
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModulation of inherent dynamical tendencies of the bisabolyl cation via preorganization in epi-isozizaene synthase†
Ryan P.
Pemberton
,
Krystina C.
Ho
and
Dean J.
Tantillo
*
Department of Chemistry, University of California, Davis, One Shields Avenue, Davis, CA 95616, USA. E-mail: djtantillo@ucdavis.edu
First published on 2nd February 2015
Abstract
The relative importance of preorganization, selective transition state stabilization and inherent reactivity are assessed through quantum chemical and docking calculations for a sesquiterpene synthase (epi-isozizaene synthase, EIZS). Inherent reactivity of the bisabolyl cation, both static and dynamic, appears to determine the pathway to product, although preorganization and selective binding of the final transition state structure in the multi-step carbocation cascade that forms epi-isozizaene appear to play important roles.
Introduction
Many factors have been proposed as contributors to selectivity control in terpene synthases, e.g., reactant preorganization,1–9 geometric constraints imposed by the enzyme active site (not only on the reactant but on reactive species generated from it),3,10–15 selective oriented intermolecular interactions (primarily π⋯π, C–H⋯π and C–H⋯lone pair) with intermediates and transition state structures (TSSs),16,17 and inherent reactivity of carbocations generated from the reactant—both in terms of underlying potential energy surfaces (PESs) for carbocation rearrangements and inherent dynamical tendencies.18–23 Herein we describe computations that bear directly on the relative importance of all of these factors for a sesquiterpene synthase—epi-isozizaene (7; Fig. 1) synthase (EIZS)—that has received considerable interest from organic chemists and mechanistic enzymologists over the past decade.9,24–28 To our knowledge, this is the first report that parses out the relative contributions of these factors for any enzyme (note that inherent dynamical tendencies of a substrate is by far the least studied factor of those described). | ||
| Fig. 1 Proposed steps involved in epi-isozizane formation:9 (I) pyrophosphate removal, (II) pyrophosphate addition, (III) conformational change, (IV) pyrophosphate removal, (V) π-cation cyclization, (VI) 1,2-hydride shift, (VII) π-cation cyclization, (VIII) π-cation cyclization, (IX) asynchronous double-alkyl shift,3 (X) 1,2-methyl shift,26 (XI) deprotonation. OPP = pyrophosphate. | ||
EIZS catalyzes the polycyclization of an acyclic substrate (farnesyl diphosphate, FPP), into a complex polycycle possessing three stereogenic centers via the 11 steps shown in Fig. 1. These reaction steps are proposed on the basis of previous quantum chemical calculations on reactions of the bisabolyl cation (1).3 Note that step VII is predicted to have a low barrier or no barrier, depending on the conformation of homobisabolyl cation 2, and step IX involves the merging, asynchronously, of two 1,2-alkyl shifts into a concerted process.29–31 While several conformations of species involved in this pathway were examined previously,3 we have now performed an exhaustive conformational search on the TSS for step VI with an eye toward elucidating the consequences of conformational preorganization on mechanism and selectivity. Relevant conformations were then docked into EIZS to assess the importance of enzyme–substrate interactions.
