
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSemi-synthesis of a HGF/SF kringle one (K1) domain scaffold generates a potent in vivo MET receptor agonist†
Claire
Simonneau‡
a,
Bérénice Leclercq‡
a,
Alexandra
Mougel
a,
Eric
Adriaenssens
a,
Charlotte
Paquet
b,
Laurent
Raibaut
a,
Nathalie
Ollivier
a,
Hervé
Drobecq
a,
Julien
Marcoux
c,
Sarah
Cianférani
c,
David
Tulasne
a,
Hugo
de Jonge
d,
Oleg
Melnyk
 *a and
Jérôme
Vicogne
*a
*a and
Jérôme
Vicogne
*a
aUMR CNRS 8161 CNRS, Université de Lille, Institut Pasteur de Lille, 1 rue du Pr Calmette, 59021 Lille Cedex, France. E-mail: jerome.vicogne@ibl.cnrs.fr; oleg.melnyk@ibl.cnrs.fr
bSIRIC ONCOLille, Maison Régionale de la Recherche Clinique, 6 rue du Pr. Laguesse, 59037 Lille Cedex, France
cUMR 7178 CNRS, Laboratoire de Spectrométrie de Masse BioOrganique (LSMBO), IPHC-DSA, Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg, France
dDivision of Immunology and General Pathology, Department of Molecular Medicine, University of Pavia, 9 via A Ferrata, 27100 Pavia, Italy
First published on 29th January 2015
Abstract
The development of MET receptor agonists is an important goal in regenerative medicine, but is limited by the complexity and incomplete understanding of its interaction with HGF/SF (Hepatocyte Growth Factor/Scatter Factor). NK1 is a natural occurring agonist comprising the N-terminal (N) and the first kringle (K1) domains of HGF/SF. In the presence of heparin, NK1 can self-associate into a “head to tail” dimer which is considered as the minimal structural module able to trigger MET dimerization and activation whereas isolated K1 and N domains showed a weak or a complete lack of agonistic activity respectively. Starting from these structural and biological observations, we investigated whether it was possible to recapitulate the biological properties of NK1 using a new molecular architecture of isolated N or K1 domains. Therefore, we engineered multivalent N or K1 scaffolds by combining synthetic and homogeneous site-specifically biotinylated N and K1 domains (NB and K1B) and streptavidin (S). NB alone or in complex failed to activate MET signaling and to trigger cellular phenotypes. Importantly and to the contrary of K1B alone, the semi-synthetic K1B/S complex mimicked NK1 MET agonist activity in cell scattering, morphogenesis and survival phenotypic assays. Impressively, K1B/S complex stimulated in vivo angiogenesis and, when injected in mice, protected the liver against fulminant hepatitis in a MET dependent manner whereas NK1 and HGF were substantially less potent. These data reveal that without N domain, proper multimerization of K1 domain is a promising strategy for the rational design of powerful MET agonists.
Introduction
Hepatocyte growth factor/scatter factor (HGF/SF) and MET, a member of the receptor tyrosine kinase family (RTK),1 are frequently involved in tumorigenesis and metastasis processes,2,3 and constitute major targets for the development of cancer therapies.4 In physiological conditions, HGF/SF plays an essential role in embryogenesis and tissue regeneration5 of several tissues and organs such as the liver,6 skin7 and kidney8 due to its strong mitogenic, motogenic and morphogenic properties.9–11 HGF/SF also stimulates angiogenesis without causing vascular inflammation and permeability, and thereby stimulates wound-healing.12,13 Mature HGF/SF is a 90 kDa disulfide-linked α/β heterodimer.14,15 The α-chain is composed of an N-terminal extension (N domain), containing a high affinity heparin binding site,16 followed by four kringle domains (K1 to K4). Contiguous N and K1 domains are believed to constitute the high affinity HGF/SF binding site for MET with critical residues located within K1 domain.17,18 The β-chain consists of an enzymatically inactive serine protease homology domain (SPH) which comprises the secondary low affinity HGF/SF binding site for MET.19 NK1, a natural 20 kD HGF/SF variant, is a partial MET agonist, and requires heparan sulfate (HS) as co-factor for activity.17,20,21 The quaternary structure of NK1 dimer as determined by X-ray crystallography shows a “head-to-tail” homodimer which is believed to be responsible for MET dimerization and activation.22MET agonists could have major applications in regenerative medicine. However, several limitations emanate from using HGF/SF or NK1 as a therapeutic agents. Unfortunately, HGF/SF is difficult to produce by recombinant methods. In addition, HGF/SF and NK1 are sensitive to proteolysis and are poorly diffusible into tissues due to the N domain strong interaction with the extracellular matrix.20,23 As a consequence, the systemic activity of HGF/SF in vivo has been observed only at very high doses incompatible with a clinical development.24 Therefore, the development of novel potent MET agonists20,25 or HGF/SF activators26 acting systemically is of utmost medical importance.
The rational design of MET agonists is complicated by the only partial understanding of the MET-HGF/SF interaction. No crystal structure of NK1 in complex with a soluble MET extracellular domain is yet available. The biological data accumulated for the NK1 molecule led most investigators to consider this structure as the minimal unit for MET agonist design. Indeed, several NK1 derivatives having either altered HS binding properties25,27 or improved dimeric stability28,29 were produced with the goal to promote tissue diffusion capacities and MET agonistic potential. A similar strategy was applied for the design of a MET antagonist with potential applications in cancer targeted chemotherapy.30 Nevertheless and despite all the attempts to propose a unified and convergent NK1–MET interaction model, the minimal NK1 molecular determinants for MET dimerization and activation are still debated. In particular, several studies examining the function of monomeric N and K1 domains have been reported, with divergent conclusions on the capacity of N domain to directly bind MET.14,31 We believe that the lack of consensus on the role of N and K1 domains is due to the inability of monomeric N and K1 domains to interrogating the functioning of HGF/SF-MET, which, by nature, is a complex multivalent system.32,33
In an effort to make progress in understanding the HGF/SF agonist determinants and pave the way for novel MET agonist design, we produced site-specifically biotinylated analogs of the N and K1 domains (NB and K1B) of HFG/SF. The linear polypeptides were produced by total chemical synthesis34,35 using a one-pot three peptide segments assembly strategy36,37 and folded to produce highly homogeneous material. Chemical synthesis ensured the production of proteins with an atom-by-atom control of the protein structure and of the semisynthetic scaffolds. Moreover, synthetic proteins are devoid of biological contaminants such as HS or heparin that could generate artifacts in the binding assays. In a second step, we produced semisynthetic38,39 and multivalent N or K1 constructs by combining NB or K1B with streptavidin homotetramer, with the aim of presenting (at least) two copies of N or K1 domains to the MET receptor. Indeed, the crystal structure of streptavidin allowed us to anticipate that two biotin binding sites within the streptavidin tetramer could serve as an appropriate scaffold for presenting two NB or K1B domains with a distance and orientation mimicking those found in the crystal structure of the NK1 homodimer.
NB or NB/S complexes failed to bind and activate MET receptor or induce any phenotype in various in vitro cell assays. In contrast, the K1B/S complex bound MET and induced a strong MET activation and specific cellular phenotypes in comparison to K1B monomer. These assays enabled us to study the importance of the distance between K1B domains within the complex by comparing K1B/S to another scaffold generated using anti-biotin antibodies (K1B/Ab).
