
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Extending N-heterocyclic carbene ligands into the third dimension: a new type of hybrid phosphazane/NHC system†‡
Torsten
Roth
a,
Vladislav
Vasilenko
a,
Callum G. M.
Benson
b,
Hubert
Wadepohl
a,
Dominic S.
Wright
*b and
Lutz H.
Gade
*a
aAnorganisch-Chemisches Institut, Universität Heidelberg, Im Neuenheimer Feld 270, 69120 Heidelberg, Germany. E-mail: lutz.gade@uni-heidelberg.de
bChemistry Department, Cambridge University, Lensfield Road, Cambridge, CB2 1EW, UK. E-mail: dsw1000@cam.ac.uk
First published on 13th February 2015
Abstract
A simple, “click” synthetic approach to a new type of hybrid phosph(III)azane/NHC system is described. The presence of the phosphazane P2N2 ring unit, with P atoms flanking the NCN fragment and with this ring perpendicular to the binding site of the NHC, provides unique opportunities for modifying the electronic and steric character of these carbenes.
Introduction
N-heterocyclic carbenes have been successfully established as versatile ligands in coordination chemistry, as powerful ancillary ligands in catalysis, as organocatalysts, and for an increasing number of other applications.1–6 In view of these applications the ability to vary the steric and electronic character of these ligands systematically, using simple modular synthetic routes, is a central theme in this area. In recent years a range of approaches to modify their σ-donor/π-acceptor and coordination properties have been implemented,7 for instance different ring sizes of the backbone resulting in different NCN-angles,8 variation of the heteroatoms adjacent to carbon,9 abnormal and remote carbenes,10 acyclic carbenes,11 anti-Bredt NHCs,12 push–pull carbenes,13 redox-switchable NHCs14 or even cyclophane-derived carbenes.15 A less explored approach relies on NHCs comprising “inorganic” or organometallic fragments in their ligand backbones (relevant examples shown in Fig. 1).16 This is, however, potentially a very powerful approach to ligand modification since introducing flanking electron-donating (e.g., P) or electron-accepting (e.g., B) main group atoms should have a profound effect on the acceptor or donor ability of the NCN-unit.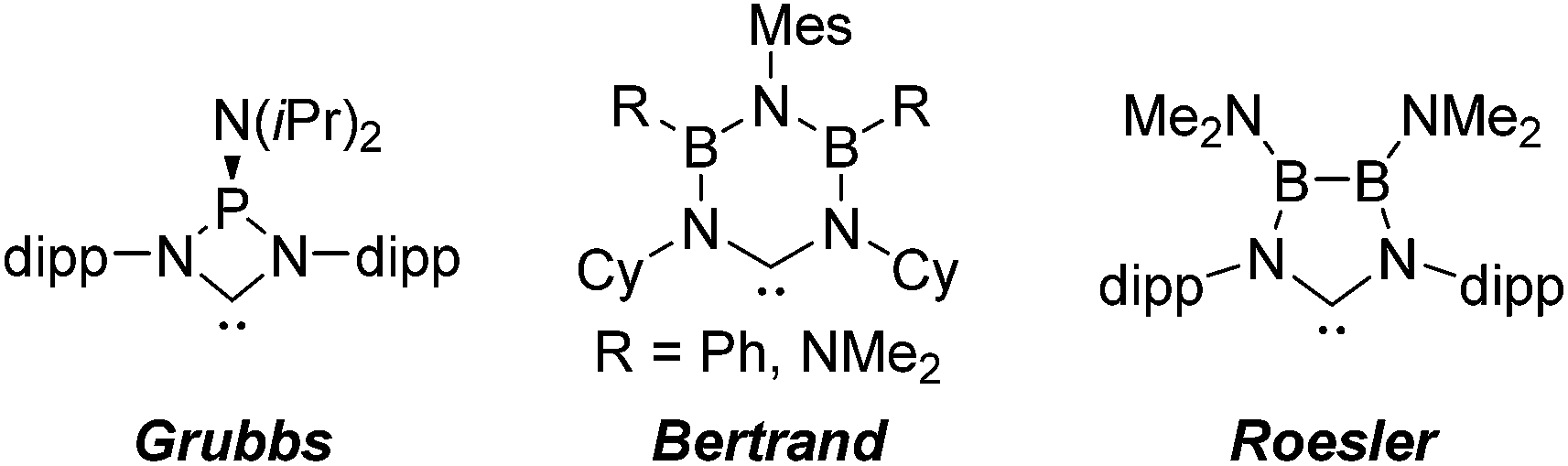 | ||
| Fig. 1 Selection of “inorganic” carbenes. | ||
In contrast, much less effort has been dedicated to the systematic development of NHC ligands with unusual and improved spatial characteristics.17 Most approaches focussed on the introduction of various bulky substituents in the wingtip position. Although a variety of backbone structures containing heteroatoms have been reported in recent years NHCs are still regarded as essentially two-dimensional ancillary ligands. Even though NHCs have proven to be superior to phosphine ligands in several respects, the latter are often chosen preferentially due to their three-dimensional shape, which is of relevance inter alia in enantioselective homogenous catalysis.18
Herein, we present a conceptually new approach for the design of the steric properties of NHC-ligands, which have so far only been perceived as essentially flat donor entities. Our system constitutes the first example of a three-dimensional arrangement encompassing the central NCN-unit that allows for straightforward control of the steric properties of the top and bottom hemispheres of the widespread NHC-ligand framework. This is achieved by a unique inorganic backbone moiety consisting of a cyclophosphazane P2N2-unit that is orthogonally attached to the NCN-fragment (Fig. 2).