Results and discussion
Conformational concerns
First, the conformational landscape associated with step VI—the step after initial formation of the 6-membered ring that is the hallmark of this branch of sesquiterpene structural space—was examined. The previously reported global minimum of the bisabolyl cation (1)3 was used as a starting point for conformational searching. Using Spartan10,32 the Merck Molecular Force Field (MMFF94)33 was employed for a systematic search in which torsions about all rotatable bonds were sampled, yielding 86 conformers of 1. These conformers were then optimized in the gas phase at the mPW1PW91/6-31+G(d,p)23a,34,35 level of theory using the Gaussian09 software suite,36 and the lowest energy minimum was designated 1a. Manual inspection of geometries and free energies led to the removal of duplicate structures, leaving 67 unique conformations. Next, scans of forming and breaking C–H bond distances were performed for each minimum to obtain approximate geometries for the 1 → 2 1,2-hydride shift TSS. These structures were then fully optimized with mPW1PW91/6-31+G(d,p). This procedure led to 67 unique TSSs covering a range of barriers of 5.6 to 12.9 kcal mol−1 (full details in ESI†). These results reveal the magnitude of the conformational problem faced by EIZS, a scenario not unique to this terpene synthase.37Each of the 67 TSSs was then docked into EIZS (PDB ID 3KB9) using the Fast Rigid Exhaustive Docking (FRED) program in the Openeye software suite.38–41 The pyrophosphate group lost in step IV was considered to be part of the enzyme active site and was held fixed in these docking simulations. Rankings and docking scores (here, reflecting primarily shape complementarity)38–40 for all TSSs can be found in the ESI,† but results for the TSSs that lead to the relative stereochemistry in epi-isozizaene if the reaction proceeds without significant post-step VI conformational changes (TS1b–TS1d, Fig. 2), along with several other representative structures, are shown in Table 1. TS1b–TS1d have some of the best docking scores (ranking 2nd–4th), despite not having the lowest free energies of the 67 TSSs; they rank 62nd, 32nd, and 65th, respectively. Note, however, that their predicted free energies are within 6 kcal mol−1 of that of the lowest energy TSS, TS1e. TS1e is not predicted to be among the best suited for docking to the EIZS active site, suggesting that the shape of the active site plays a role in selecting the TSSs for 1,2-hydride shift that have conformations productive for subsequent reactions in the epi-isozizaene-forming pathway.
 | ||
| Fig. 2 Docked structures (bottom) of 1 → 2 TSS conformations (top) with the best docking scores (TS1a–d) and the lowest energy 1 → 2 TSS conformation (TS1e). Relative energies and docking scores are shown in Table 1. The carbons between which a bond will form in the 2 → 3 reaction (C6 and C10) are labeled in each TSS. In docked structures, the substrate is blue, protein sidechains are grey, the diphosphate group is red/orange and Mg ions are purple. | ||
| Conformer | Docking score | Docking score ranking | Free energy (rel. to 1a) | Free energy ranking | Volume (Å3) |
|---|---|---|---|---|---|
| TS1a | −81.5 | 1 | 7.1 | 10 | 334 |
| TS1b | −79.7 | 2 | 9.0 | 62 | 315 |
| TS1c | −78.2 | 3 | 10.9 | 32 | 373 |
| TS1d | −77.3 | 4 | 11.3 | 65 | 327 |
| TS1e | −64.3 | 48 | 5.6 | 1 | 278 |
Product selectivity
Each carbocation (1–6) in Fig. 1, if encountered as an intermediate with a significant lifetime, represents a potential source of byproducts for EIZS. epi-Isozizaene is the predominant natural product produced by EIZS from Streptomyces coelicolor;9,24,27 at 20 °C, a product ratio of 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5:1:1 is reported for 7:8:9:10 (Fig. 3; Table 2; this table also shows product distributions at other temperatures). Although EIZS does not produce a sole product, its selectivity is impressive given the number of potential exit channels from the epi-isozizaene-forming pathway and the inherent conformational flexibility of the species encountered en route to epi-isozizaene.
:5:1:1 is reported for 7:8:9:10 (Fig. 3; Table 2; this table also shows product distributions at other temperatures). Although EIZS does not produce a sole product, its selectivity is impressive given the number of potential exit channels from the epi-isozizaene-forming pathway and the inherent conformational flexibility of the species encountered en route to epi-isozizaene.