Importantly, the K1B/S complex also displayed a potent MET agonist activity in vivo. It induced local angiogenesis and protected mice from Fas-induced fulminant hepatitis, in comparison to NK1, which had a limited effect, and HGF/SF, which had no effect. These data demonstrate for the first time that N domain is not required for engineering a strong MET agonist and that scaffolds based on K1 domain alone could promote tissue protection and/or regeneration.
Results
Total chemical synthesis of biotinylated K1 and N domains
The K1 domain (HGF/SF 125–209) is composed of 85 amino acid residues, and its tertiary structure is stabilized by three disulfide bonds (Fig. 1A). In K1B, the K1 primary structure was extended at the C-terminus by addition of two glycine residues and a lysine residue modified on its side chain with a biotin group. | ||
| Fig. 1 K1B total chemical synthesis. (A) Structure of the K1 domain of HGF/SF (residues 125–209, extracted from PDB 1BHT). The annotation was done according to UniProt database (entry P14210) with the 3 internal cysteine bridges and C-term biotin. (B) Scheme of one-pot assembly and folding of K1B. (C) RP-HPLC characterization of the crude linear K1B domain (left), the purified K1B domain (center) and MS analysis of folded K1B domain (right). | ||
The chemical synthesis of K1B was performed using a one-pot sequential three peptide segments assembly process,36,37 which required the preparation of HGF/SF segments 125–148 (segment 1), 149–176 (segment 2) and 177–209 (segment 3), the latter with the lysine-biotin extension (Fig. 1B). A thioester and bis(2-sulfanylethyl)amido cyclic disulfide (SEAoff) group were introduced on the C-terminus of peptide segments 1 and 2 respectively. Assembly of K1B linear polypeptide started by joining thioester segment 1 with segment 2 using the Native Chemical Ligation reaction.40 The reaction led to the successful formation of segment 1–2 featuring C-terminal SEAoff group acting as a blocked thioester moiety.36,41,42 Then, activation of the SEAoff group by reduction with tris(2-carboxyethyl)phosphine (TCEP) and addition of biotinylated segment 3 triggered the SEA native peptide ligation step43 and the successful formation of linear K1B domain as shown by the LC-MS of the crude reaction mixture (Fig. 1C, left). Linear K1B was further purified by HPLC to give 3.6 mg (40% overall) of homogeneous material and folded using the glutathione–glutathione disulfide redox system (Fig. 1C, center & right). Proteomic analysis of the folded K1B domain demonstrated the formation of the native disulfide bond pattern.
Interestingly, a MET phosphorylation assay using HeLa cells (Fig. S1A†) and cell scattering assay using MDCK cells (Fig. S1B†) showed that K1B activity was indistinguishable from unmodified synthetic K1 domain and behaved as a micromolar MET agonist, as already reported for recombinant K1 domain.14 Thus, biotinylation had no detectable influence on the biological activity of K1B at this stage.
The N domain of HGF/SF is composed of 97 residues (31–127) and is stabilized by two disulfide bonds.15 The total chemical synthesis of N domain modified with a C-terminal biotin group (NB) was performed using the same strategy as described in detail elsewhere.44 NB showed a disulfide bond pattern and a secondary structure similar to those reported for the recombinant N domain. Although synthetic NB was functional in regard to its capacity to bind heparin or heparan sulfate molecules,44 a property which is critically dependent on the tertiary structure of the protein, it had no detectable MET agonistic activity in cell assays (Fig. S1C†) in agreement with previous studies.14,31
Design of N and K1 multivalent complexes
Analysis of the relative positions of N and K1 domains in the NK1 homodimer crystal structure reveals that the C-termini of the two N domains and the C-termini of the two K1 domains are separated by only ∼1.3–2 nm (Fig. 2A).45 Interestingly, the individual biotin binding sites within a streptavidin homotetramer (S) are separated by distances of ∼2.0–3.5 nm (see Fig. S2A†). Therefore, we anticipated that the formation of K1B/S or NB/S complexes might recapitulate the distances and positions of N and K1 domains found in NK1 dimer independently of each other.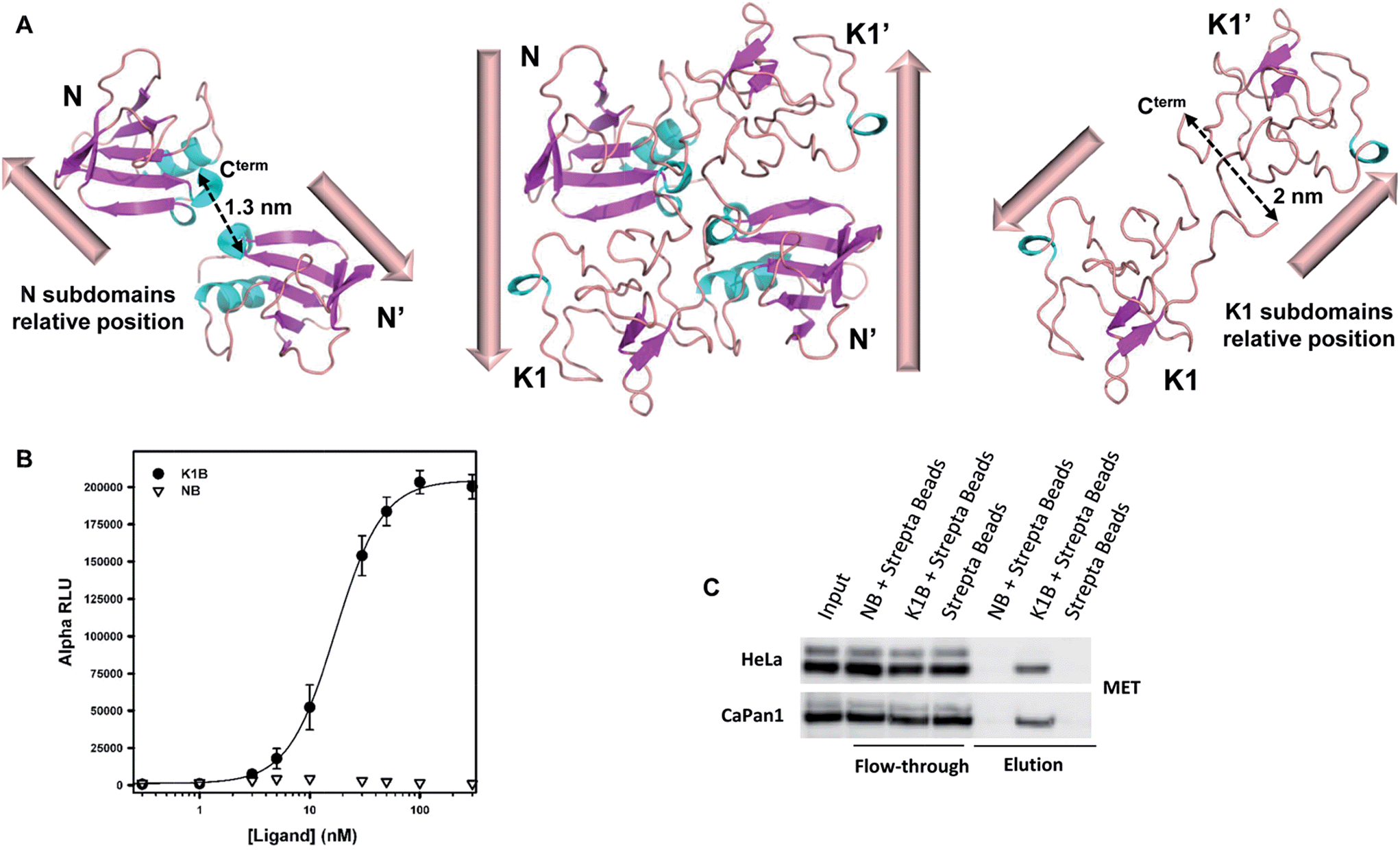 | ||
| Fig. 2 K1B and NB MET binding properties. (A) Structure of NK1 dimer (center, PDB 1BHT) and spatial relative orientation of each N (left) and K1 (right) monomers within the dimer. Dashed arrows indicate distances between subdomain C-termini. (B) NB, K1B and MET-Fc binding assay. Increasing concentrations of NB or K1B were mixed with extracellular MET domain fused with human IgG1-Fc (MET-Fc), and incubated with streptavidin AlphaScreen donor beads and Protein A acceptor beads. Error bars correspond to standard error (±SD) of triplicates. (C) Endogenous MET capture. Streptavidin coated beads loaded with NB or K1B were incubated with HeLa or CaPan1 total cell lysates. Input, flow-through and elution fractions from NB or K1 loaded beads were analyzed by specific total MET Western blot. | ||
The binding of K1B/S and NB/S complexes to MET was examined using AlphaScreen® technology. NB or K1B was loaded on streptavidin-coated donor beads and incubated with recombinant extracellular MET-Fc chimera loaded on Protein A-coated acceptor beads. If K1B/S or NB/S donor beads interact with MET-Fc/Protein A acceptor beads, a chemical energy transfer is possible between the beads, leading to fluorescence emission upon laser excitation.