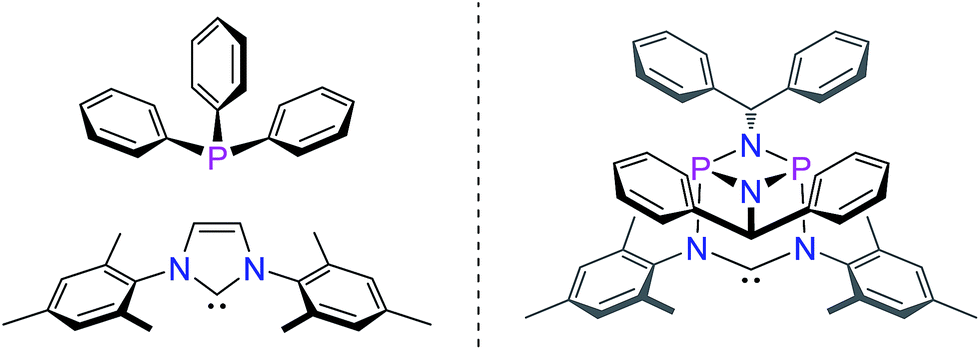 | ||
| Fig. 2 Spatial characteristics of phosphines, NHCs (left) and the targeted 3-dimensional hybrid phosphazane/NHC ligand (right). | ||
Stemming from our interest in the modular synthesis of cyclophosph(III)azane ligands for catalysis,19 we have begun to explore the possibility of developing a simple “click-type” approach to ambidentate20 hybrid phosphazane/NHC ligands as shown in Fig. 2. We reasoned that the presence of the P2N2 ring unit (perpendicular and adjacent to the NCN-fragment) would provide a unique strategy for modifying the electronic and spatial characteristics of the NHC system.
Results
The preparation of the new ligand system starts from simple Me3Si-protected diarylformamidines and dichloro-cyclophosphazanes, both of which are readily accessible on a multi-gram scale. Condensation of the two components and subsequent ring-closure using Et3Si-OTf furnishes cationic azolium salts,21 a key example of which is 2 (Fig. 3). The solid state structure of 2 confirms the bicyclic cage structure in which the NCN-unit bridges the P2N2-ring. The P2N2 moiety and its substituents are orthogonal to the carbene plane, thus effectively shrouding the posterior of the carbene system.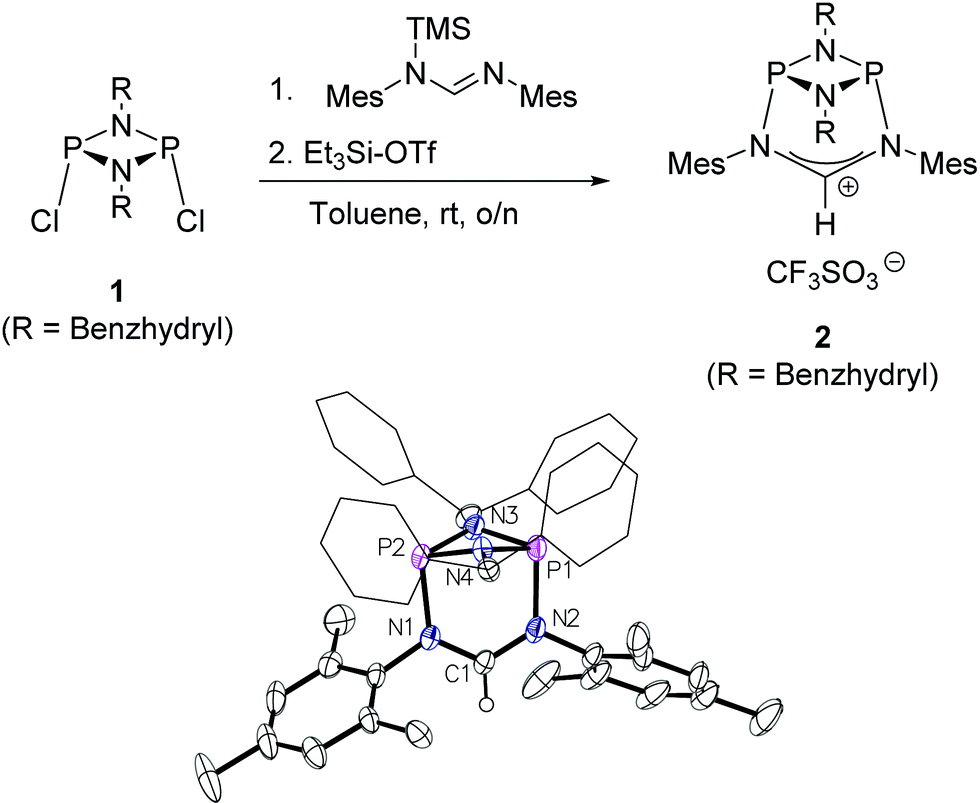 | ||
| Fig. 3 Top: one-pot access to the cationic cyclophosphazane cage compound 2. Bottom: molecular structure of 2, thermal ellipsoids set at the 50% probability level. H-atoms except H(1) and the trifluoromethanesulfonate counterion have been omitted for clarity. Phenyl rings of the benzhydryl substituents are drawn as wireframes. Selected bond lengths [Å] and angles [°]: P1–N2 1.837(3), P1–N3 1.704(3), N1–C1 1.323(4), N2–C1–N1 122.7(3). | ||
Notably, quantitative formation of carbene species 3 was observed upon addition of KHMDS to 2, exhibiting a single 31P NMR signal (197 ppm) and a characteristic 13C NMR resonance at 260 ppm, corresponding to its carbene carbon atom, in the in situ recorded spectrum (Scheme 1). Although carbene species 3 is stable in solution for several hours, attempts to isolate the free carbene invariably led to partial decomposition.