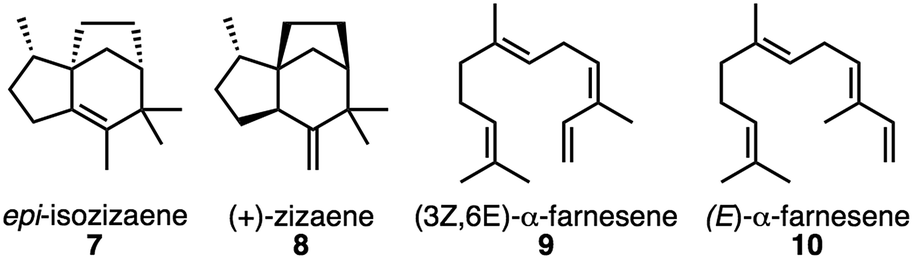 | ||
| Fig. 3 Natural products produced by EIZS.27 | ||
9 and 2014.27 Remaining rows: product distributions (%) from dynamics calculations. For EIZS, products correspond to species shown in Fig. 1 and 3 or species derived from deprotonation of indicated carbocations (1, 4, 11). For dynamics calculations (rows with bold TSS labels in first column), products correspond to carbocations or carbocation precursors to neutral species (7–10) and are listed as a percent to allow a direct comparison to experiment; 50 trajectories were generated for each transition state
| Experiment | 1 | 2 | 3 | 4 | 7 | 8 | 9 | 10 | 11 | 12 | 13 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| EIZS 2010 (30 °C) | 1 | 2 | 79 | 9 | 5 | 2 | |||||
| EIZS 2014 (20 °C) | 93 | 5 | 1 | 1 | |||||||
| EIZS 2014 (4 °C) | 99 | ||||||||||
| TS1a | 16 | 84 | |||||||||
| TS1b | 20 | 80 | |||||||||
| TS1c | 4 | 66 | 2 | 28 | |||||||
| TS1d | 6 | 24 | 20 | 38 | 10 | 2 | |||||
| TS1e | 20 | 80 |
 | ||
| Fig. 4 Carbocations produced during dynamics trajectories. | ||
The product distributions from our dynamics calculations reveal: (1) TSS conformations TS1a, TS1b, and TS1e are not predisposed to proceed along the reaction coordinate towards 7, since trajectories for none passed the homobisabolyl cation (2) or its non-productive cyclization product 11. The inability of TS1a and TS1e to proceed toward product was expected on conformational grounds, while the inability of TS1b to do so was not so certain at the outset. The position of the “free” isoprenyl group in TS1b is apparently far enough away from the 6-membered ring (Fig. 2; note relative position of C6 and C10) that bringing these two groups close enough together for reaction requires intervention. (2) TSS conformations TS1c and TS1d, which differ from each other in the puckering of their cyclohexenyl rings and which display a more productive orientation of their isoprenyl groups than does TS1b (Fig. 2), proceeded readily to the acorenyl cation (3) region and some trajectories made it to the cedryl cation (4) region (a greater number for the more compact TS1e; molecular volumes in Table 1). (3) Dynamics trajectories for no TSSs examined reached the prezizyl (5) or zizyl (6) cation regions.
These results suggest that conformationally preorganized (in this case, compact) TSSs can proceed directly along the pathway to the cedryl cation (4) region without spending any significant amount of time near cations 2 or 3, consistent with the absence of byproducts expected to be derived from these two cations in the experimental product distributions. The fact that cations 4 and 11 are observed in our dynamics simulations is consistent with the observation of sesquiterpenes derived from them in the 2010 report on EIZS.9 Minor products 9 and 10 would not show up in our dynamics studies, since these would be formed from deprotonation of species preceding the bisabolyl cation on the epi-isozizaene-forming reaction coordinate. The observation of cations 12 and 13 for TS1d is consistent with: (a) products of their deprotonation be formed in very small amounts, (b) their conversion, given more time and/or direct enzymatic intervention, to experimentally detected products, or (c) prevention of their formation by EIZS.
The remainder of our discussion is focused on TS1d, since this TSS appears to best suited for epi-isozizaene formation, being the TSS that produced the most trajectories that passed the cation 3 region. For TS1d, approximately half of the trajectories exited the cation 2 region within 500 fs (some proceeding towards 3, others towards 11). Approximately 80% of the trajectories that ultimately reached cation 4 exited the cation 2 region within this first 500 fs, proceeding directly to the cation 4 region.