NB showed background signal intensities in this saturation experiment, and thus was unable to bind MET in this assay. In contrast, K1B induced strong signal intensities with an apparent dissociation constant KD (∼16 nM) about 100-fold lower than the KD reported14 for monomeric K1 protein–MET interaction (Fig. 2B). Since the bead-based AlphaScreen® assay can generate avidity and thus introduce a bias in the estimation of the apparent KD in saturation experiments, we performed the reciprocal competition assay by adding increasing concentrations of preformed K1B/S complex (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio) into the K1B/MET-Fc/AlphaScreen bead mixture (Fig. S2B†). With this competition assay, we estimated a KD of ∼14.6 nM in good agreement with the apparent K1B/MET-Fc KD from the saturation assay.
:1 molar ratio) into the K1B/MET-Fc/AlphaScreen bead mixture (Fig. S2B†). With this competition assay, we estimated a KD of ∼14.6 nM in good agreement with the apparent K1B/MET-Fc KD from the saturation assay.
We complemented this study by examining the binding of NB/S and K1B/S complexes to endogenous MET from a whole cell lysate (Fig. 2C). Streptavidin-coated agarose beads were incubated with NB or K1B to form immobilized complexes, which were subsequently incubated with whole lysate from HeLa or CaPan1 cells.
Western blot analysis of the eluted material showed that only K1B/S complexes were able to capture MET from cell lysates.
Collectively, these data show that N domain does not bind endogenous or recombinant MET receptor to the contrary of K1B/S complex which binds MET at low nanomolar concentration. Importantly, the high affinity of K1B/S for MET shows the critical role played by multivalency in the K1–MET interaction system.
K1B/S but not NB/S activates MET and downstream signaling
These results set the stage for evaluating the K1B/S complex agonistic activity using in vitro cell assays in the human HeLa cell line. For this, we fixed the stoichiometry for K1B/S complex formation to 2:1, which generates several species varying in the number of K1B proteins bound per streptavidin tetramer. With this molar ratio, and by assuming that each biotin binding unit is independent, the probability of having 0, 1, 2, 3 or 4 K1B proteins bound per streptavidin should correspond to 6%, 25%, 38%, 25% and 6% respectively, meaning 70% of K1B/S multimers in theory. These K1B/S multimers were indeed identified by SDS-PAGE analysis (Fig. S3A†)46 and by native mass spectrometry analysis (Fig. S3B–D†). Using the latter technique, we estimated that the 2:1 K1B:S molar ratio resulted in 75% of the K1 domain presented at least as pairs within K1B/S multimers. In practice, we noticed that a 2:1 K1B:S molar ratio was sufficient to achieve a maximum cellular response, since a higher proportion of K1B in the mixture from 3:1 up to 8:1 led to no improvement in potency (Fig. S3E†).
We also designed another complex produced by mixing K1B with an anti-biotin antibody (Ab) in a 2:1 molar ratio. The antibody is expected to produce consistent K1B dimers, albeit with a distance of ∼6–13 nm between each K1B protein, which is much larger than those found between the K1 domains in the NK1 crystal structure or in K1B/S complexes (Fig. S3F†).47,48
MET activation and downstream signaling in HeLa cells upon HGF/SF, K1B, K1B/S, K1B/Ab or recombinant NK1 incubation was analyzed by Western blot and quantified by HTRF approaches (Fig. 3A and B). Typically, HGF/SF triggered maximal ERK and Akt activation down to pM concentrations. Impressively, K1B/S complexes were able to trigger ERK and Akt phosphorylation levels down to a low nM range, and thus displayed an agonist activity similar to NK1 protein. Moreover, K1B/S but not K1B induced a strong MET phosphorylation at 100 nM. The fact that activation of MET by monomeric K1B was detected only for μM concentrations, as reported in the literature, highlights again the critical role played by multivalency for achieving strong receptor activation.
 | ||
| Fig. 3 MET signaling analysis upon K1B/S stimulation. (A) HeLa cells were treated for 7 min with 50 nM streptavidin (S), 50 nM anti-biotin antibody (Ab), 500 pM mature HGF/SF (HGF), 100 nM and 1 μM K1B, 100 nM K1B/S, 100 nM K1B/Ab and 100 nM NK1. Cell lysates were then analyzed by specific total MET, Akt and ERK or phospho-MET, phospho-Akt and phospho-ERK Western blot. Ctrl: vehicle, MW: molecular weight. (B) HeLa cells were treated with increasing concentrations of mature HGF/SF, K1B/S, NK1 and K1B/Ab for 7 min. Activation levels of ERK and Akt were measured using HTRF technology, and plotted as the 665/620 nm HTRF signal ratio. (C) K1B/S and NK1, K1B/Ab kinetic analysis. HeLa cells were treated with 100 nM K1B/S or NK1, for 1, 5, 10, 20, 30, 40 or 90 min. Cell lysates were then analyzed by specific total MET, Akt and ERK or phospho-MET, phospho-Akt and phospho-ERK Western blot. (D) HGF/SF, K1B/S, NK1 and K1B/Ab kinetic analysis. HeLa cells were treated with optimal concentration of 100 pM HGF/SF, 50 nM K1B/S, 50 nM NK1 or 400 nM K1B/Ab for 1, 3, 5, 7, 10, 15, 20, 30, 60 or 90 min. Activation levels of ERK and Akt were measured using HTRF technology and plotted as the 665/620 nm HTRF signal ratio. | ||
A similar multivalent process was evident for the K1B/Ab complex, which unlike K1B, also induced a MET phosphorylation at 100 nM. However, K1B/Ab was substantially less active than K1B/S for triggering ERK and Akt downstream signaling (Fig. 3A and B). Intrigued by this difference, we analyzed the MET phosphorylation pattern at the tyrosine level. Indeed, auto-phosphorylation of tyrosines 1234 and 1235 is the first event leading to MET activation. It is crucial for unlocking and maintaining sustained kinase activity. Subsequently, phosphorylation of C-terminal tyrosines 1349 and 1356 is required to provide recognition sites for scaffolding partners that propagate, amplify and diversify MET signaling.49 Both K1B/S and K1B/Ab activated MET auto-phosphorylation onto tyrosines 1234 and 1235. However, and unlike K1B/S, K1B/Ab failed to trigger phosphorylation of tyrosines 1349 and 1356 (Fig. S3G†), and thus, to trigger the critical downstream signaling cascade. This fact might be due to the large distance between K1B domains in the antibody complex and thus to the suboptimal stabilization of MET dimers.