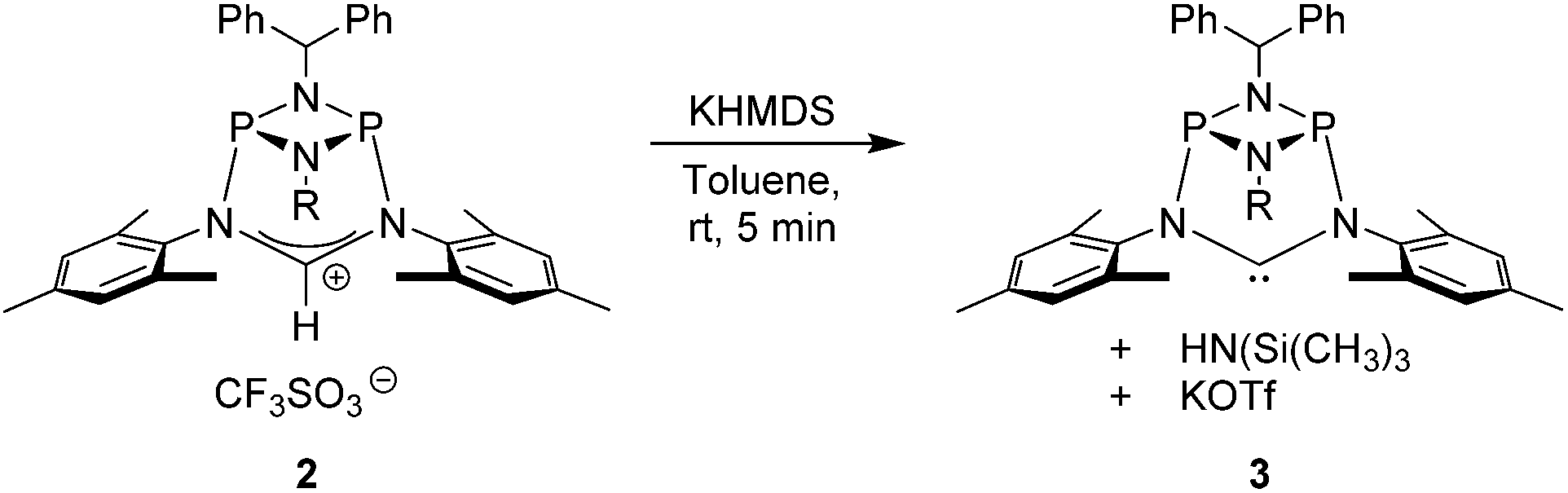 | ||
| Scheme 1 In situ generation of carbene 4. | ||
We then turned to probing the reactivity of carbene 3 employing Lewis-acidic metal-fragments. Deprotonation of 2 and subsequent addition of half an equivalent of [Ir(cod)Cl]2 led to quantitative formation of the robust22 NHC–metal complex 4 (Scheme 2, Fig. 4, left). To classify the electron-donor properties of the carbene unit we determined the electrochemical characteristics of 4 and the TEP (Tolman Electronic Parameter)23 of the [Ir(CO)2Cl] carbene complex 5 (for the solid-state structure of 5 see ESI†).
 | ||
| Scheme 2 Metalation of carbene 3. | ||
 | ||
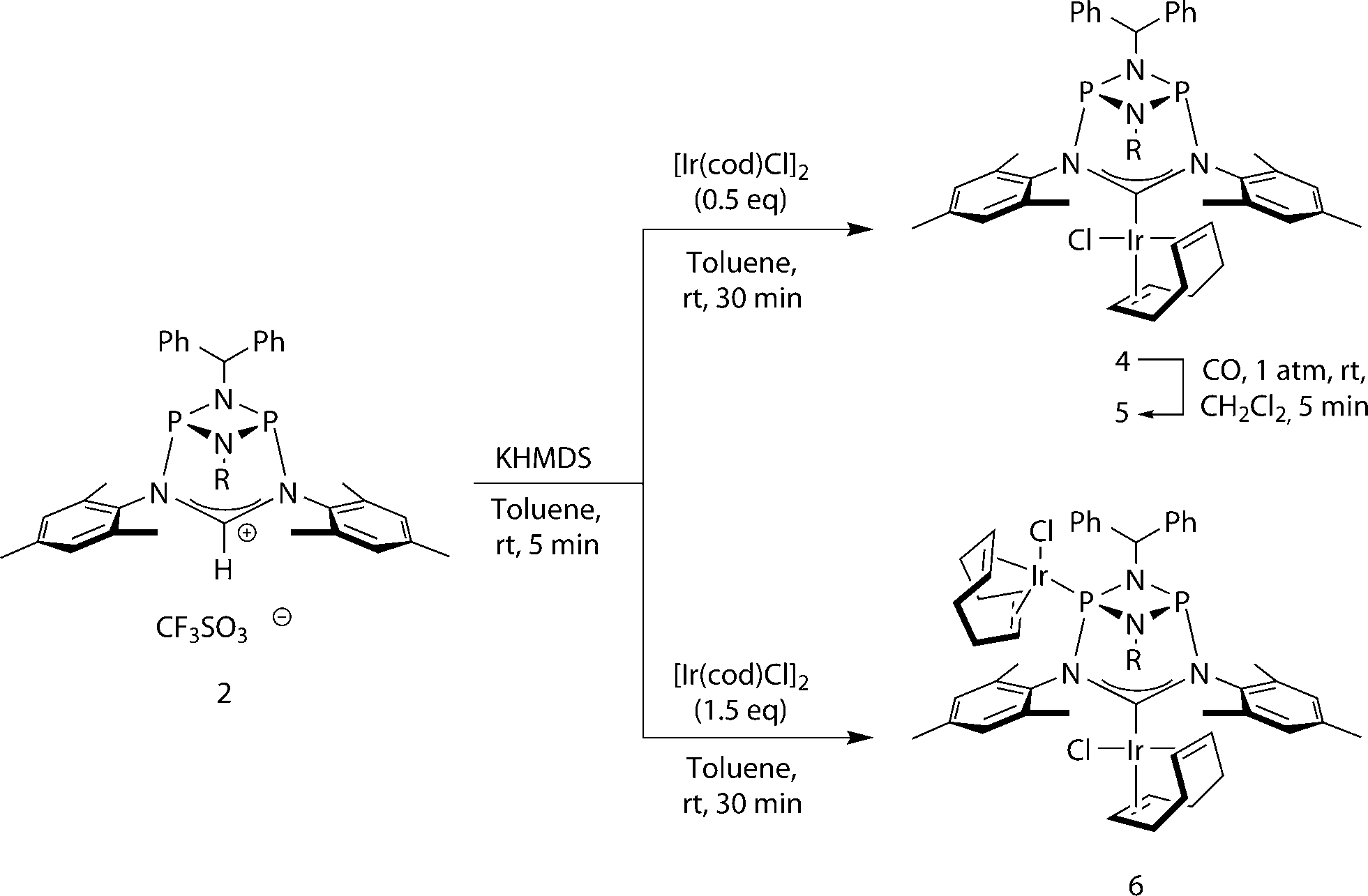
| Fig. 4 Left: molecular structure of 4, thermal ellipsoids set at the 50% probability level. H-atoms have been omitted for clarity. Phenyl rings of the benzhydryl substituents are drawn as wireframes. Selected bond lengths [Å] and angles [°]: Ir–C1 2.040(2), P1–N3 1.7658(19), N3–C1 1.372(3), N3–C1–N4 114.06(19). Right: space-filling representation of one of the C3-symmetric binding pockets. Phenyl and mesityl rings marking the boundary of the cavity are highlighted in red. | ||
The IR spectrum of 5 displays CO stretching vibrations at 1976 cm−1 and 2060 cm−1 with a calculated24 TEP value of 2045 cm−1 representing a two-electron donor capacity comparable to other six-membered “inorganic” carbenes (Fig. 1, Bertrand et al.).16c Moreover, the cyclic voltammogram of 4 features a single quasi-reversible oxidation step (E1/2 = +0.653 V, ΔE = 159 mV),25 suggesting that the cyclophosphazane carbene is a powerful electron donor ligand. The strong σ-donor character of 3 is consistent with the presence of the donor-P atoms adjacent to the NCN-unit.