The free energy surface for conversion of bisabolyl cation 1 to zizyl cation 6viaTS1d (Fig. 5, solid lines) is consistent with dynamics trajectories not passing cedryl cation 4. The following three factors are expected to correlate with whether or not a barrier will “block” a direct trajectory:43,52 (1) the height of the barrier, (2) the height of the downslope preceding the barrier, and (3) whether or not the vibrations occurring along the downslope are coupled to those needed to surpass the barrier. Intrinsic reaction coordinate (IRC) calculations61,62 connect TS1d (called TS1-3 in Fig. 5) to the acorenyl cation (3), with the homobisabolyl geometry existing on the downslope rather than as a discrete minimum. The subsequent barrier of only 2.2 kcal mol−1, approached from a height of 12.8 kcal mol−1, does not provide a significant impediment to formation of 4. No trajectories surpassed the next free energy barrier, however, which is larger—6.2 kcal mol−1—and approached from a lesser height—7.4 kcal mol−1. Although firm guidelines as to the magnitudes of the downslope height and forward barrier necessary for preventing passage are not yet available, previous work by Carpenter and co-workers showed that a change in downslope height/barrier size from 7.5/3.6 kcal mol−1 to 5.1/2.2 kcal mol−1 (the result of calculations with two different levels of theory on the same reaction) increased the percentage of direct trajectories.43 With regard to factor 3, Carpenter and co-workers showed, for ring-opening/1,5-hydride shift of [2.1.0]bicyclopentene, that kinetic energy associated with traversing an initial barrier was only directly accessible to vibrations of the same symmetry in subsequent steps.52 It is difficult to determine how much dynamic matching of vibrational modes contributes to the viability of the 4 to 5 reaction (a dyotropic or “double-shift” reaction).3,21,63,64 While formation of both involves stretching/compressing of the same bond (the C2–C11 bond that is made in forming 4 breaks in forming 5), the imaginary frequency for TS3-4 corresponds primarily to a twisting of the molecule around the C6–C10 bond while that for TS4-5 corresponds primarily to formation of the C3–C11 bond (Fig. 1).
 | ||
| Fig. 5 Computed mPW1PW91/6-31+G(d,p) free energy profile for formation of 6viaTS1d (here, labeled TS1-3) without conformational changes of intermediates (energies are relative to the global minimum of 1; note that for this series of conformations, 2 is not a minimum), along with productive docking poses (substrate is blue, protein sidechains are grey, the diphosphate group is red/orange, Mg ions are purple, groups involved in bond making/breaking are highlighted in transition state structures). Dotted lines and arrows indicate the qualitative effects of binding to EIZS, estimated based on computed docking scores for productive poses. | ||
| Stationary point | Pose with best docking score | Productive pose | Volume (Å3) |
|---|---|---|---|
| 1 | −79.1 | Same | 377 |
| TS1-3 | −77.3 | Same | 327 |
| 3 | −77.8 | −65.9 | 315 |
| TS3-4 | −72.3 | −68.2 | 313 |
| 4 | −76.8 | −75.9 | 279 |
| TS4-5 | −79.4 | Same | 273 |
| 5 | −80.0 | Same | 270 |
| TS5-6 | −84.9 | Same | 295 |
| 6 | −78.6 | −74.9 | 303 |
Although FRED docking scores are not predicted binding energies,38–40 if one assumes that they correlate qualitatively with binding energies then effects on the energetics of epi-isozizaene formation can be assessed. As shown in Fig. 5 (dotted lines; only effects corresponding to differences in docking scores of >0.5 are shown), the first two TSSs following the bisabolyl cation, along with the minima directly following them, are selectively destabilized relative to the bisabolyl cation when considering the best productive pose (also true for the best docked pose). Conversely, the final TSS, leading to the zizyl cation, is selectively stabilized. The net effect of this modulation in relative energies is to promote passage from 4 to 5 to 6. Both the 4 → 5 and 5 → 6 barriers would be lowered by selective complexation, and the height of the downslope leading to 4 would be increased, thereby reducing the lifetime of 4 and perhaps allowing for the direct passage to 5 not observed in our dynamics calculations in the absence of EIZS. The docking approach used scores primarily on the basis of the complementarity of cation and active site shape,38–40 consistent with the idea that terpene synthase active sites resemble structures occurring later along carbocation cyclization/rearrangement reaction coordinates.65 Note also that the substrate volume decreases monotonically from 1 to 5, but then increases for the final two stationary points (Table 3). Although one can identify specific C–H/H–C, C–H⋯π and C–H⋯O contacts in docked structures (see ESI†), quantification of their effects on the reaction pathway will require more advanced methods (vide infra).