We also determined the MET and downstream signaling activation kinetics (0–90 min) using Western blot (Fig. 3C and S3H†) and HTRF analytical techniques (Fig. 3D). Typically, HGF/SF induced a maximum of MET auto-phosphorylation between 5 and 30 min (Fig. S3H†) that induced a strong downstream Akt and ERK phosphorylation after ∼5–10 min followed by a slow decrease over time. In comparison, Western blot analysis revealed that MET phosphorylation proceeded faster with K1B/S or NK1 than with HGF/SF, i.e. within the very first minute of incubation (Fig. 3C). A strong downstream phosphorylation of ERK and Akt as quantified by HTRF was observed after 5–7 min (Fig. 3D), that is faster than for HGF/SF too, but faded rapidly to become below HGF/SF levels. Finally, the kinetics for MET activation and downstream Akt and ERK phosphorylation by K1B/Ab were similar to those observed for K1B/S or NK1 (Fig. 3D and S3H†), albeit with much lower levels.
Importantly and as expected from binding experiments, NB/S complex showed no agonistic activity (Fig. S4A†), and did not promote any cellular phenotypes (Fig. S4B†).
K1B/S promotes strong and specific cell phenotypes and angiogenesis
We next evaluated the ability of MET agonists to induce cell scattering in MDCK cells, the reference cell line for this phenotypic assay (Fig. 4A).50 In the presence of HGF/SF or NK1 for 18 h, MDCK cells acquired a mesenchymal-like phenotype and scattered as expected.51 Impressively, K1B/S complex induced a similar phenotype, whereas scattering with K1B and K1B/Ab was weak. Obviously, the result of the scattering assay was strongly correlated with the capacity of the agonists to induce a sustained phosphorylation of MET, ERK and Akt kinases (see Fig. 3). Further cell assays were performed using lumina basal like matrix (Matrigel®) as a mimic of basement extracellular matrix. In these conditions, untreated MDCK cells form spontaneously tight spherical clusters within 24 h. In contrast, stimulation of MDCK cells with HGF/SF or NK1 triggered their self-organization into branched and connected structures (Fig. 4B). Likewise, K1B/S widely promoted the formation of such structures while K1B and K1B/Ab were unable to do so. To extend this observation in vivo, the different agonists were injected subcutaneously with Matrigel plugs into immunodeficient SCID mice to induce angiogenesis. Indeed, HGF/SF is a potent angiogenic factor that stimulates endothelial cell proliferation and migration.52 The plugs were extracted after 11 days to determine the quantity of hemoglobin infiltrated into the plug as a measure of angiogenesis induced (Fig. 4C). As expected, VEGF or HGF/SF showed potent angiogenic properties compared to control plugs. Remarquably, K1B/S induced the formation of vessels with a hemoglobin content comparable to that of VEGF and significantly higher than those induced by NK1 or K1B. Thus, while NK1 and K1B/S displayed similar potencies in in vitro cell assays, their angiogenic properties were significantly different in vivo in favor of the semi-synthetic construct. | ||
| Fig. 4 Cellular phenotypes induced by K1B/S. (A) Cell scattering assay. MDCK isolated cell islets were incubated for 18 h in culture media with 50 nM streptavidin (S), 50 nM anti-biotin antibody (Ab), 500 pM mature HGF/SF (HGF), 100 nM K1B, 100 nM K1B/S, 100 nM NK1 and 100 nM K1B/Ab. Cells were then stained and observed under microscope (40×). (B) Matrigel morphogenesis assay. MDCK cells were seeded onto a layer of Matrigel and treated for 18 h with 50 nM streptavidin (S), 50 nM anti-biotin antibody (Ab), 500 pM mature HGF/SF (HGF), 100 nM K1B, 100 nM K1B/S, 100 nM NK1 and 100 nM K1B/Ab. Cells were then observed under microscope (40×). (C) Angiogenesis. Mice were injected with a mixture of Matrigel and 1 nM HGF/SF (HGF), 10 nM VEGF, 100 nM NK1, 100 nM K1B/S, 100 nM K1B or 50 nM S. Hemoglobin absorbance was measured and concentration was determined using a rate hemoglobin standard curve and plug weight. ANOVA tests were performed to compare all the means, and a P-value < 0.001 was considered to indicate a statistically significant difference. (D) MTT assay. MDCK cells were cultured overnight (15 h) in medium containing 0.1% FBS with or without anisomycin (0.7 μM) and in the presence of 500 pM mature HGF/SF (HGF), 100 nM K1B, 100 nM K1B/S, 100 nM NK1 and 100 nM K1B/Ab. An MTT assay was then performed to evaluate cell survival. Results are expressed as the percentage of untreated control. An ANOVA test was performed to compare the 3 means, with a P-value < 0.05 considered statistically significant. | ||
Finally, we examined the capacity of the agonists to promote the survival of cells after apoptotic stress. This phenotype is a hallmark of HGF/SF, which can protect many cell types against death induced by serum depletion, ultra-violet radiation or exposure to some chemical substances. We used anisomycin as stress-inducer, a DNA and protein synthesis inhibitor which induces cell death by apoptosis.53 Anisomycin treatment induced ∼90% of cell death after 16 h, but only 50% of cell death when pre-treated with HGF/SF (Fig. 4D). Interestingly, K1B/S displayed a survival rate similar to NK1, whereas K1B or K1B/Ab complex failed to protect the cells to a significant extent.
Taken together, these results tell us that K1B/S complex fully mimics, in vitro, the properties of NK1 as a potent MET agonist. When properly dimerized, K1 domain alone is sufficient for strong MET activation. The reduced activity of K1B/Ab suggests that the distance and/or orientation which separate the two K1 domains are crucial to induce full MET activation. Another important conclusion is the structural role played by N domain in NK1 molecule. N domain does not bind MET directly and probably pre-organize two K1 domains for optimal presentation to MET receptor.