One of the most notable features of the solid-state structures of the iridium complexes 4 and 5 is the presence of two conical binding pockets of local C3 symmetry around the uncoordinated phosphorus atoms, the borders of these cavities being defined by the benzhydryl phenyl and mesityl rings (Fig. 4, right).
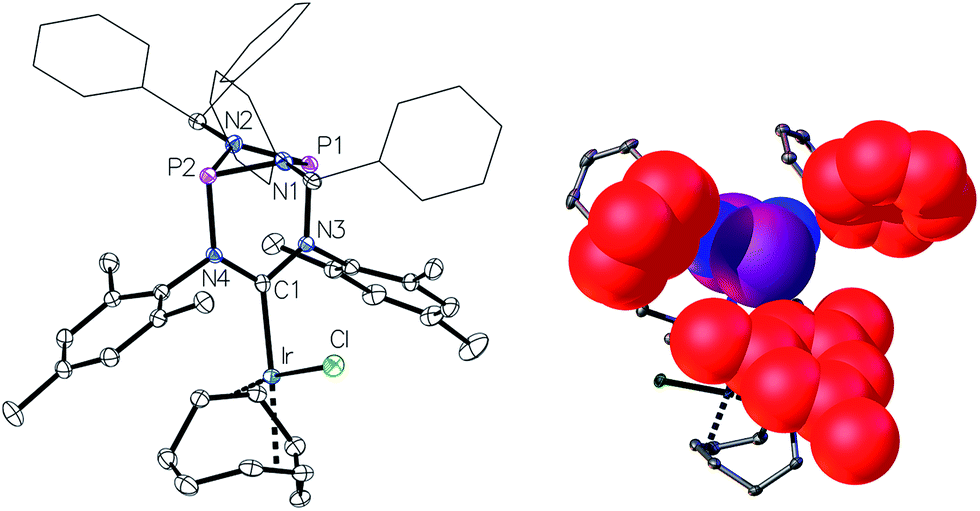
This led us to repeat the complexation reaction used in the formation of 4 employing 1.5 equivalents [Ir(cod)Cl]2. Surprisingly, the 31P NMR spectrum showed two doublets (JP–P = 30 Hz), suggesting a dimetallic complex in which there is unsymmetrical coordination of the P2N2 ring. Even after the addition of an excess of the iridium precursor no further species were detected by 31P NMR, indicating that the lone pair of the second phosphorus atom is not accessible for coordination. The solid-state structure of the product 6 reveals that the coordination of the second iridium fragment by one phosphorus lone pair induces a slight internal rearrangement within the core structure, thus effectively sterically-blocking the second phosphorus coordination site (Fig. 5). Since 6 could only be isolated in pure form in the solid state and 31P NMR spectra showed a mixture of 4 and 6 in solution, a VT NMR study was conducted to probe the coordination behavior of the P2N2 phosphorus atoms. Crystals of complex 6 were dissolved in THF and the resulting equilibrium was studied in the temperature range −30 to 50 °C. A labile P–Ir bond and an entropically driven reversible dissociation process was found (6 → 4 + 1/2[Ir(cod)Cl]2, ΔH = +30 kJ mol−1, ΔS = +77 J mol−1).