Conclusions
On the basis of our results, avoidance of the previously postulated secondary carbocation between 4 and 5 can be ascribed to the nature of the PES in the absence of the enzyme,3,24,66 and avoidance of byproducts derived from putative intermediates between 1 and 4 can be ascribed to inherent dynamical tendencies.67 Enzyme-enforced conformational restriction (of reactant and subsequent species) clearly also plays a role in directing the reactant toward TSSs that are productive for epi-isozizaene formation and appears also to play a role in promoting the conversion of 4 to 5 to 6. That leaves only fine-tuning to be ascribed to specific intermolecular electrostatic interactions (with OPP and/or active site aromatics) and/or effects of enzyme dynamics (rather than inherent substrate dynamics; tunneling may also play a role37); these issues will be addressed in future quantum mechanical molecular mechanics (QM/MM)68–70 and theozyme18,71–73 studies on EIZS. Note how little is left to explain; while EIZS clearly plays keys roles in promoting pyrophosphate dissociation and preventing premature quenching of carbocations, we show here that its product distribution can be rationalized in large part on the basis of inherent carbocation reactivity and shape selection.74Acknowledgements
We gratefully acknowledge support from the National Science Foundation (CHE-0957416 and CHE030089 for supercomputing resources via XSEDE) and the United States Department of Education (GAANN fellowship to RPP). We thank Dan Singleton (TAMU) for helpful advice and for providing Progdyn and OpenEye for providing software.References
- G. Stork and A. W. Burgstahler, J. Am. Chem. Soc., 1955, 77, 5068–5077 CrossRef CAS.
- A. Eschenmoser, L. Ruzicka, O. Jeger and D. Arigoni, Helv. Chim. Acta, 1955, 38, 1890–1904 CrossRef CAS.
- Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2009, 131, 7999–8015 CrossRef CAS PubMed.
- Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2014, 136, 2450–2463 CrossRef CAS PubMed.
- Y. J. Hong and D. J. Tantillo, J. Am. Chem. Soc., 2011, 133, 18249–18256 CrossRef CAS PubMed.
- J. P. Noel, N. Dellas, J. A. Faraldos, M. Zhao, B. A. Hess, L. Smentek, R. M. Coates and P. E. O'Maille, ACS Chem. Biol., 2010, 5, 377–392 CrossRef CAS PubMed.
- T. Gaich and P. S. Baran, J. Org. Chem., 2010, 75, 4657–4673 CrossRef CAS PubMed.
- R. J. Felix, C. Munro-Leighton and M. R. Gagné, Acc. Chem. Res., 2014, 47, 2319–2331 CrossRef CAS PubMed.
- J. A. Aaron, X. Lin, D. E. Cane and D. W. Christianson, Biochemistry, 2010, 49, 1787–1797 CrossRef CAS PubMed.
- Y. J. Hong and D. J. Tantillo, Org. Lett., 2011, 13, 1294–1297 CrossRef CAS PubMed.
- Y. J. Hong and D. J. Tantillo, J. Org. Chem., 2007, 72, 8877–8881 CrossRef CAS PubMed.
- L. S. Vedula, M. J. Rynkiewicz, H.-J. Pyun, R. M. Coates, D. E. Cane and D. W. Christianson, Biochemistry, 2005, 44, 6153–6163 CrossRef CAS PubMed.
- D. E. Cane, G. Yang, R. M. Coates, H. J. Pyun and T. M. Hohn, J. Org. Chem., 1992, 57, 3454–3462 CrossRef CAS.
- P. A. Sigala, D. A. Kraut, J. M. M. Caaveiro, B. Pybus, E. A. Ruben, D. Ringe, G. A. Petsko and D. Herschlag, J. Am. Chem. Soc., 2008, 130, 13696–13708 CrossRef CAS PubMed.
- A. Warshel, P. K. Sharma, M. Kato, Y. Xiang, H. Liu and M. H. M. Olsson, Chem. Rev., 2006, 106, 3210–3235 CrossRef CAS PubMed.
- L. Pauling, Nature, 1948, 161, 707–709 CrossRef CAS PubMed.
- Y. J. Hong and D. J. Tantillo, Chem. Sci., 2013, 4, 2512–2518 RSC.
- R. P. Pemberton and D. J. Tantillo, Chem. Sci., 2014, 5, 3301–3308 RSC.
- M. R. Siebert, J. Zhang, S. V. Addepalli, D. J. Tantillo and W. L. Hase, J. Am. Chem. Soc., 2011, 133, 8335–8343 CrossRef CAS PubMed.
- M. R. Siebert, P. Manikandan, R. Sun, D. J. Tantillo and W. L. Hase, J. Chem. Theory Comput., 2012, 8, 1212–1222 CrossRef CAS.
- Y. J. Hong and D. J. Tantillo, Org. Biomol. Chem., 2010, 8, 4589–4600 CAS.
- R. P. Pemberton, Y. J. Hong and D. J. Tantillo, Pure Appl. Chem., 2013, 85, 1949–1957 CrossRef CAS.
- (a) D. J. Tantillo, Nat. Prod. Rep., 2011, 28, 1035–1053 RSC; (b) This manuscript is part 14 of our series on theoretical studies of sesquiterpene forming carbocation rearrangements. For part 13, see: M. Isegawa, S. Maeda, D. J. Tantillo and K. Morokuma, Chem. Sci., 2014, 5, 1555–1560 RSC.
- X. Lin, R. Hopson and D. E. Cane, J. Am. Chem. Soc., 2006, 128, 6022–6023 CrossRef CAS PubMed.
- B. Zhao, X. Lin, L. Lei, D. C. Lamb, S. L. Kelly, M. R. Waterman and D. E. Cane, J. Biol. Chem., 2008, 283, 8183–8189 CrossRef CAS PubMed.
- X. Lin and D. E. Cane, J. Am. Chem. Soc., 2009, 131, 6332–6333 CrossRef CAS PubMed.
- R. Li, W. K. W. Chou, J. A. Himmelberger, K. M. Litwin, G. G. Harris, D. E. Cane and D. W. Christianson, Biochemistry, 2014, 53, 1155–1168 CrossRef CAS PubMed.
- Zizaene derivatives are key components in the scent of vetiver: E. Belhassen, N. Baldovini, H. Brevard, U. J. Meierhenrich and J.-J. Filippi, Chem. Biodiversity, 2014, 11, 1821–1842 CAS.
- A. Williams, Concerted Organic and Bio-Organic Mechanisms, CRC Press, 2000 Search PubMed.
- B. A. Hess, J. Am. Chem. Soc., 2002, 124, 10286–10287 CrossRef CAS PubMed.
- D. J. Tantillo, J. Phys. Org. Chem., 2008, 21, 561–570 CrossRef CAS.
- Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio Jr, R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Yeh Lin, T. Van Voorhis, S. Hung Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. Min Rhee, J. Ritchie, E. Rosta, C. David Sherrill, A. C. Simmonett, J. E. Subotnik, H. Lee Woodcock Iii, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer Iii, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172–3191 RSC.
- T. A. Halgren, J. Comput. Chem., 1996, 17, 490–519 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1998, 108, 664–675 CrossRef CAS.
- S. P. T. Matsuda, W. K. Wilson and Q. Xiong, Org. Biomol. Chem., 2006, 4, 530–543 CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Wallingford CT, 2009 Search PubMed.
- (a) A striking example: B. A. Hess, L. Smentek, J. P. Noel and P. E. O'Maille, J. Am. Chem. Soc., 2011, 133, 12632–12641 CrossRef CAS PubMed; (b) While tunneling likely plays a role in navigating the potential energy surface, the barriers for hydride transfer are inherently very small.
- (a) FRED (docking): M. McGann, J. Comput.-Aided Mol. Des., 2012, 26, 897–906 CrossRef CAS PubMed; (b) OMEGA (conformational searching): P. C. D. Hawkins, A. G. Skillman, G. L. Warren, B. A. Ellingson and M. T. Stahl, J. Chem. Inf. Model., 2010, 50, 572–584 CrossRef CAS PubMed.
- Openeye Scientific Software, Version 1.7.4, Santa Fe, NM, http://www.eyesopen.com Search PubMed.
- Docked poses were scored and ranked using the Chemgauss3 scoring function, which scores poses on the basis of shape, hydrogen bonding between ligand and protein, hydrogen bonding interactions with implicit solvent, and metal–chelator interactions; for the cases described herein, the latter three are not relevant.
- After initial submission of this manuscript, a study on a related but different docking approach for carbocation transition state structures and intermediates formed in triterpene synthases was reported: B.-X. Tian, F. H. Wallrapp, G. L. Holiday, J.-Y. Chow, P. C. Babbitt, C. D. Poulter and M. P. Jacobson, PLoS Comput. Biol., 2014, 10, e1003874 Search PubMed.
- R. Car and M. Parrinello, Phys. Rev. Lett., 1985, 55, 2471–2474 CrossRef CAS PubMed.
- B. K. Carpenter, J. Am. Chem. Soc., 1996, 118, 10329–10330 CrossRef CAS.
- C. Doubleday, K. Bolton and W. L. Hase, J. Am. Chem. Soc., 1997, 119, 5251–5252 CrossRef CAS.
- L. Sun, K. Song and W. L. Hase, Science, 2002, 296, 875–878 CrossRef CAS PubMed.
- D. A. Singleton, C. Hang, M. J. Szymanski and E. E. Greenwald, J. Am. Chem. Soc., 2003, 125, 1176–1177 CrossRef CAS PubMed.
- D. Townsend, S. A. Lahankar, S. K. Lee, S. D. Chambreau, A. G. Suits, X. Zhang, J. Rheinecker, L. B. Harding and J. M. Bowman, Science, 2004, 306, 1158–1161 CrossRef CAS PubMed.
- B. R. Ussing, C. Hang and D. A. Singleton, J. Am. Chem. Soc., 2006, 128, 7594–7607 CrossRef CAS PubMed.
- A. E. Litovitz, I. Keresztes and B. K. Carpenter, J. Am. Chem. Soc., 2008, 130, 12085–12094 CrossRef CAS PubMed.
- Y. Oyola and D. A. Singleton, J. Am. Chem. Soc., 2009, 131, 3130–3131 CrossRef CAS PubMed.
- L. Xu, C. E. Doubleday and K. N. Houk, J. Am. Chem. Soc., 2010, 132, 3029–3037 CrossRef CAS PubMed.
- L. M. Goldman, D. R. Glowacki and B. K. Carpenter, J. Am. Chem. Soc., 2011, 133, 5312–5318 CrossRef CAS PubMed.