K1B/S complex activates MET in the liver and impairs Fas-induced fulminant hepatitis
The design of MET agonists acting potently in vivo is highly challenging. Given the above results, we were wondering if the K1B/S complex could act in vivo on distant tissues when injected systemically, and thus could constitute a basis for designing potent MET agonists of potential therapeutic interest.In a first approach, the different agonists were injected intravenously to see if they could activate MET and downstream pathways in the liver, an organ well known to strongly express MET receptor. After 10 min, livers were extracted and MET, ERK and Akt phosphorylation status was determined by Western blot (Fig. 5). K1B/S, NK1 and HGF/SF injection induced a clear MET phosphorylation associated with a strong Akt and ERK activation in the liver. Importantly, activation by K1B/S was detectable at doses as low as 2.5 pmol (250 ng) per g of body weight (Fig. S5A†) and even up to 30 min post-injection (Fig. S5B†). In contrast, K1B and streptavidin control led to no detectable signal.
 | ||
| Fig. 5 In vivo MET activation assays. FVB mice were injected intravenously with PBS (ctrl), 25 pmol K1B (250 ng), 25 pmol K1B/S complex (250 ng K1/700 ng S), 25 pmol NK1 (500 ng) or 2.5 pmol mature HGF/SF (250 ng) per g of body weight. After 10 min, livers were extracted, snap frozen and crushed. MET, Akt and ERK phosphorylation status in cell lysates was analyzed by Western blot. The data shown are representative of three independent experiments which were performed using two mice per condition. | ||
Motivated by the fact that K1B/S complex is able to diffuse into the liver through the blood circulation and induce MET activation, we further examined whether the complex could promote hepatocyte survival when an apoptotic stress was induced in the liver. Indeed, injection of an anti-Fas antibody (anti-CD95) in mice quickly induces a massive hepatocellular apoptosis leading to fulminant hepatitis and death of the animals.54 Previous studies showed that HGF/SF was able to abrogate Fas-induced fulminant hepatitis, but required prohibitive amounts to show significant effects (usually 1 nmol, i.e. ∼100 μg per mouse).24 In our assay, anti-Fas antibody was co-injected with 25 pmol of K1B, K1B/S or NK1, or 2.5 pmol of mature HGF/SF per g of body weight as these agonist concentrations were sufficient to promote strong MET signaling for at least 30 min. After 90 min, a second injection of each protein was performed to sustain signaling. Livers were extracted after 3 additional hours for histological and molecular analysis.
Macroscopically, mice treated with anti-Fas antibody and K1B, NK1 or mature HGF/SF presented an altered liver, retaining a deep brown color even after PBS perfusion and elimination of vascular blood content (Fig. 6A). Remarkably, mice treated with K1B/S maintained a clear liver, almost intact. Histological analysis demonstrated that this dark color was mostly induced by a vascular congestion attributable to a massive hepatocyte loss and subsequent blood infiltration (Fig. 6B). Controls and HGF/SF treated mice showed totally disorganized livers with significant blood infiltration. In contrast, K1B/S mice kept well organized structures, although some blood infiltration could be visualized too. NK1 treated mice presented an intermediate phenotype, retaining some organized areas but with massive blood infiltration. Further analysis confirmed that these disorganized regions corresponded to large clusters of apoptotic hepatocytes (Fig. 6C). Interestingly, all the mice challenged with anti-Fas antibody showed the early molecular markers characteristic for apoptosis such as cleaved caspase 3 and PARP1/2, even for the animals which were protected by K1B/S complex (Fig. S5C†). These results show that K1B/S doesn't act on the initial steps following Fas receptor activation but rather on downstream intracellular apoptotic signaling.24,55
 | ||
| Fig. 6 (A) FVB mice were injected intravenously with 125 ng anti-Fas monoclonal antibody (aFas) mixed with 25 pmol K1B (250 ng), 25 pmol K1B/S complex (250 ng/700 ng), 25 pmol NK1 (500 ng) or 2.5 pmol mature HGF/SF (250 ng) per g of body weight, or PBS. A second injection without anti-Fas was performed 90 min later. Livers were extracted and fixed in formalin after 3 additional hours. (B) Frozen liver sections were stained with hematoxylin–eosin for histological observation (40×). (C) Frozen liver sections were treated with Apoptag® Kit for apoptotic nuclei labelling (green) and counterstained with DAPI for total nuclei labelling (blue) (100×, insert: 200× on apoptotic cells). | ||
These histological and molecular analyses demonstrated that K1B/S complex acts systematically, efficiently activates MET signaling in the liver and is a potent survival factor even in extreme apoptotic stress conditions. The fact that K1B/S was more potent than NK1 highlights the significance of these findings for future MET agonist design.
Discussion
A multivalent interaction is defined as an interaction in which two or more molecular recognition events take place simultaneously between the two interacting bodies. Biological systems make extensive use of multivalency to achieve high specificities and apparent affinities, i.e. avidities, compared to their component monovalent interactions.56,57 The HGF/SF-MET interaction belongs to the family of multivalent interactions and is regulated by proteolytic maturation.This maturation converts the inactive pro-HGF/SF form into a heterobivalent ligand which can stabilize MET dimer formation, trans-phosphorylation and intracellular signaling. Recent work showed that synthetic peptides can bind endogenous HGF/SF beta chain and induce conformational changes that mimic mature HGF/SF.26
This work uncovers another of its essential components by showing that proper dimerization of K1 domain in the absence of N domain enables the reconstitution of a potent MET agonist. It gives a deeper understanding of MET activation mechanisms by HGF/SF which is crucial for the rational design of future drugs. To date, NK1 was considered the minimal HGF/SF-derived fragment able to activate MET signaling at “physiological concentrations”,58 apart from K1 alone, which is active, but only at very high concentration.14 Earlier studies suggested that, within NK1, N is the high affinity HS binding domain20 and K1 is the major subdomain of HGF/SF involved in MET binding and activation.17,18 However, attempts to reproduce potent MET agonistic activities based on K1 domain alone have failed until now. Previous studies using recombinant17 or synthetic K1 protein36 showed a weak micromolar agonist activity which relies on the presence of heparan sulfates which might cluster several K1 molecules and thus enable a multivalent presentation of this domain to MET receptor, apparently with poor efficacy.
Note that another approach to generate MET agonist used dimerized engineered internalin B (InlB) from Listeria monocytogenes,59 a surface bacterial protein that binds and activates MET60 but in a different way than HGF/SF.61
Our experiments clarify also the role of the N domain which is not involved in direct MET binding but rather organizes NK1 protein in a dimeric structure. Therefore, multivalent presentation of K1 domain is sufficient to achieve high potency in various in vitro cellular and in vivo assays. We noticed also that the distance between the K1 domains seems to be a critical parameter to obtain full MET activation. The two K1 domains within the NK1 dimer are very close and separated by 2 nm. The distances between two K1B within the K1B/S complex range from 2.5 to 3.5 nm, and are thus close to the K1–K1 distances measured in the native NK1 structure. The streptavidin tetramer linking two K1B domains can also be considered as a rigid linker. In contrast, the two paratopes of the anti-biotin antibody are separated by 6–13 nm and subject to significant conformational flexibility. In all our experiments, the K1B/Ab complex induced only a partial MET phosphorylation and a weak signaling induction while K1B/S complex was able to induce activation comparable to that of NK1. We believe that the high agonist activity of K1B/S relies on a better distance fit and rigidity of the K1 proteins within the complex.
Dose-response and kinetics experiments clearly indicated that K1B/S and NK1 possess similar agonistic activities in cell assays, and are efficient up to the low nanomolar range. However, mature HGF/SF still remains the best MET agonist in cell-based assays, being ∼100 to 200 times more potent. Our work suggests that for achieving such a high affinity, two K1 domains and one or two SPH domains synergize in a hetero-multivalent interaction to stabilize a MET dimer.
NK1 and K1B/S induced similar cell survival, scattering and morphogenesis phenotypes, but presented unexpected and distinct properties when used in vivo. K1B/S complex strongly promoted angiogenesis whereas NK1 failed. These data suggest that NK1 and K1B/S possess distinct diffusion and/or dimerization properties in the Matrigel® plug and/or probably different half-lives when injected in vivo. In particular, the sensitivity of the basic N domain within NK1 to proteolytic degradation might explain the poor pro-angiogenic activity of this protein in assays that span several days. In contrast, NK1 and K1B/S triggered a strong MET and downstream cascade in hepatocytes 10 min after intravenous injection, showing that these proteins circulated in the blood and could activate MET in a short period of time.
