIS (67.4 mg, 0.3 mmol) and AgOTf (15.4 mg, 0.06 mmol) were added and the reaction mixture was stirred for additional 30 min at −20 °C before the addition of Et3N (1 mL). The solution was concentrated to a crude residue that was purified by chromatography (2:1 hexane–EtOAc) to give 29 (122 mg, 69%) as an amorphous solid: Rf 0.38 (2:1 hexane–EtOAc); [α]D +28.5 (c 1.2, CHCl3); 1H NMR (500 MHz, CDCl3, δH) 8.09–8.07 (m, 2H, Ar), 7.58–7.55 (m, 1H, Ar), 7.47–7.44 (m, 2H, Ar), 7.30–7.09 (m, 10H, Ar), 7.01–6.95 (m, 2H, Ar), 6.85–6.80 (m, 2H, Ar), 5.94–5.86 (m, 1H, CH2CH), 5.67 (dd, 1H, J1‴,2‴ = 1.8 Hz, J2‴,3‴ = 3.1 Hz, H-2‴), 5.39 (d, 1H, J1,2 = 1.8 Hz, H-1), 5.28–5.27 (m, 1H, CH2CH), 5.25–5.24 (m, 1H, CH2CH), 5.17–5.14 (m, 3H, H-1′, H-1″, H-1‴), 5.13, 4.52 (ABq, 2H, J = 11.3 Hz, ArCH2), 4.78.459 (ABq, 2H, J = 11.5 Hz, ArCH2), 4.34–4.30 (m, 1H, –CH2O), 4.23–4.20 (m, 2H, H-3″, H-5″), 4.10–4.06 (m, 2H, H-3′, CH2O), 4.00 (dd, 1H, J2,3 = 3.2 Hz, J3,4 = 9.5 Hz, H-3), 3.94 (dq, 1H, J4′,5′ = 9.5 Hz, J5′,6′ = 6.2 Hz, H-5′), 3.90 (dd, 1H, J2‴,3‴ = 3.1 Hz, J3‴,4‴ = 9.6 Hz, H-3‴), 3.84–3.78 (m, 1H, H-2), 3.76 (s, 3H, OCH3), 3.75–3.68 (m, 4H, H-2′, H-2″, H-5, H-5‴), 3.59 (s, 3H, OCH3), 3.54 (s, 3H, OCH3), 3.59 (s, 3H, OCH3), 3.54 (s, 3H, OCH3), 3.53–3.46 (m, 2H, H-4′, H-4″), 3.50 (s, 3H, OCH3), 3.48 (s, 3H, OCH3), 3.31 (s, 3H, OCH3), 3.26 (app t, 1H, J3‴,4‴ = J4‴,5‴ = 9.6 Hz, H-4‴), 3.22 (app t, 1H, J3,4 = J4,5 = 9.5 Hz, H-4), 1.38 (d, 3H, J5‴,6‴ = 6.3 Hz, H-6‴), 1.33 (d, 3H, J5″,6″ = 6.5 Hz, H-6″), 1.27 (d, 3H, J5′,6′ = 6.2 Hz, H-6′), 1.24 (d, 3H, J5,6 = 6.2 Hz, H-6); 13C NMR (125.7 MHz, CDCl3, δC) 165.6 (CO), 154.9 (Ar), 150.5 (Ar), 139.1 (Ar), 138.1 (Ar), 135.1 (CH), 133.1 (Ar), 130.1 (Ar × 2), 129.9 (Ar × 2), 128.4 (Ar × 2), 128.3 (Ar × 2), 128.1 (Ar × 2), 127.9 (Ar × 2), 127.6 (Ar), 127.5 (Ar), 127.1 (Ar), 117.5 (Ar × 2), 117.3 (CH2), 114.6 (Ar × 2), 99.7, 99.1, 98.5, 95.6, 82.4, 82.0, 81.5, 80.7, 80.2, 80.0, 79.5, 79.4, 79.0, 75.5, 75.1, 74.4, 71.3, 69.3, 68.7, 68.6(5), 68.5, 66.9, 61.3, 61.2 (OCH3), 58.9 (OCH3), 58.6 (OCH3), 57.8 (OCH3), 55.7 (OCH3), 55.6 (OCH3), 18.2, 18.2, 17.9, 16.8. HRMS (ESI) calcd for (M + Na)+ C60H78NaO19: 1125.5030. Found 1125.5020.
p-Methoxyphenyl 3-benzyl-6-deoxy-2,4-di-O-methyl-α-D-mannopyranosyl-(1 → 3)-4-O-allyl-2-O-methyl-α-L-fucopyranosyl-(1 → 3)-4-O-benzyl-2-O-methyl-α-L-rhamnopyranosyl-(1 → 3)-2,4-di-O-methyl-α-L-rhamnopyranoside (30)
To a solution of 29 (100 mg, 0.09 mmol) in 1:1 CH3OH–CH2Cl2 (20 mL), 1 M NaOCH3 (0.2 mL) was added and the reaction mixture was stirred for 5 h at rt. The reaction mixture was then neutralized by the addition of Amberlite IR-120 H+ resin, filtered and concentrated. The resulting residue was dissolved in DMF (3 mL), and then CH3I (0.1 mL) and NaH (60% in mineral oil, 10 mg) were added. The reaction mixture was stirred for 1 h at rt before chilled water (8 mL) was added. The solution was diluted with CH2Cl2 (15 mL), washed with water (2 × 10 mL) and finally brine (10 mL). The organic layer was separated, concentrated and the resulting residue was purified by chromatography (3:1 hexane–EtOAc) to give 30 (83.6 mg, 91%) as a colorless oil: Rf 0.45 (3:1 hexane–EtOAc); [α]D −28.1 (c 1.1, CHCl3); 1H NMR (500 MHz, CDCl3, δH) 7.40–7.22 (m, 10H, Ar), 7.00–6.96 (m, 2H, Ar), 6.83–6.81 (m, 2H, Ar), 5.92–5.84 (m, 1H, CH), 5.39 (d, 1H, J1,2 = 1.8 Hz, H-1), 5.26–5.22 (m, 1H, CH2CH), 5.17–5.14 (m, 5H, H-1′, H-1″, H-1‴, CH2CH, ArCH2), 4.72, 4.68 (ABq, 2H, J = 12.5 Hz, ArCH2), 4.56 (d, 1H, J = 11.5 Hz, ArCH2), 4.31–4.27 (m, 1H, CH2O), 4.21–4.14 (m, 2H, H-3″, H-5″), 4.07–4.02 (m, 2H, H-3′, CH2O), 3.99 (dd, 1H, J2,3 = 3.3 Hz, J3,4 = 9.6 Hz, H-3), 3.94 (dq, 1H, J4′,5′ = 9.5 Hz, J5′,6′ = 6.2 Hz, H-5′), 3.76 (s, 3H, OCH3), 3.75–3.72 (m, 2H, H-3‴, H-2), 3.70–3.67 (m, 2H, H-2′, H-2″), 3.66–3.60 (m, 2H, H-5, H-5‴), 3.58 (s, 3H, OCH3), 3.53 (s, 3H, OCH3), 3.50 (s, 3H, OCH3), 3.48–3.46 (m, 3H, H-2‴, H-4′, H-4″), 3.47 (s, 3H, OCH3), 3.40 (s, 3H, OCH3), 3.22–3.18 (m, 2H, H-4, H-4‴), 3.17 (s, 3H, OCH3), 1.33–1.31 (m, 6H, H-6, H-6′), 1.26 (d, 3H, J5‴,6‴ = 6.2 Hz, H-6‴), 1.21 (d, 3H, J5″,6″ = 6.7 Hz, H-6″); 13C NMR (125.7 MHz, CDCl3, δC) 154.9 (Ar), 150.5 (Ar), 139.3 (Ar), 138.5 (Ar), 135.1 (CH), 128.3 (Ar × 2), 128.1 (Ar × 2), 127.9 (Ar × 2), 127.6 (Ar), 127.3 (Ar × 2), 127.2 (Ar), 117.5 (Ar × 2), 117.3 (CH2), 114.6 (Ar × 2), 99.6, 98.5, 98.4, 95.5, 82.2, 82.0, 80.7, 80.2, 80.1, 79.6, 79.2, 79.0, 78.9, 78.4, 75.9, 75.0, 74.3, 72.2, 68.7(0), 68.6(5), 66.9, 61.2, 61.1, 58.8 (OCH3), 58.7 (OCH3), 58.1 (OCH3), 57.6 (OCH3), 57.6 (OCH3), 55.7 (OCH3), 55.6 (OCH3), 18.2, 17.9, 17.8, 16.8. HRMS (ESI) calcd for (M + Na)+ C54H76NaO18: 1035.4924. Found 1035.4920.
General methods for immunoassays
Acknowledgements
We acknowledge NSERC, Alberta Innovates and the Alberta Glycomics Centre for financial support of this work. We thank Professor David R. Bundle (University of Alberta) for providing the lipopolysaccharide from E. coli (ATCC 12408) used in these experiments.Notes and references
- S. Chobot, J. Malis, H. Sebakova, M. Pelikan, O. Zatloukal, P. Palicka and D. Kocurova, Cent. Eur. J. Public Health, 1997, 5, 164–173 CAS.
- J. O. Falkinham III, Clin. Microbiol. Rev., 1996, 9, 177–215 Search PubMed.
- S. M. Arend, E. Cerda de Palou, P. De Haas, R. Janssen, M. A. Hoeve, E. M. Verhard, T. H. M. Ottenhoff, D. Van Soolingen and J. T. van Dissel, Clin. Microbiol. Infect., 2004, 10, 738–748 CrossRef CAS PubMed.
- R. E. Campo and C. E. Campo, Clin. Infect. Dis., 1997, 24, 1233–1238 CAS.
- C. Taillard, G. Greub, R. Weber, G. E. Pfyffer, T. Bodmer, S. Zimmerli, R. Frei, S. Bassetti, P. Rohner, J. C. Piffaretti, E. Bernasconi, J. Bille, A. Telenti and G. Prod'hom, J. Clin. Microbiol., 2003, 41, 1240–1244 CrossRef PubMed.
- G. Blaauw and B. J. Appelmelk, in Protein–Carbohydrate Interactions in Infectious Diseases, ed. C. Bewley, RSC, Cambridge, 2006, vol. 6, pp. 6–29 Search PubMed.
- J.-J. Fournie, M. Riviere, F. Papa and G. Puzo, J. Biol. Chem., 1987, 26, 3180–3184 Search PubMed.
- M. Watanabe, Y. Aoyagi, A. Ohta and D. E. Minnikin, Eur. J. Biochem., 1997, 248, 93–98 CAS.
- M. B. Reed, P. Domenech, C. Manca, H. Su, A. K. Barczak, B. N. Kreiswirth, G. Kaplan and C. E. Barry, Nature, 2004, 431, 84–87 CrossRef CAS PubMed.
- C. J. Cambier, K. K. Takaki, R. P. Larson, R. E. Hernandez, D. M. Tobin, K. B. Urdahl, C. L. Cosma and L. Ramakrishnan, Nature, 2014, 505, 218–222 CrossRef CAS PubMed.
- A. Arbues, G. Lugo-Villarino, O. Neyrolles, C. Guilhot and C. Astarie-Dequeker, Front. Cell. Infect. Microbiol., 2014, 4, 137 Search PubMed.
- H. R. H. Elsaidi, D. R. Barreda, C. W. Cairo and T. L. Lowary, ChemBioChem, 2013, 14, 2153–2159 CrossRef CAS PubMed.
- H. R. H. Elsaidi and T. L. Lowary, ChemBioChem, 2014, 15, 1176–1182 CrossRef CAS PubMed.
- I. B. McInnes and G. Schett, Nat. Rev., 2007, 7, 429–442 CAS.
- S. Sakaguchi, H. Benham, A. P. Cope and R. Thomas, Immunol. Cell Biol., 2012, 90, 277–287 CrossRef CAS PubMed.
- R. N. Maini and P. C. Taylor, Annu. Rev. Med., 2000, 51, 207–229 CrossRef CAS PubMed.
- A. E. Koch, Arthritis Rheum., 2005, 52, 710–721 CrossRef PubMed.
- J. J. Haringman and P. P. Tak, Arthritis Res. Ther., 2004, 6, 93–97 CAS.
- L. Šenolt, J. Vencovský, K. Pavelka, C. Ospelt and S. Gay, Autoimmun. Rev., 2009, 9, 102–107 CrossRef PubMed.
- M. J. Kaplan, Rheumatic Diseases Clinics of North America, 2010, 36, 405–426 CrossRef PubMed.
- G. S. Firestein, Nature, 2003, 423, 356–361 CrossRef CAS PubMed.
- C. Book, T. Saxne and L. T. Jacobsson, J. Rheumatol., 2005, 32, 430–443 Search PubMed.
- (a) K. Nakayama, K. Uoto, K. Higashi, T. Soga and T. Kusama, Chem. Pharm. Bull., 1992, 40, 1718–1720 CrossRef CAS; (b) D. Hou and T. L. Lowary, J. Org. Chem., 2009, 74, 2278–2289 CrossRef CAS PubMed.
- (a) K. Bock and C. Pedersen, J. Chem. Soc., Perkin Trans. 2, 1974, 293–299 RSC; (b) K. Bock, I. Lundt and C. Pedersen, Tetrahedron Lett., 1973, 14, 1037–1040 CrossRef.
- (a) S. Józefowski, A. Sobota and K. Kwiatkowska, BioEssays, 2008, 30, 943–954 CrossRef PubMed; (b) C. H. Ladel, C. Blum, A. Dreher, K. Reifenberg, M. Kopf and S. H. E. Kaufmann, Infect. Immun., 1997, 65, 4843–4849 CAS; (c) H. Yamada, S. Mizumo, R. Horai, Y. Iwakura and I. Sugawara, Lab. Invest., 2000, 80, 759–767 CrossRef CAS PubMed; (d) W. Peters, H. M. Scott, H. F. Chambers, J. L. Flynn, I. F. Charo and J. D. Ernst, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 7958–7963 CrossRef CAS PubMed.
- (a) S. Tsuchiya, M. Yamabe, Y. Yamaguchi, Y. Kobayashi, T. Konno and K. Tada, Int. J. Cancer, 1980, 26, 171–176 CrossRef CAS PubMed; (b) J. Auwerx, Experientia, 1991, 47, 22–31 CrossRef CAS PubMed; (c) R. W. Stokes and D. Doxsee, Cell. Immunol., 1999, 197, 1–9 CrossRef CAS PubMed; (d) C. J. Riendeau and H. Kornfeld, Infect. Immun., 2003, 71, 254–259 CrossRef CAS PubMed; (e) M. Miyazawa, Y. Ito, Y. Yoshida, H. Sakaguchi and H. Suzuki, Toxicol. In Vitro, 2007, 21, 428–437 CrossRef CAS PubMed; (f) E. K. Park, H. S. Jung, H. I. Yang, M. C. Yoo, C. Kim and K. S. Kim, Inflammation Res., 2007, 56, 45–50 CrossRef CAS PubMed.
- U. V. D. Esche, M. Ayoub, S. D. C. Pfannes, M. R. Müller, M. Huber, K.-H. Wiesmüller, T. Loop, M. Humar, K.-F. Fischbach, M. Strünkelnberg, P. Hoffmann, W. G. Bessler and K. Mittenbühler, Int. J. Immunopharmacol., 2000, 22, 1093–1102 CrossRef.
- A. K. Mishra, J. E. Alves, K. Krumbach, J. Nigou, A. G. Castro, J. Geurtsen, L. Eggeling, M. Saraiva and G. S. Besra, J. Biol. Chem., 2012, 287, 44173–44183 CrossRef CAS PubMed.
- S. I. Miller, R. K. Ernst and M. W. Bader, Nat. Rev. Microbiol., 2005, 3, 36–46 CrossRef CAS PubMed.
- Y. Wada, R. Lu, D. Zhou, J. Chu, T. Przewloka, S. Zhang, L. Li, Y. Wu, J. Qin, V. Balasubramanyam, J. Barsoum and M. Ono, Blood, 2007, 109, 1156–1164 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc04004j |
| This journal is © The Royal Society of Chemistry 2015 |