- L. Xu, C. E. Doubleday and K. N. Houk, J. Am. Chem. Soc., 2011, 133, 17848–17854 CrossRef CAS PubMed.
- K. Black, P. Liu, L. Xu, C. Doubleday and K. N. Houk, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12860–12865 CrossRef CAS PubMed.
- S. C. Ammal, H. Yamataka, M. Aida and M. Dupuis, Science, 2003, 299, 1555–1557 CrossRef CAS PubMed.
- G. Ghigo, A. Maranzana and G. Tonachini, J. Org. Chem., 2013, 78, 9041–9050 CrossRef CAS PubMed.
- L. Salem, Acc. Chem. Res., 1971, 4, 322–328 CrossRef CAS.
- D. Marx and J. Hutter, Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge Univ. Press, 2009 Search PubMed.
- M. Paranjothy, R. Sun, Y. Zhuang and W. L. Hase, WIREs Comput. Mol. Sci., 2013, 3, 296–316 CrossRef CAS.
- Y. J. Hong and D. J. Tantillo, Nat. Chem., 2014, 6, 104–111 CrossRef CAS PubMed.
- K. Fukui, Acc. Chem. Res., 1981, 14, 363–368 CrossRef CAS.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem., 1990, 94, 5523–5527 CrossRef CAS.
- O. Gutierrez and D. J. Tantillo, J. Org. Chem., 2012, 77, 8845–8850 CrossRef CAS PubMed.
- R. L. Davis and D. J. Tantillo, J. Org. Chem., 2010, 75, 1693–1700 CrossRef CAS PubMed.
- M. Koksal, Y. Jin, R. M. Coates, R. Croteau and D. W. Christianson, Nature, 2011, 469, 116–120 CrossRef PubMed.
- D. J. Tantillo, Chem. Soc. Rev., 2010, 39, 2847–2854 RSC.
- The idea that thermodynamically disfavored TSSs can be accessed under kinetic conditions in a terpene synthase, i.e., “the reaction coordinate from one intermediate to the next being governed by the barrier height separating intermediates and not the relative binding energies of the intermediates,” was suggested previously: M. Chen, N. Al-lami, M. Janvier, E. L. D'Antonio, J. A. Faraldos, D. E. Cane, R. K. Allemann and D. W. Christianson, Biochemistry, 2013, 52, 5441–5453 CrossRef CAS PubMed.
- M. Weitman and D. T. Major, J. Am. Chem. Soc., 2010, 132, 6349–6360 CrossRef CAS PubMed.
- D. T. Major and M. Weitman, J. Am. Chem. Soc., 2012, 134, 19454–19462 CrossRef CAS PubMed.
- D. T. Major, Y. Freud and M. Weitman, Curr. Opin. Chem. Biol., 2014, 21, 25–33 CrossRef CAS PubMed.
- D. J. Tantillo, J. Chen and K. N. Houk, Curr. Opin. Chem. Biol., 1998, 2, 743–750 CrossRef CAS PubMed.
- D. J. Tantillo and K. N. Houk, in Stimulating Concepts in Chemistry, Wiley-VCH, Weinheim, Germany, 2000 Search PubMed.
- J. DeChancie, F. R. Clemente, A. J. T. Smith, H. Gunaydin, Y.-L. Zhao, X. Zhang and K. N. Houk, Protein Sci., 2007, 16, 1851–1866 CrossRef CAS PubMed.
- A fascinating interchange between Robinson and Eschenmoser in which the former proposes that inherent reactivity is key: G. E. W. Wolstenholme and M. O'Connor, in CIBA Foundation on the Biosynthesis of Terpenes and Sterols, Little Brown and Company, Boston, MA, 1958, p. 299 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Movies of representative trajectories, structures of all relevant stationary points, table of electronic and free energies, relative free energies, docking scores, and frequency information for transition state structures. See DOI: 10.1039/c4sc03782k |
| This journal is © The Royal Society of Chemistry 2015 |