HGF/SF can abrogate a Fas-induced fulminant hepatitis62 but only at very high doses in animals (>100 μg per mouse) which are not compatible with a clinical development.24 Importantly, K1B/S complex could efficiently protect the liver from Fas-induced apoptosis at very low doses (<5 μg per mouse) compared to NK1 and HGF/SF. This potent protective effect is probably generated by an efficient presentation of a stable dimer (or multimer) within the complex in the absence of the “sticky” N domain.
Materials and methods
Chemical protein synthesis
Total chemical synthesis of K1 C-terminal biotin (K1B) and N C-terminal biotin (NB) was performed using 3 fragments in a one-pot protocol process, as described for the synthesis of biologically active K1 domain of HGF-SF.36 Final purification of the full length synthetic 88 residues polypeptide and folding with concomitant formation of the 3 disulfide bridges gave synthetic biologically active K1B. The protein was aliquoted and stored at −80 °C. Total chemical synthesis of NB was also performed using 3 fragments in a one-pot protocol process, as described in ref. 44.Design of K1B/S complex
NK1 (entry 1BHT) and streptavidin (entry 1SWE) structures were obtained from the PDB database (http://www.rcsb.org/). Extraction of K1 domain portion, visualization and distance measurements were performed on PyMol v1.7 software (http://www.pymol.org).Preparation of K1B/S, NB/S or K1B/Ab complexes
A typical procedure is illustrated with the preparation of a 100 nM K1B/S complex at 2:1 molar ratio.
Solutions of K1B (10 μM) and streptavidin (5 μM) were prepared in PBS. The protein solutions (5 μL for each) were mixed a few seconds in a low binding Eppendorf tube and incubated without agitation at 20 °C for 15 min to allow K1B/S complex formation (final concentration 5 μM). This solution was further diluted in Dulbecco's Modified Eagle's Medium (DMEM, pre-warmed at 37 °C, 490 μL) to a 100 nM final concentration. The same procedure was used for NB/S or K1B/Ab complex formation.
These solutions were used immediately after preparation.
AlphaScreen® saturation and competition assay
Saturation assays for binding of K1B to recombinant MET-Fc protein were performed in 384-well microtiter plates (OptiPlate™-384, PerkinElmer©, CA, USA, 50 μL of final reaction volume). Final concentrations were 0–300 nM for K1B, 2.5 nM for MET-Fc, 10 μg mL−1 for streptavidin coated donor beads and protein A-conjugated acceptor beads. The buffer used for preparing all protein solutions and the bead suspensions was: PBS, 5 mM HEPES pH 7.4, 0.1% BSA.For K1B and MET-Fc binding assay, K1B (10 μL, 0–1.5 μM) was mixed with solutions of hMET-Fc (10 μL, 10 nM). The mixture was incubated for 10 min (final volume 15 μL). Protein A-conjugated acceptor beads (10 μL, 50 μg mL−1) were then added to the vials. The plate was incubated at 23 °C for 30 min in a dark box. Finally, streptavidin coated donor beads (10 μL, 50 μg mL−1) were added and the plate was further incubated at 23 °C for 30 min in a dark box. The emitted signal intensity was measured using standard Alpha settings on an EnSpire® Multimode Plate Reader (PerkinElmer). For the competition assay: increasing concentrations of K1B/S complex (ratio 2:1) were added to pre-mixed K1B (20 nM)/MET-Fc (2 nM)/ALPHA bead (10 μg mL−1) complex. In this situation, IC50 approximates KD according to Cheng and Prusoff equation (See details in Fig. S2B†).
Endogenous MET capture
Streptavidin coated beads loaded with NB or K1B were incubated with HeLa or CaPan1 total cell lysates. Input, flow-through and elution fractions from NB or K1 loaded beads were analyzed by specific total MET Western blot.Cell culture and drug treatments
Madin Darby Canine Kidney (MDCK, kind gift of Dr Jacqueline Jouanneau, Institut Curie, Paris, France) and Human cervical cancer HeLa cells, purchased from ATCC® (American Type Culture Collection, Rockville, MD, USA), were cultured in DMEM medium (Dulbecco's Modified Eagle's Medium, Gibco, Karlsruhe, Germany), supplemented with 10% FBS (Fetal Bovine Serum, Gibco®, Life technologies, Grand Island, NY, USA) and 5 mL of ZellShield™ (Minerva Biolabs GmbH, Germany). Twenty-four hours before drug treatment, the medium was exchanged with DMEM containing 0.1% FBS, and cells were then treated for different times with different drugs.Akt and ERK phosphorylation assay by HTRF method
The assay was performed according to the manufacturer's protocol mentioned in HTRF® (Cisbio bioassays©, Bedford, MA, USA). Briefly, cells were plated, stimulated with different agonists (HGF/SF, NK1, K1B/S and K1B/Ab), and then lysed in the same 96-well culture plate. Lysates (16 μL) were transferred to 384-well microplates for the detection of phosphorylated Akt (Ser473) and ERK (Thr202/Tyr204) by HTRF® reagents via a sandwich assay format using 2 different specific monoclonal antibodies: an antibody labelled with d2 (acceptor) and an antibody labelled with Eu3+-cryptate (donor). Antibodies were pre-mixed (2 μL of each antibody) and added in a single dispensing step. When the dyes are in close proximity, the excitation of the donor with a light source (laser) triggers a Fluorescence Resonance Energy Transfer (FRET) towards the acceptor, which in turn fluoresces at a specific wavelength (665 nm). Upon laser excitation, energy transfer between d2 and Eu3+-cryptate molecules occurs and fluorescence is detected at 620 and 665 nm on an EnVision® Multilabel reader (PerkinElmer©). Data are presented as a 665/620 nm ratio for signal normalization.Angiogenesis
Immunodeficient SCID mice weighing 19–21 g (from Institut Pasteur of Lille, France) were used for this experiment. Mice were housed in a facility with a 12 h light/dark cycle at 22 °C and had free access to food and water. Mature HGF/SF, VEGF-A, NK1, K1B, streptavidin and K1/S complexes were added to growth factor reduced Matrigel® (BD Biosciences, Becton Dickinson, Belgium). Mice (n = 6) were injected subcutaneously in the flank with 400 μL of Matrigel. After 11 days, mice were sacrificed, Matrigel plugs were removed and weighed, and 300 μL of water was added to induce hypotonic red blood cell lysis and hemoglobin release. Hemoglobin absorbance (405 nm) was measured, and concentration was determined against a hemoglobin standard curve and plug weight.All experimental procedures were conducted with the approval of the Ethics Committee for Animal Experimentation of the Nord Pas de Calais Region (CEEA 75).
Fas-induced fulminant hepatitis
FVB mice weighing 19–21 g (Charles River) were used for this experiment. After anesthesia with isoflurane (Aerrane, Baxter, USA), mice (n = 3) were given intravenous injections of 125 ng per g body weight of anti-Fas antibody (Clone Jo-2, CD95, Pharmingen, BD Biosciences) mixed with different agonists (HGF/SF, NK1, and K1/S) in PBS. The mice were injected a second time with each agonist 90 min after the first injection. The mice were sacrificed after 3 additional hours, and their livers perfused with PBS supplemented with protease and phosphatase inhibitors.In parallel, to visualize MET activation in the liver, mice (n = 2) were given intravenous injections of each agonist for 10 min.