 | ||
| Fig. 5 Molecular structure of 6, thermal ellipsoids set at the 50% probability level. H-atoms have been omitted for clarity. Phenyl rings of the benzhydryl substituents are drawn as wireframes. Selected bond lengths [Å] and angles [°]: Ir1–C1 2.046(3), Ir2–P1 2.2359(8), P1–N1 1.756(3), N1–C1 1.385(4), Ir2–P1–P2 158.50(4), N2–C1–N1 114.9(3). | ||
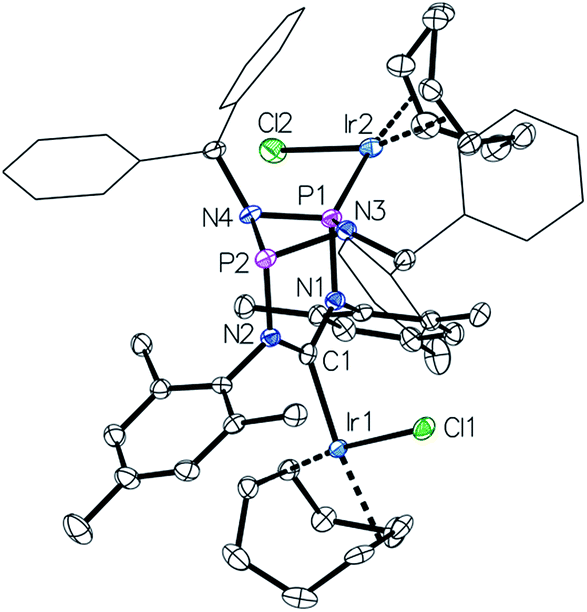
Moreover, to preclude electronic effects as the cause for the bismetalation product 6, we exchanged the square-planar [Ir]-precursor for a sterically less demanding linear gold(I) fragment. By a procedure analogous to the preparation of 6 the heterotrimetallic complex 7 was obtained (Fig. 6). Consequently, the 13C NMR shift of the carbene carbon atom of δ = 234 ppm (t, JC–P = 4.5 Hz) corroborates coordination of the [Ir(cod)Cl] fragment and the single 31P NMR resonance of 128 ppm establishes metalation of both phosphorus atoms. Details of the molecular structure have also been established by X-ray diffraction (Fig. 6). Complex 7 was also converted to its corresponding carbonyl complex by the previously mentioned procedure for compound 5. However, significant decomposition precluded its isolation in a pure state. The carbonyl complex exhibits a 31P NMR resonance at 131 ppm and the IR spectrum shows two CO stretching vibrations at 2071 cm−1 and 1986 cm−1 (νav(CO) = 2029 cm−1; TEP = 2054 cm−1).
 | ||
| Fig. 6 Top: access to the trimetallic complex 7. Bottom: molecular structure of 7, thermal ellipsoids set at the 50% probability level. H-atoms have been omitted for clarity. Phenyl rings of the benzhydryl substituents are drawn as wireframes. Selected bond lengths [Å] and angles [°]: Au1–P1 2.1839(10), Ir–C1 2.020(4), P1–N1 1.719(4), P1–N3 1.712(4), N1–C1 1.390(5), N2–C1–N1 115.3(4). | ||
Finally, the steric properties of the ligand system were analyzed. The extended heteroatomic backbone leads to a more confined metal coordination site compared to the standard SIMes NHC (Fig. 7). This effect is accomplished by a smaller angle between the mesityl substituents and the presence of the bulky P2N2 substituents. Moreover the CPK model demonstrates the close spatial proximity of the benzhydryl phenyl groups to the iridium binding pocket, thus effectively shielding the backside of the NHC.
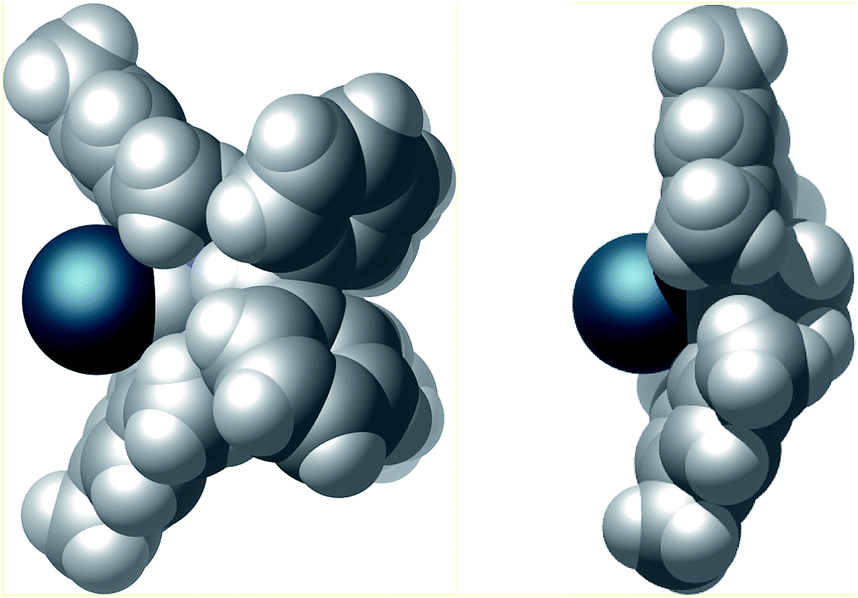 | ||
| Fig. 7 CPK model of the van der Waals surface of complex 4 (left) and, for comparison complex SIMes·[Ir(cod)Cl] (right).26 Cyclooctadiene and chlorido ligands have been omitted for clarity. | ||
To obtain a quantitative measure for the steric congestion of the metal centre we have determined buried volumes %Vbur of all isolated complexes for the NHC and the phosphorus donor sites (Table 1).27 As expected, all phosphazane–NHC complexes reported in this work feature increased buried volumes compared to the SIMes reference system. Remarkably, the steric properties of the NHC–metal unit are unaffected by the coordination of metal fragments to the P2N2 backbone. This offers a convenient way to modify the NHC donor strength independently from the sterics.
Conclusion
We have established a cyclic P2N2 unit as a stable “inorganic” carbene backbone moiety within a new type of polytopical hybrid phosph(III)azane/NHC system. The new ligand is structurally rigid and a strong σ-donor. Importantly, the essentially flat spatial arrangement of conventional NHCs, often described as fan-, wedge- or fence-like, is extended into the third dimension in our new system, in which the orthogonal disposition of the P2N2 ring unit provides a unique architecture for tuning the steric and spatial properties of the carbene binding site. The ability of these new ligands to coordinate metal centres using their P-atoms provides a further means by which the steric and electronic character of the carbene fragment can be modified. In addition, the simple “click” approach may allow a broad range of hybrid NHCs of this type to be prepared, with (for example) the future potential for readily introduced chiral functionality at the phosphazane-N atom. Further studies in this area are ongoing.Acknowledgements
We gratefully acknowledge the award of a Ph.D. grant to T.R. from the Landesgraduiertenförderung (LGF Funding Program of the state of Baden-Württemberg), the award of the national scholarship “Deutschlandstipendium” to V.V., the University of Heidelberg for generous funding, the EU (Erasmus-Program for V.V., C.G.M.B.) and the ERC-Advanced Investigator Grant (C.G.M.B., D.S.W.).Notes and references
- Monographs: (a) S. P. Nolan, N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis, Wiley-VCH, 2014 Search PubMed; (b) R. A. Moss and M. P. Doyle, Contemporary Carbene Chemistry, Wiley, Hoboken, 2014 Search PubMed; (c) S. Díez-González, N-Heterocyclic Carbenes: From Laboratory Curiosities to Efficient Synthetic Tools, Royal Society of Chemistry, 2011 Search PubMed; (d) C. S. J. Cazin, N-Heterocyclic Carbenes in Transition Metal Catalysis and Organocatalysis, Springer, 2011, vol. 32 Search PubMed; (e) O. Kühl, Functionalised N-Heterocyclic Carbene Complexes, Wiley, 2010 Search PubMed; (f) F. Glorius, N-Heterocyclic Carbenes in Transition Metal Catalysis, Springer, vol. 21, 2007 Search PubMed; (g) S. P. Nolan, N-Heterocyclic Carbenes in Synthesis, Wiley-VCH, 2006 Search PubMed; (h) G. Bertrand, Carbene Chemistry: From Fleeting Intermediates to Powerful Reagents, Marcel Dekker Inc., 2002 Search PubMed; (i) E. Kolychev, E. Theuergarten and M. Tamm, in Frustrated Lewis Pairs II, ed. G. Erker and D. W. Stephan, Springer, Berlin, Heidelberg, 2013, vol. 334, pp. 121–155 Search PubMed.
- General review articles: (a) M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed; (b) D. Bourissou, O. Guerret, F. P. Gabbaï and G. Bertrand, Chem. Rev., 2000, 100, 39–91 CrossRef CAS PubMed; (c) A. J. Arduengo III, Acc. Chem. Res., 1999, 32, 913–921 CrossRef.
- Coordination chemistry review articles: (a) F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed; (b) D. Martin, M. Soleilhavoup and G. Bertrand, Chem. Sci., 2011, 2, 389–399 RSC; (c) J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS PubMed; (d) Y. Wang and G. H. Robinson, Dalton Trans., 2012, 41, 337–345 RSC; (e) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2009, 253, 862–892 CrossRef.
- Catalysis review articles: (a) S. Díez-González, N. Marion and S. P. Nolan, Chem. Rev., 2009, 109, 3612–3676 CrossRef PubMed; (b) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS; (c) V. César, S. Bellemin-Laponnaz and L. H. Gade, Chem. Soc. Rev., 2004, 33, 619–636 RSC; (d) F. Wang, L.-j. Liu, W. Wang, S. Li and M. Shi, Coord. Chem. Rev., 2012, 256, 804–853 CrossRef CAS.
- Organocatalysis review articles: (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS PubMed; (b) X. Bugaut and F. Glorius, Chem. Soc. Rev., 2012, 41, 3511–3522 RSC.
- Other review articles: (a) W. Liu and R. Gust, Chem. Soc. Rev., 2013, 42, 755–773 RSC; (b) M. Fèvre, J. Pinaud, Y. Gnanou, J. Vignolle and D. Taton, Chem. Soc. Rev., 2013, 42, 2142–2172 RSC; (c) L. Mercs and M. Albrecht, Chem. Soc. Rev., 2010, 39, 1903–1912 RSC; (d) R. Visbal and M. C. Gimeno, Chem. Soc. Rev., 2014, 43, 3551–3574 RSC; (e) K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS PubMed; (f) A. Gautier and F. Cisnetti, Metallomics, 2012, 4, 23–32 RSC.
- (a) D. J. Nelson and S. P. Nolan, Chem. Soc. Rev., 2013, 42, 6723–6753 RSC; (b) T. Dröge and F. Glorius, Angew. Chem., Int. Ed., 2010, 49, 6940–6952 CrossRef PubMed; (c) S. Díez-González and S. P. Nolan, Coord. Chem. Rev., 2007, 251, 874–883 CrossRef; (d) O. Kühl, Coord. Chem. Rev., 2005, 249, 693–704 CrossRef.
- (a) D. Martin, Y. Canac, V. Lavallo and G. Bertrand, J. Am. Chem. Soc., 2014, 136, 5023–5030 CrossRef CAS PubMed; (b) C. C. Scarborough, M. J. W. Grady, I. A. Guzei, B. A. Gandhi, E. E. Bunel and S. S. Stahl, Angew. Chem., Int. Ed., 2005, 44, 5269–5272 CrossRef CAS PubMed; (c) W. Y. Lu, K. J. Cavell, J. S. Wixey and B. Kariuki, Organometallics, 2011, 30, 5649–5655 CrossRef CAS; (d) M. Iglesias, D. J. Beetstra, B. Kariuki, K. J. Cavell, A. Dervisi and I. A. Fallis, Eur. J. Inorg. Chem., 2009, 1913–1919 CrossRef CAS.
- (a) M. Melaimi, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2010, 49, 8810–8849 CrossRef CAS PubMed; (b) V. Lavallo, Y. Canac, C. Präsang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705–5709 CrossRef CAS PubMed; (c) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256–266 CrossRef CAS PubMed.
- (a) R. H. Crabtree, Coord. Chem. Rev., 2013, 257, 755–766 CrossRef CAS; (b) O. Schuster, L. Yang, H. G. Raubenheimer and M. Albrecht, Chem. Rev., 2009, 109, 3445–3478 CrossRef CAS PubMed; (c) P. L. Arnold and S. Pearson, Coord. Chem. Rev., 2007, 251, 596–609 CrossRef CAS.
- V. P. Boyarskiy, K. V. Luzyanin and V. Y. Kukushkin, Coord. Chem. Rev., 2012, 256, 2029–2056 CrossRef CAS.
- D. Martin, N. Lassauque, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2012, 51, 6172–6175 CrossRef CAS PubMed.
- (a) A. Igau, H. Grützmacher, A. Baceiredo and G. Bertrand, J. Am. Chem. Soc., 1988, 110, 6463–6466 CrossRef CAS; (b) G. Bertrand and R. Reed, Coord. Chem. Rev., 1994, 137, 323–355 CrossRef CAS.
- For a selection see: (a) D. M. Khramov, E. L. Rosen, V. M. Lynch and C. W. Bielawski, Angew. Chem., Int. Ed., 2008, 47, 2267–2270 CrossRef CAS PubMed; (b) E. L. Rosen, C. D. Varnado, A. G. Tennyson, D. M. Khramov, J. W. Kamplain, D. H. Sung, P. T. Cresswell, V. M. Lynch and C. W. Bielawski, Organometallics, 2009, 28, 6695–6706 CrossRef CAS; (c) A. G. Tennyson, V. M. Lynch and C. W. Bielawski, J. Am. Chem. Soc., 2010, 132, 9420–9429 CrossRef CAS PubMed; (d) B. M. Neilson and C. W. Bielawski, ACS Catal., 2013, 3, 1874–1885 CrossRef CAS.
- A. Fürstner, M. Alcarazo, H. Krause and C. W. Lehmann, J. Am. Chem. Soc., 2007, 129, 12676–12677 CrossRef PubMed.
- (a) E. Despagnet-Ayoub and R. H. Grubbs, J. Am. Chem. Soc., 2004, 126, 10198–10199 CrossRef CAS PubMed; (b) K. E. Krahulic, G. D. Enright, M. Parvez and R. Roesler, J. Am. Chem. Soc., 2005, 127, 4142–4143 CrossRef CAS PubMed; (c) C. Präsang, B. Donnadieu and G. Bertrand, J. Am. Chem. Soc., 2005, 127, 10182–10183 CrossRef PubMed; (d) Y. Ishida, B. Donnadieu and G. Bertrand, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13585–13588 CrossRef CAS PubMed; (e) T. D. Forster, K. E. Krahulic, H. M. Tuononen, R. McDonald, M. Parvez and R. Roesler, Angew. Chem., Int. Ed., 2006, 45, 6356–6359 CrossRef CAS PubMed; (f) J. Ruiz, L. García, B. F. Perandones and M. Vivanco, Angew. Chem., Int. Ed., 2011, 50, 3010–3012 CrossRef CAS PubMed; (g) A. J. Arduengo III, D. Tapu and W. J. Marshall, Angew. Chem., Int. Ed., 2005, 44, 7240–7244 CrossRef PubMed; (h) A. Kausamo, H. M. Tuononen, K. E. Krahulic and R. Roesler, Inorg. Chem., 2008, 47, 1145–1154 CrossRef CAS PubMed; (i) A. El-Hellani and V. Lavallo, Angew. Chem., Int. Ed., 2014, 53, 4489–4493 CrossRef CAS PubMed; (j) M. J. Asay, S. P. Fisher, S. E. Lee, F. S. Tham, D. Borchardt and V. Lavallo, Chem. Commun., 2015 10.1039/c4cc08267b.
- D. Katayev, Y.-X. Jia, A. K. Sharma, D. Banerjee, C. Besnard, R. B. Sunoj and E. P. Kündig, Chem.–Eur. J., 2013, 19, 11916–11927 CrossRef CAS PubMed and references therein.
- (a) R. H. Crabtree, J. Organomet. Chem., 2005, 690, 5451–5457 CrossRef CAS; (b) P. W. N. M. van Leeuwen and P. C. J. Kamer, Phosphorus Ligand Effects in Homogeneous Catalysis and Rational Catalyst Design, in Phosphorus(III) Ligands in Homogeneous Catalysis: Design and Synthesis, John Wiley & Sons, Ltd, Chichester, UK, 2012. See also ref. 1–6 for a broader overview.
- T. Roth, H. Wadepohl, D. S. Wright and L. H. Gade, Chem.–Eur. J., 2013, 19, 13823–13837 CrossRef CAS PubMed.
- For a review of polytopical NHC ligands see M. Poyatos, J. A. Mata and E. Peris, Chem. Rev., 2009, 109, 3677–3707 CrossRef CAS PubMed . For a selection of hybrid ambidentate NHC ligands see (a) D. M. Khramov, A. J. Boydston and C. W. Bielawski, Angew. Chem., Int. Ed., 2006, 45, 6186–6189 CrossRef CAS PubMed; (b) A. Zanardi, J. A. Mata and E. Peris, J. Am. Chem. Soc., 2009, 131, 14531–14537 CrossRef CAS PubMed; (c) N. Vujkovic, V. César, N. Lugan and G. Lavigne, Chem.–Eur. J., 2011, 17, 13151–13155 CrossRef CAS PubMed; (d) D. Mendoza-Espinosa, B. Donnadieu and G. Bertrand, Chem.–Asian J., 2011, 6, 1099–1103 CrossRef CAS PubMed; (e) J. Ruiz and A. F. Mesa, Chem.–Eur. J., 2012, 18, 4485–4488 CrossRef CAS PubMed; (f) P. L. Arnold and S. T. Liddle, Organometallics, 2006, 25, 1485–1491 CrossRef CAS; V. César, V. Mallardo, A. Nano, G. Dahm, N. Lugan, G. Lavigne and S. Bellemin-Laponnaz, Chem. Commun., 2015 10.1039/c4cc08641d ; and references therein.
- L. Benhamou, E. Chardon, G. Lavigne, S. Bellemin-Laponnaz and V. César, Chem. Rev., 2011, 111, 2705–2733 CrossRef CAS PubMed.
- No signs of decomposition were observed upon heating a toluene solution of 5 to 100 °C.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- The expression TEP = 0.847νav(CO) + 336 cm−1 was used. (See ref. (7a)).
- For CV and IR data of a range of iridium NHC complexes see (a) S. Leuthäußer, D. Schwarz and H. Plenio, Chem.–Eur. J., 2007, 13, 7195–7203 CrossRef PubMed; (b) R. A. Kelly III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202–210 CrossRef; (c) S. Wolf and H. Plenio, J. Organomet. Chem., 2009, 694, 1487–1492 CrossRef CAS; (d) A. R. Chianese, X. Li, M. C. Janzen, J. W. Faller and R. H. Crabtree, Organometallics, 2003, 22, 1663–1667 CrossRef CAS.
- Crystal structure of SIMes·[Ir(cod)Cl] taken from R. A. Kelly III, H. Clavier, S. Giudice, N. M. Scott, E. D. Stevens, J. Bordner, I. Samardjiev, C. D. Hoff, L. Cavallo and S. P. Nolan, Organometallics, 2008, 27, 202 CrossRef.
- The calculations of the buried volume %Vbur were carried out using the SambVca Web Application (https://www.molnac.unisa.it/OMtools/sambvca.php) by Cavallo and coworkers: A. Poater, B. Cosenza, A. Correa, S. Giudice, F. Ragone, V. Scarano and L. Cavallo, Eur. J. Inorg. Chem., 2009, 13, 1759–1777 CrossRef . Parameters used: sphere radius 3.5 Å, distance from the center of the sphere: 2.10 Å, mesh spacing: 0.05 Å, H atoms were not included, Bondi radii were scaled by 1.17.
Footnotes |
| † Electronic supplementary information (ESI) available: Methods and additional data. CCDC 10401231040125–1040128. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03966a |
| ‡ Dedicated to Professor Manfred Scheer on the occasion of his 60th birthday. |
| This journal is © The Royal Society of Chemistry 2015 |