For histological analysis, liver tissue was collected, fixed overnight in 4% paraformaldehyde, and snap frozen in isopentane, submerged in liquid nitrogen, and embedded in OCT (Tissue-Tek®, VWR, PA, USA). Frozen liver sections (5 μm) were stained with hematoxylin and eosin (HE) for general morphology. TUNEL staining for apoptosis was also performed on liver sections according to the manufacturer's instructions (Apoptag® Fluorescein Direct In Situ kit, Merck Millipore, Billerica, MA, USA). For molecular analysis, extracted liver tissue was immediately frozen in liquid nitrogen. Livers were crushed in lysis buffer supplemented with freshly added protease and phosphatase inhibitors.
Other methods
Descriptions of reagents, antibodies, immunoblotting, MTT assay, scattering assay, morphogenesis assay and statistical analysis, see ESI.†Conclusion
This study reveals that N domain is not necessary to engineer strong MET agonists and that two K1 domains are sufficient by themselves to allow strong MET binding and activation.Moreover, due to its strong in vivo activity in promoting angiogenesis and liver protection, the K1 dimer structural motif can serve as a new basis for the rational development of potent MET agonists useful for liver regeneration or neovascularization enhancement. Future work will address the challenge of producing covalent K1 dimers as potent in vivo MET agonists.
Authors' contributions
C.S. and B.L. designed the research, performed the experiments and wrote the paper. A.M. performed the experiments and analyzed data. E.A and C.P. performed the animal experiments and histological analysis. L.R., N.O. and H.D. carried out total protein synthesis, purification and characterization. J.M. and S.C. carried out native mass spectrometry analysis. D.T. guided the research and analyzed data. H. G. provided recombinant NK1, analyzed data and wrote the paper. O.M. and J.V. conceived the project, guided the research, analyzed data and wrote the paper.Acknowledgements
We sincerely thank Dr Véronique Fafeur, Dr Roland Bourette and Giovanni De Nola for their fruitful and inspiring discussions, Pr Michel Raibaut and Dr Phillippe Briand for K1B/S assembly statistical models and probability analysis, and Prof. Ermanno Gherardi for his expert advice and material support. We thank the Chemistry Systems Biology (CSB) platform for technical advice. This work has been supported by CNRS, GEFLUC Flandres-Artois, Lille Métropole Communauté Urbaine, la Ligue Contre le Cancer du Septentrion, Institut Pasteur de Lille and by SIRIC ONCOLille, Grant INCa-DGOS-Inserm 6041.Notes and references
- M. A. Lemmon and J. Schlessinger, Cell, 2010, 141, 1117–1134 CrossRef CAS PubMed.
- M. G. Ponzo and M. Park, Cell Cycle, 2010, 9, 1043–1050 CrossRef CAS.
- Z. S. Zeng, M. R. Weiser, E. Kuntz, C. T. Chen, S. A. Khan, A. Forslund, G. M. Nash, M. Gimbel, Y. Yamaguchi, A. T. t. Culliford, M. D'Alessio, F. Barany and P. B. Paty, Cancer Lett., 2008, 265, 258–269 CrossRef CAS PubMed.
- E. Gherardi, W. Birchmeier, C. Birchmeier and G. Vande Woude, Nat. Rev. Cancer, 2012, 12, 89–103 CrossRef CAS PubMed.
- L. Trusolino, A. Bertotti and P. M. Comoglio, Nat. Rev. Mol. Cell Biol., 2010, 11, 834–848 CrossRef CAS PubMed.
- S. Mizuno and T. Nakamura, Regener. Med., 2007, 2, 161–170 CrossRef CAS PubMed.
- D. Bevan, E. Gherardi, T. P. Fan, D. Edwards and R. Warn, J. Pathol., 2004, 203, 831–838 CrossRef CAS PubMed.
- K. Kawaida, K. Matsumoto, H. Shimazu and T. Nakamura, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 4357–4361 CrossRef CAS.
- T. Nakamura, K. Nawa and A. Ichihara, Biochem. Biophys. Res. Commun., 1984, 122, 1450–1459 CrossRef CAS.
- R. Montesano, K. Matsumoto, T. Nakamura and L. Orci, Cell, 1991, 67, 901–908 CrossRef CAS.
- G. Maulik, A. Shrikhande, T. Kijima, P. C. Ma, P. T. Morrison and R. Salgia, Cytokine Growth Factor Rev., 2002, 13, 41–59 CrossRef CAS.
- R. Gong, Curr. Opin. Invest. Drugs, 2008, 9, 1163–1170 CAS.
- T. Kaga, H. Kawano, M. Sakaguchi, T. Nakazawa, Y. Taniyama and R. Morishita, Vasc. Pharmacol., 2012, 57, 3–9 CrossRef CAS PubMed.
- O. Holmes, S. Pillozzi, J. A. Deakin, F. Carafoli, L. Kemp, P. J. Butler, M. Lyon and E. Gherardi, J. Mol. Biol., 2007, 367, 395–408 CrossRef CAS PubMed.
- T. Nakamura, Prog. Growth Factor Res., 1991, 3, 67–85 CrossRef CAS.
- M. Lyon, J. A. Deakin, K. Mizuno, T. Nakamura and J. T. Gallagher, J. Biol. Chem., 1994, 269, 11216–11223 CAS.
- J. S. Rubin, R. M. Day, D. Breckenridge, N. Atabey, W. G. Taylor, S. J. Stahl, P. T. Wingfield, J. D. Kaufman, R. Schwall and D. P. Bottaro, J. Biol. Chem., 2001, 276, 32977–32983 CrossRef CAS PubMed.
- N. A. Lokker, L. G. Presta and P. J. Godowski, Protein Eng., 1994, 7, 895–903 CrossRef CAS PubMed.
- J. Stamos, R. A. Lazarus, X. Yao, D. Kirchhofer and C. Wiesmann, EMBO J., 2004, 23, 2325–2335 CrossRef CAS PubMed.
- D. Lietha, D. Y. Chirgadze, B. Mulloy, T. L. Blundell and E. Gherardi, EMBO J., 2001, 20, 5543–5555 CrossRef CAS PubMed.
- M. Lyon, J. A. Deakin, D. Lietha, E. Gherardi and J. T. Gallagher, J. Biol. Chem., 2004, 279, 43560–43567 CrossRef CAS PubMed.
- E. Gherardi, S. Sandin, M. V. Petoukhov, J. Finch, M. E. Youles, L. G. Ofverstedt, R. N. Miguel, T. L. Blundell, G. F. Vande Woude, U. Skoglund and D. I. Svergun, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 4046–4051 CrossRef CAS PubMed.
- G. Hartmann, T. Prospero, V. Brinkmann, C. Ozcelik, G. Winter, J. Hepple, S. Batley, F. Bladt, M. Sachs, C. Birchmeier, W. Birchmeier and E. Gherardi, Curr. Biol., 1998, 8, 125–134 CrossRef CAS.
- K. Kosai, K. Matsumoto, S. Nagata, Y. Tsujimoto and T. Nakamura, Biochem. Biophys. Res. Commun., 1998, 244, 683–690 CrossRef CAS.
- J. Ross, E. Gherardi, N. Mallorqui-Fernandez, M. Bocci, A. Sobkowicz, M. Rees, A. Rowe, S. Ellmerich, I. Massie, J. Soeda, C. Selden and H. Hodgson, Gastroenterology, 2012, 142, 897–906 CrossRef CAS PubMed.
- K. E. Landgraf, M. Steffek, C. Quan, J. Tom, C. Yu, L. Santell, H. R. Maun, C. Eigenbrot and R. A. Lazarus, Nat. Chem. Biol., 2014, 10, 567–573 CrossRef CAS PubMed.
- R. Sinha Roy, S. Soni, R. Harfouche, P. R. Vasudevan, O. Holmes, H. de Jonge, A. Rowe, A. Paraskar, D. M. Hentschel, D. Chirgadze, T. L. Blundell, E. Gherardi, R. A. Mashelkar and S. Sengupta, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13608–13613 CrossRef PubMed.
- M. Cassano, S. Biressi, A. Finan, L. Benedetti, C. Omes, R. Boratto, F. Martin, M. Allegretti, V. Broccoli, G. Cusella De Angelis, P. M. Comoglio, C. Basilico, Y. Torrente, P. Michieli, G. Cossu and M. Sampaolesi, PLoS One, 2008, 3, e3223 Search PubMed.
- D. S. Jones 2nd, P. C. Tsai and J. R. Cochran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 13035–13040 CrossRef PubMed.
- F. Cecchi, D. C. Rabe and D. P. Bottaro, Expert Opin. Ther. Targets, 2012, 16, 553–572 CrossRef CAS PubMed.
- T. Merkulova-Rainon, P. England, S. Ding, C. Demerens and G. Tobelem, J. Biol. Chem., 2003, 278, 37400–37408 CrossRef CAS PubMed.
- W. D. Tolbert, J. Daugherty-Holtrop, E. Gherardi, G. Vande Woude and H. E. Xu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13264–13269 CrossRef CAS PubMed.
- D. Kirchhofer, M. T. Lipari, L. Santell, K. L. Billeci, H. R. Maun, W. N. Sandoval, P. Moran, J. Ridgway, C. Eigenbrot and R. A. Lazarus, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5306–5311 CrossRef CAS PubMed.
- S. B. Kent, Chem. Soc. Rev., 2009, 38, 338–351 RSC.
- L. Raibaut, N. Ollivier and O. Melnyk, Chem. Soc. Rev., 2012, 41, 7001–7015 RSC.
- N. Ollivier, J. Vicogne, A. Vallin, H. Drobecq, R. Desmet, O. El-Mahdi, B. Leclercq, G. Goormachtigh, V. Fafeur and O. Melnyk, Angew. Chem., Int. Ed., 2012, 51, 209–213 CrossRef CAS PubMed.
- E. Boll, H. Drobecq, N. Ollivier, L. Raibaut, R. Desmet, J. Vicogne and O. Melnyk, Chem. Sci., 2014, 5, 2017–2022 RSC.
- T. W. Muir, Biopolymers, 2008, 90, 743–750 CrossRef CAS PubMed.
- J. P. Pellois and T. W. Muir, Curr. Opin. Chem. Biol., 2006, 10, 487–491 CrossRef CAS PubMed.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. Kent, Science, 1994, 266, 776–779 CAS.
- O. Melnyk and V. Agouridas, e-EROS Encycl. Reagents Org. Synth., 2014 DOI:10.1002/047084289X.rn01723.
- L. Raibaut, H. Adihou, R. Desmet, A. F. Delmas, V. Aucagne and O. Melnyk, Chem. Sci., 2013, 4, 4061–4066 RSC.
- N. Ollivier, J. Dheur, R. Mhidia, A. Blanpain and O. Melnyk, Org. Lett., 2010, 12, 5238–5241 CrossRef CAS PubMed.
- L. Raibaut, J. Vicogne, B. Leclercq, H. Drobecq, R. Desmet and O. Melnyk, Bioorg. Med. Chem., 2013, 21, 3486–3494 CrossRef CAS PubMed.
- M. Ultsch, N. A. Lokker, P. J. Godowski and A. M. de Vos, Structure, 1998, 6, 1383–1393 CrossRef CAS.
- T. Sano and C. R. Cantor, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3180–3184 CrossRef CAS.
- Y. H. Tan, M. Liu, B. Nolting, J. G. Go, J. Gervay-Hague and G. Y. Liu, ACS Nano, 2008, 2, 2374–2384 CrossRef CAS PubMed.
- L. Bongini, D. Fanelli, F. Piazza, P. De Los Rios, S. Sandin and U. Skoglund, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 6466–6471 CrossRef CAS PubMed.
- C. Ponzetto, A. Bardelli, Z. Zhen, F. Maina, P. dalla Zonca, S. Giordano, A. Graziani, G. Panayotou and P. M. Comoglio, Cell, 1994, 77, 261–271 CrossRef CAS.
- C. Birchmeier and E. Gherardi, Trends Cell Biol., 1998, 8, 404–410 CrossRef CAS.
- I. Royal and M. Park, J. Biol. Chem., 1995, 270, 27780–27787 CrossRef CAS PubMed.
- F. Bussolino, M. F. Di Renzo, M. Ziche, E. Bocchietto, M. Olivero, L. Naldini, G. Gaudino, L. Tamagnone, A. Coffer and P. M. Comoglio, J. Cell Biol., 1992, 119, 629–641 CrossRef CAS.
- T. Hori, T. Kondo, Y. Tabuchi, I. Takasaki, Q. L. Zhao, M. Kanamori, T. Yasuda and T. Kimura, Chem.-Biol. Interact., 2008, 172, 125–140 CrossRef CAS PubMed.
- J. Ogasawara, R. Watanabe-Fukunaga, M. Adachi, A. Matsuzawa, T. Kasugai, Y. Kitamura, N. Itoh, T. Suda and S. Nagata, Nature, 1993, 364, 806–809 CrossRef CAS PubMed.
- K. Kosai, K. Matsumoto, H. Funakoshi and T. Nakamura, Hepatology, 1999, 30, 151–159 CrossRef CAS PubMed.
- C. Fasting, C. A. Schalley, M. Weber, O. Seitz, S. Hecht, B. Koksch, J. Dernedde, C. Graf, E. W. Knapp and R. Haag, Angew. Chem., Int. Ed., 2012, 51, 10472–10498 CrossRef CAS PubMed.
- M. Mammen, S. K. Choi and G. M. Whitesides, Angew. Chem., Int. Ed., 1998, 37, 2754–2794 CrossRef.
- S. J. Stahl, P. T. Wingfield, J. D. Kaufman, L. K. Pannell, V. Cioce, H. Sakata, W. G. Taylor, J. S. Rubin and D. P. Bottaro, Biochem. J., 1997, 326(Pt 3), 763–772 CAS.
- F. Kolditz, J. Krausze, D. W. Heinz, H. H. Niemann and C. C. Muller-Goymann, Eur. J. Pharm. Biopharm., 2014, 86, 277–283 CrossRef CAS PubMed.
- Y. Shen, M. Naujokas, M. Park and K. Ireton, Cell, 2000, 103, 501–510 CrossRef CAS.
- H. Bierne and P. Cossart, J. Cell Sci., 2002, 115, 3357–3367 CAS.
- H. Schulze-Bergkamen, D. Brenner, A. Krueger, D. Suess, S. C. Fas, C. R. Frey, A. Dax, D. Zink, P. Buchler, M. Muller and P. H. Krammer, Hepatology, 2004, 39, 645–654 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Descriptions of reagents, antibodies, immunoblotting, MTT assay, scattering assay, morphogenesis assay and statistical analysis. See DOI: 10.1039/c4sc03856h |
| ‡ C.S. and B.L. contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |