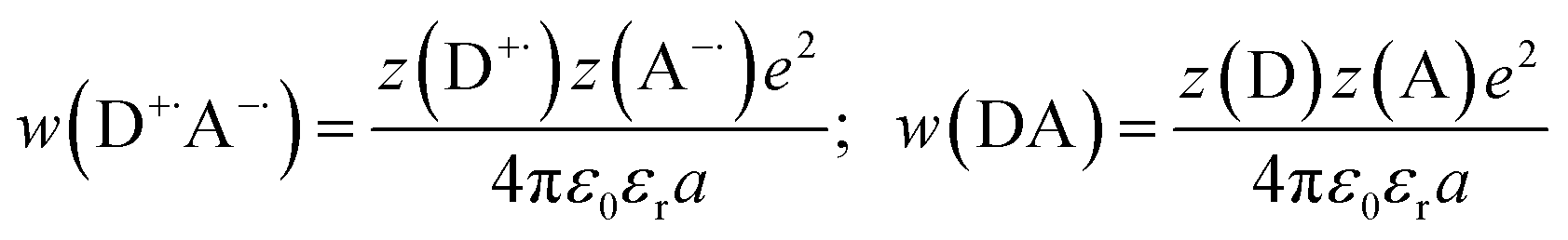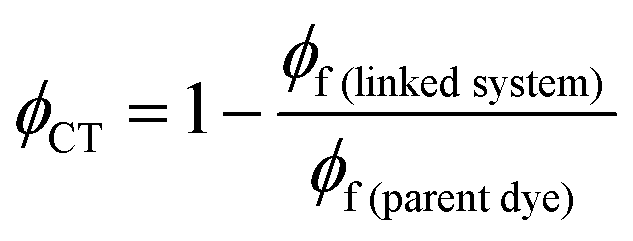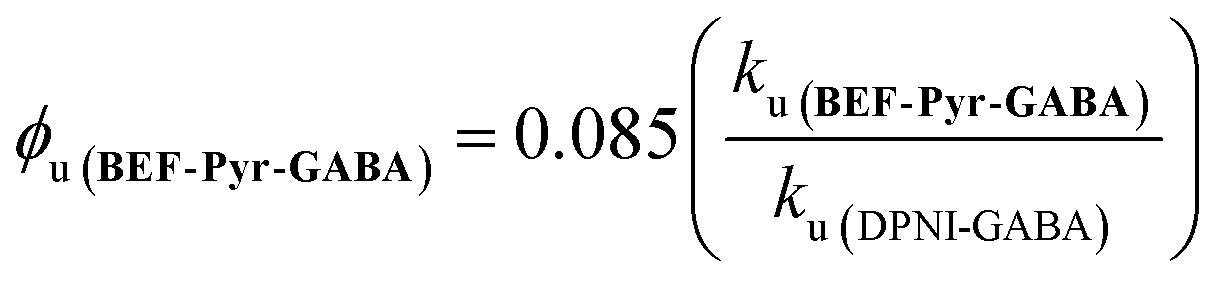DOI:
10.1039/C4SC03775H
(Edge Article)
Chem. Sci., 2015,
6, 2419-2426
Two-photon sensitive protecting groups operating via intramolecular electron transfer: uncaging of GABA and tryptophan†
Received
6th December 2014
, Accepted 2nd February 2015
First published on 3rd February 2015
Abstract
Improved photo-labile protecting groups, with high sensitivity to two-photon excitation, are needed for the controlled release of drugs, as tools in neuroscience and physiology. Here we present a new modular approach to the design of caging groups based on photoinduced electron transfer from an electron-rich two-photon dye to an electron acceptor, followed by scission of an ester to release a carboxylic acid. Three different electron acceptors were tested: nitrobenzyl, phenacyl and pyridinium. The nitrobenzyl system was ineffective, giving only photochemical decomposition and no release of the carboxylic acid. The phenacyl system also performed poorly, liberating the carboxylic acid in 20% chemical yield and 0.2% photochemical yield. The pyridinium system was most successful, and was tested for the release of two carboxylic acids: γ-amino butyric acid (GABA) and tryptophan. The caged GABA undergoes photochemical cleavage with a chemical yield of >95% and a photochemical yield of 1%; it exhibits a two-photon absorption cross section of 1100 GM at 700 nm, corresponding to a two-photon uncaging cross section of 10 ± 3 GM.
Introduction
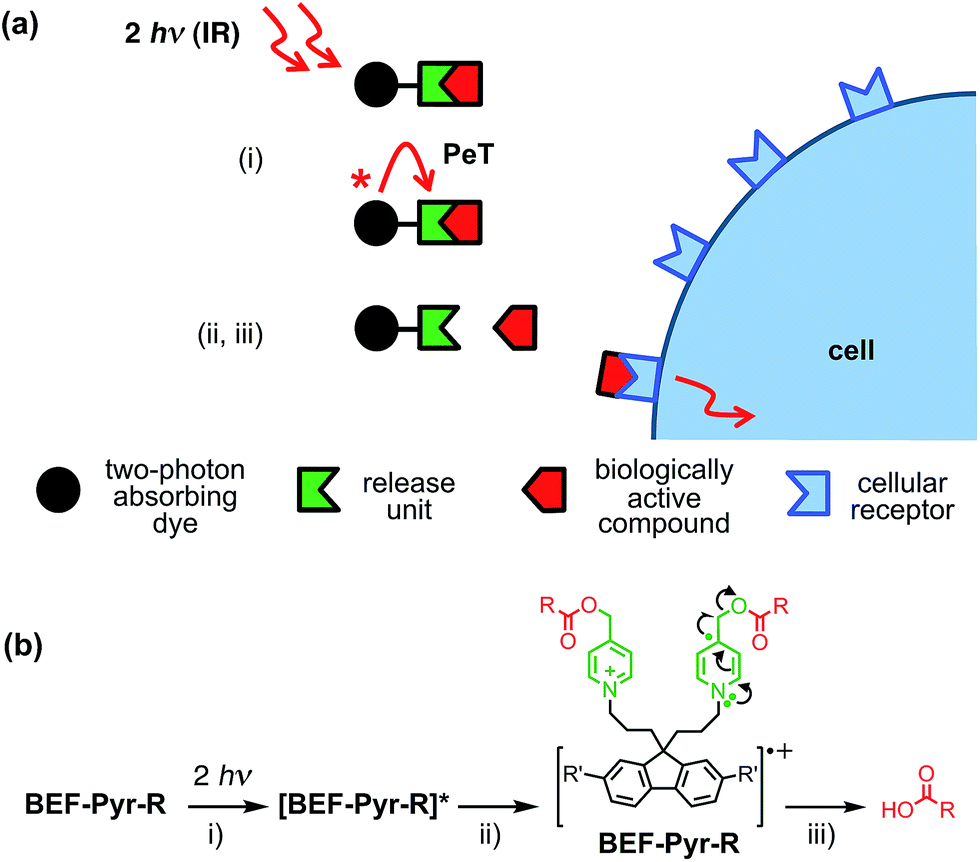
Light-sensitive protecting groups have continued to gain importance as tools for investigating the role of physiologically active compounds, ever since they were first applied in biological system in 1978.1 A number of UV- and visible-light cleavable cages have been developed2 to allow rapid spatially and temporally controlled photo-release of various biomolecules within living cells.3 In the 1990s, the concept of uncaging was extended to take advantage of two-photon absorption (TPA),4,5 a nonlinear optical phenomenon in which excitation occurs by the simultaneous absorption of two photons, each having half the energy of the corresponding one-photon process. The main advantage of TPA is that excitation is effectively restricted to the focal volume, giving tight spatial control. Furthermore, TPA allows the use of near-IR wavelengths which diminishes photo-damage and improves tissue penetration by reducing scattering and avoiding absorption by natural pigments. Unfortunately, most protecting groups optimized for one-photon uncaging display low efficiency under two-photon excitation, because their chromophores lack the specific features required for efficient TPA. The process of uncaging can be divided into two steps: light absorption followed by bond scission. The efficiency of TPA is quantified by the TPA cross-section (δa), while the quantum yield of uncaging (ϕu) measures the efficiency of photo-induced bond scission. The product of these two parameters, the two-photon uncaging cross-section (δu = δaϕu) is a figure of merit reflecting the overall sensitivity of a protecting group to two-photon uncaging. Typically, δu values reported to date lie between 0.05 and 2.5 GM (ref. 2c, 6, 7) (1 GM = 1050 cm4 s per photon) whereas δu > 3 GM is desired for efficient uncaging in living cells.8 A common strategy for enhancing the TPA cross-section of a pre-existing protecting platform is to extend the π-system and modulate the strength of electron donating/accepting substituents. Dipolar, quadrupolar and octupolar architectures have been explored for functionalizing established caging groups such as coumarine,9o-nitrobenzyl,10,11c 2-(o-nitrophenyl)propyl,11 phenacyl11c and quinoline12 with vinyl, phenyl, styryl, dihydronaphthalenyl, thienyl, fluorenyl and triphenylamine groups. The strategy of incorporating an existing protecting group into a conjugated donor–π–acceptor system led to a dipolar protecting group within the 2-(o-nitrophenyl)propyl series with a record δu of 11 GM at 800 nm.11a However, as absorption and bond scission are inherently related, any alteration in the caging group influences both δa and ϕu. Therefore some structurally modified cages, such as BNSF (2,7-bis-{4-nitro-8-[3-(2-propyl)-styryl]}-9,9-bis-[1-(3,6-dioxaheptyl)]-fluorene, δu of 5 GM at 800 nm, 65% yield of uncaging),11b suffer from light-induced side reactions or decreased yield of release, compared to their parent protecting units. Since it proves extremely difficult to enhance δa while preserving a high value of δu, several attempts have been made to explore an alternative approach, in which the absorption and release steps are decoupled and occur in different parts of the caging platform. A modular design allows each process to be optimized independently. This concept was first demonstrated for one-photon photolysis, where two spatially separated steps of uncaging were linked by intramolecular photoinduced electron transfer (PeT)13 or triplet sensitization.14 Recently this strategy has been implemented in two-photon uncaging systems: the fluorenyl–nitroindolinyl derived protecting group (δu of 0.5 GM at 730 nm), where the absorption step is followed by intramolecular energy transfer-mediated release15 and 2-(o-nitrophenyl)propyl–thioxanthone with intermolecular FRET (δu of 0.86 GM at 766 nm).16 Here, we report a study of PeT-mediated uncaging in a two-photon excitable system.17 Drawing upon previously reported designs,13 we devised a protecting group, the removal of which operates via intramolecular PeT between a photoexcited electron-donor (a TPA dye with high δa) and an electron-acceptor (pre-existing release unit) to achieve efficient release of physiologically active compounds (Fig. 1a).
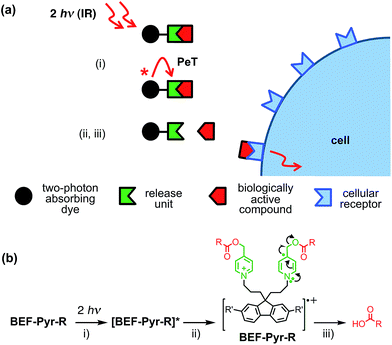 |
| | Fig. 1 Concept of a photolabile protecting group operating via photoinduced electron transfer. Upon two-photon excitation (i) the dye unit donates an electron to the release unit (ii), which undergoes photochemical reaction and liberates the drug (iii). | |
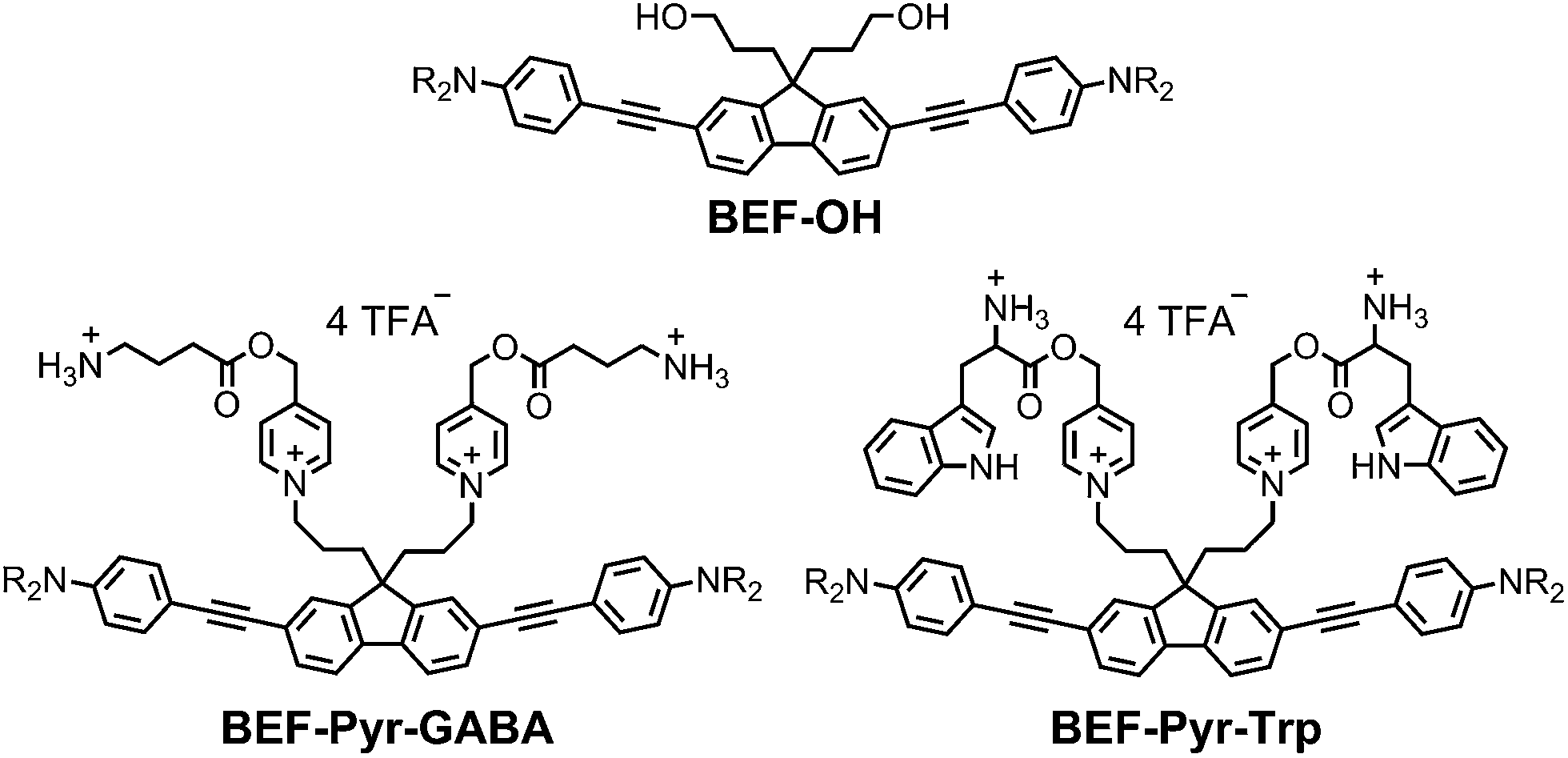
Our studies began with choosing a suitable electron-donor with high δa. We selected a symmetric banana-shaped bisethynyl fluorene (BEF) dye, in which the core is extended with substituted anilines via acetylene bridges.18 A pyridinium salt was chosen as a potential electron-acceptor, since it has been demonstrated to release carboxylic acids upon PeT.13b,c,19Fig. 1b shows the proposed mechanism of photo-deprotection. Light absorption generates a photosensitizer-based singlet state, which is quenched by electron transfer to the release group. The resulting charge-shifted state decays by σ-bond cleavage to liberate the physiologically active carboxylic acid. The symmetrical design was chosen to simplify the synthesis, while the aniline unit was substituted with heptaethyleneglycol chains to promote solubility in aqueous media. A model dye-unit BEF-OH was synthesized for the purpose of photophysical and electrochemical studies (Fig. 2). We used caged tryptophan, BEF-Pyr-Trp, for testing the intramolecular PeT mediated uncaging mechanism. L-Tryptophan (Trp) was selected as a model amino acid due to the presence of an indole chromophore that allows release to be quantified by HPLC, with UV detection. We also prepared caged γ-amino butyric acid (GABA), BEF-Pyr-GABA, to explore the utility of our protecting group for release of an inhibitory neurotransmitter.
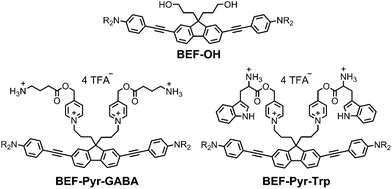 |
| | Fig. 2 Structure of model electron-donor BEF-OH, caged tryptophan, BEF-Pyr-Trp, and caged GABA, BEF-Pyr-GABA; R = (CH2CH2O)7CH3. | |
Results and discussion
Synthesis
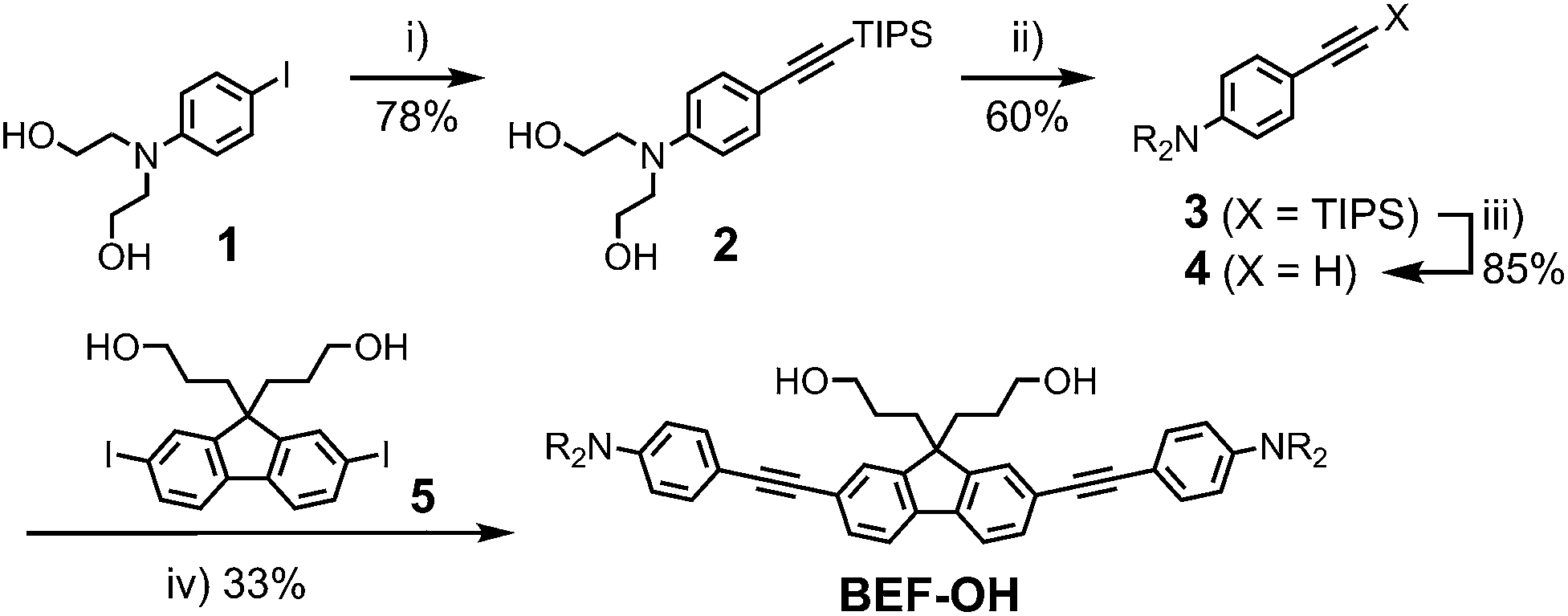
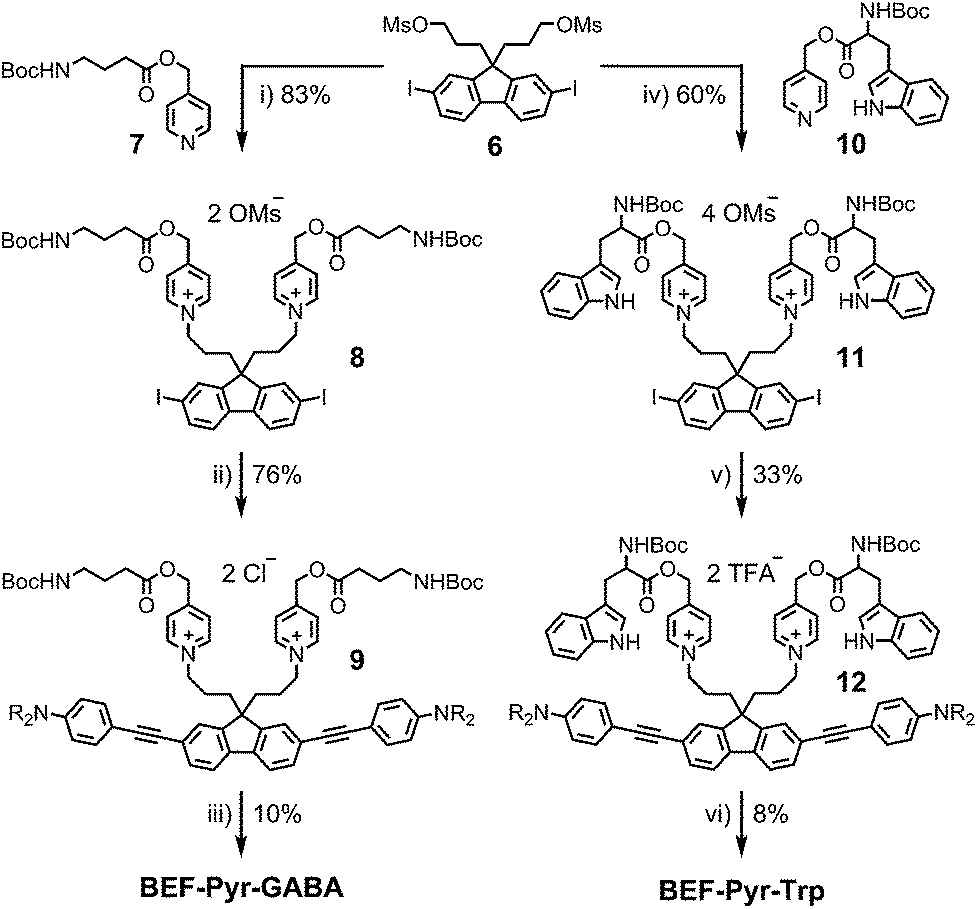
The reference dye BEF-OH was prepared as shown in Scheme 1. The synthesis started from N-(4-iodophenyly)diethanolamine20 (1) by coupling with TIPS-acetylene to give intermediate 2, which was subsequently alkylated with hexaethyleneglycol monomethyl ether tosylate. Removal of TIPS group from 3 resulted in the key aniline intermediate 4, which was coupled with the central fluorene diiodide 5 (see ESI†) under Sonogashira conditions to give BEF-OH. In a convergent synthesis towards GABA and tryptophan derivatives, the fluorene core was appended with the pyridinium esters before conjugation with the substituted aniline units 4 using Sonogashira cross-coupling (Scheme 2). The final step involved removal of the Boc protecting groups to yield the trifluoroacetate salts BEF-Pyr-GABA and BEF-Pyr-Trp.
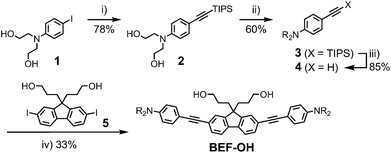 |
| | Scheme 1 Reagents: (i) TIPS-acetylene, Pd(OAc)2, PPh3, CuI, DIPA, 50 °C, 16 h, (ii) CH3(OCH2CH2)6OTs, NaH, THF, reflux, 48 h, (iii) TBAF, THF, 20 °C, 12 h, (iv) 4, Pd(OAc)2, PPh3, CuI, DIPA, MeCN, 20 °C, 3 h; Ts = p-toluenesulfonate, TIPS = triisopropylsilyl, THF = tetrahydrofuran, DIPA = diisopropylamine, TBAF = tetra-n-butylammonium fluoride, R = (CH2CH2O)7CH3. | |
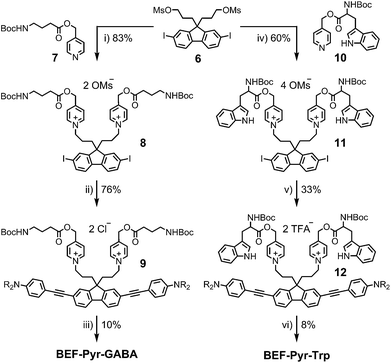 |
| | Scheme 2 Reagents: (i) MeCN, reflux, 36 h, (ii) 4, Pd(OAc)2, PPh3, CuI, DIPA, 20 °C, 1 h, brine wash, (iii) BF3·OEt2, DCM, 0 °C → 20 °C, 5 h, (iv) MeCN, 100 °C, 24 h, (v) 4, Pd(OAc)2, PPh3, CuI, DIPA, DCM, MeCN, 20 °C, 2 h, then purification in TFA-buffered solvent, (vi) HCO2H, TIPS-H, 20 °C, 1 h. | |
One- and two-photon absorption and fluorescence spectra
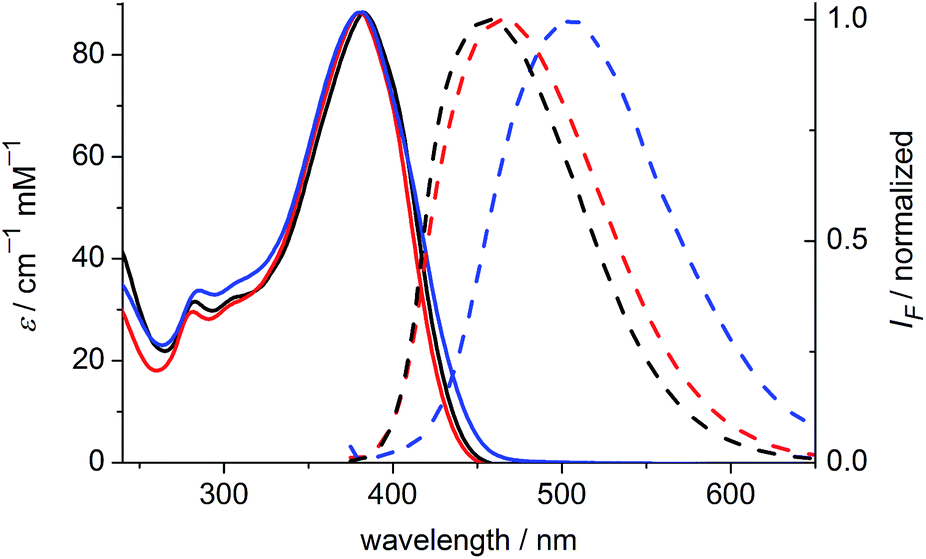
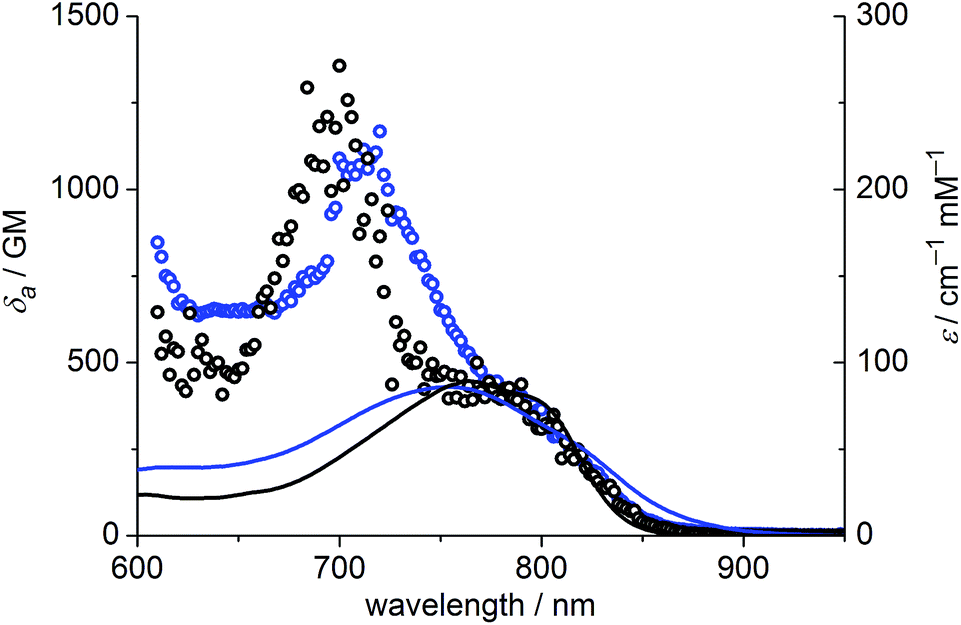
The one-photon absorption and emission spectra of BEF-OH in water and polar organic solvents (EtOH and THF) are shown in Fig. 3. The fluorene-based dye exhibits a strong absorption at 300–400 nm (maximum: 380 nm; ε380 = 9.0 × 104 M−1 cm−1). Comparison of the absorption spectra in water and organic solvents reveals that the one-photon absorption properties of BEF-OH are independent of the solvent polarity. However, the fluorescence spectrum is sensitive to the environment, being red-shifted and broadened in polar solvents. The Stokes shifts are 78, 83 and 128 nm in THF, EtOH and water, respectively. Bathochromic shifts in the luminescence spectra of banana-shaped fluorene-dyes have previously been attributed to symmetry breaking and the formation of a polar excited state, which is stabilized in polar solvents.21 The fluorescence quantum yield (ϕf) of BEF-OH is about 0.42 in organic solvents (0.43 in THF, 0.41 in EtOH, referenced to quinine in 0.5 M H2SO4), but it falls to 0.1 in water. The TPA spectra of BEF-OH and its t-butyldimethylsilyl ether derivative, BEF-OTBDMS (Fig. S28†), in water and EtOH, respectively, are compared in Fig. 4. The TPA maxima are 1150 GM at 700 nm (for BEF-OTBDMS in EtOH) and 1100 GM at 715 nm (for BEF-OH in water). These cross-sections are similar to those reported previously for closely related dyes.18,22 The spectrum is slightly broader and red-shifted in water, but the spectra are similar, revealing that the TPA is insensitive to the solvent environment. In both solvents, there is a shoulder in the TPA spectrum at twice the wavelength of the one-photon allowed S0 → S1 transition. However, the TPA spectra are dominated by peaks corresponding to the one-photon forbidden, two-photon allowed higher-energy electronic or vibronic transitions. This behavior is similar to that reported for slightly non-centrosymmetric D–π–D quadrupolar chromophores.18
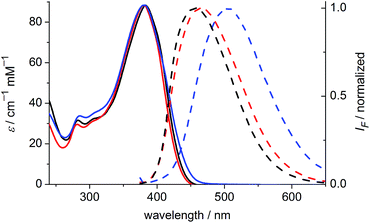 |
| | Fig. 3 Absorption (solid) and emission (dashed) spectra of BEF-OH in THF (black), EtOH (red) and water (blue). Excitation wavelength: 366 nm. | |
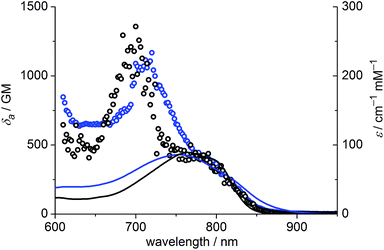 |
| | Fig. 4 TPA spectra (circles) overlaid with double-wavelength one-photon absorption spectra (lines); blue – BEF-OH recorded in water, black – BEF-OTBDMS (t-butyldimethylsilyl ether derivative) recorded in EtOH. | |
The model electron acceptor, N-methyl pyridinium hexafluorophosphate (Pyr) (Fig. 5), displays weak absorption in the UV region (λmax = 270 nm, ε270 = 0.5 × 104 M−1 cm−1 in THF, Fig. S26†), with no significant absorption at wavelengths greater than 300 nm. The difference between the absorption spectra of the dye unit BEF-OH and release platform Pyr, mean that the fluorene dye is the only absorbing species at wavelengths longer than 300 nm.
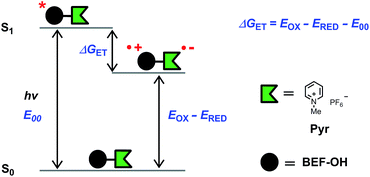 |
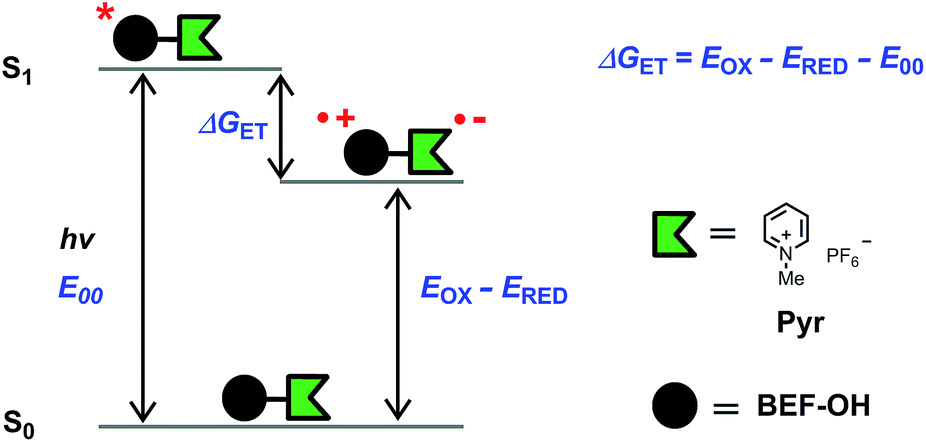
| | Fig. 5 Diagram representing the energy levels of the species involved in electron transfer. Right: structure of N-methyl pyridinium hexafluorophosphate (Pyr), the model compound used in electrochemical measurements. | |
Thermodynamics of electron transfer
The fundamental requirement for efficient PeT is that the Gibbs free energy (ΔGET) for the process must be negative. The energy of the singlet excited state of the dye (E00) must be greater than the energy cost of transferring an electron from the donor to the acceptor, i.e. greater than the difference between the oxidation potential of the donor (EOX) and the reduction potential of the acceptor (ERED), corrected by the Coulombic stabilization of the charges, as summarized by Fig. 5 and eqn (1),23| | | ΔGET = NA{e[EOX − ERED] + w(D+˙A−˙) − w(DA)} − E00 | (1) |
where w(D+˙A−˙) and w(DA) are terms that factor in electrostatic interaction in the products and reactants:| | 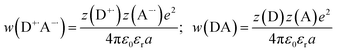 | (2) |
and NA is the Avogadro constant, e is the elementary charge, ε0 is the vacuum permittivity, εr is the dielectric constant of the solvent, a is the distance of charge separation and z(D/A) is charge of the species (D: donor; A: acceptor). The excited state energy (E00) is defined as the energy of transition between the lowest vibrational level of the ground and excited states, and can be estimated from the point of overlap between the absorption and emission spectra. The oxidation potential of the donor (EOX) and reduction potential of the acceptor (ERED) can be measured electrochemically. The excited state energy of BEF-OH in THF was estimated at 2.96 eV (418 nm) and the first oxidation potential was determined to be 0.36 V (relative to ferrocene in THF with 0.1 M Bu4PF6). The reduction potential of Pyr relative to ferrocene is −1.76 V under the same conditions. The free Gibbs energy (ΔGET) for PeT calculated according to eqn (1) for the pair BET-OH and Pyr is −0.84 eV indicating that PeT is strongly favorable. The Coulombic term is zero in the case of the pyridinium-based systems because electron transfer does not result information of a charge-separated state but only in the migration of a pre-existing charge. An extended study of photoinduced electron transfer in model dyads, in which the BEF electron donor is covalently linked to a variety of electron acceptors, is reported separately.24
Fluorescence quenching
The efficiency of electron transfer in the systems reported here can be evaluated from their fluorescence quantum yields, because PeT competes directly with fluorescence. Comparison of the fluorescence quantum yields of the free donor unit (BEF-OH; ϕ = 0.10 in water) and the donor incorporated into the dyad (BEF-Pyr-GABA; ϕ = 0.001 in water) shows that the fluorescence of the fluorene dye is severely quenched by the presence of acceptor, implying that intramolecular PeT is fast and efficient. Using this information and eqn (3), the quantum yield of charge transfer is estimated to be near unity (ϕCT = 0.98).| | 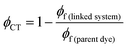 | (3) |
Hydrolytic stability
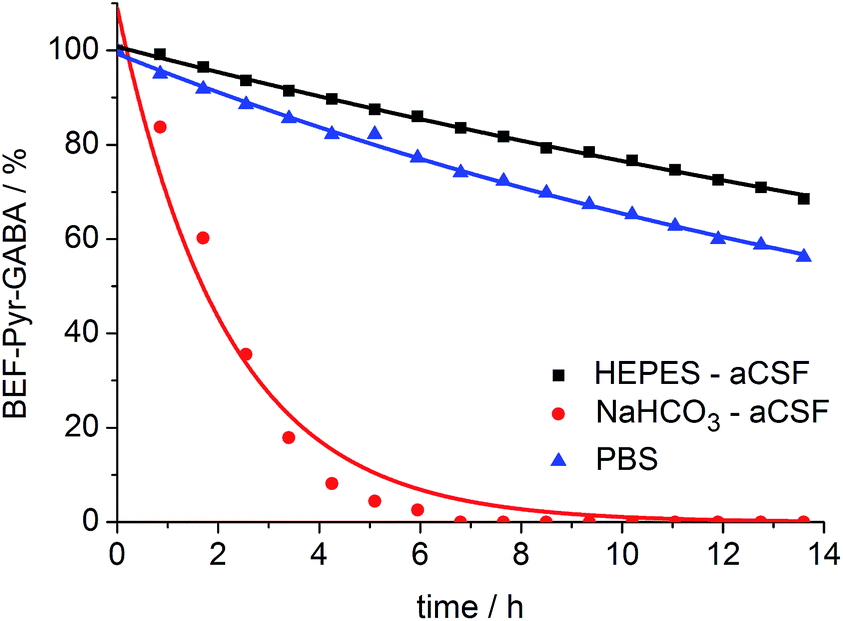
A prerequisite for applications of a caged drug in physiological experiments is that it must be stable in aqueous media, in the absence of light, at least for a few hours. The stability of BEF-Pyr-GABA was assessed in aqueous buffers at pH 7.4 by HPLC and in non-buffered D2O by NMR at 20 °C. We found that its stability is sensitive to pH and to the composition of the buffer. The half-life of BEF-Pyr-GABA in phosphate-buffered saline (PBS) was found to be ca. 15 h (Fig. 6; [BEF-Pyr-GABA]: 20 μM). In contrast, in NaHCO3-based artificial cerebrospinal fluid (aCSF buffer, pH 7.4, which is routinely used as a medium in experiments with neurons) the half-life was reduced to 1.5 h. An extensive set of troubleshooting experiments, in which eight versions of aCSF-buffer were prepared (each missing one different component) allowed us to identify HCO3− as the detrimental component. When NaHCO3 was replaced with HEPES [4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid, an alternative component of standard buffers used in neuroscience] the half-life of BEF-Pyr-GABA rose to ca. 50 h. No hydrolysis was detected in non-buffered D2O (Fig. S31†). These observations highlight the necessity of evaluating the stability of caged compounds under conditions identical to those of the final target application. We have not investigated how bicarbonate catalyzes this hydrolysis reaction, but the hydrolysis of α-amino acid esters under similar conditions has been attributed to CO2-mediated carbamate formation and intramolecular cyclization.25 The low hydrolytic stability of GABA-pyridinium esters in standard aCSF buffer poses a limitation for the use of these compounds under strictly physiological conditions that will need to be addressed in future molecular designs.
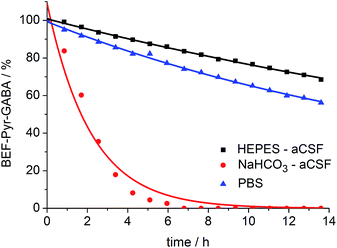 |
| | Fig. 6 Stability of BEF-Pyr-GABA (20 μM) in aqueous buffers at pH 7.4 at 20 °C: HEPES-based aCSF – black, NaHCO3 based aCSF – red, PBS – blue; HEPES: 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid, aCSF: artificial cerebrospinal fluid, PBS: phosphate-buffered saline. Changes in concentration of BEF-Pyr-GABA were determined by HPLC. Mono-exponential fitting curves were applied to the data. | |
The hydrolytic stability of BEF-Pyr-Trp was monitored only by HPLC and is different from that of BEF-Pyr-GABA. BEF-Pyr-Trp has a half-life of only about 2 h at pH 7.4 (in both PBS and NaHCO3 based aCSF buffers). The hydrolysis proceeds more slowly at pH 3.0 (citric acid/citrate buffer), with a half-life of 3 h (Fig. S36†). The close proximity of the protonated amino group to the ester functionality probably increases the electrophilicity of the carbonyl center, enhancing the hydrolytic instability of the tryptophan derivative. BEF-Pyr-Trp also undergoes decomposition at low pH, in a reaction that appears to be associated with the fluorene dye rather than ester hydrolysis (Fig. S37†).
Uncaging studies
We evaluated the uncaging properties of BEF-Pyr-Trp and BEF-Pyr-GABA in a series of one-photon irradiation experiments. The method used to monitor release of the amino acid was dictated by properties of caged species. The high aqueous solubility of BEF-Pyr-GABA facilitated photolysis at mM concentration and we followed the reaction by 1H NMR spectroscopy. It was not possible to monitor the release of GABA by HPLC because this amino acid lacks a UV chromophore. In contrast, BEF-Pyr-Trp gave broad 1H NMR spectra at mM concentrations, presumably due to aggregation. Photolysis studies were conducted at μM concentration and the indole motif in the side chain of tryptophan enabled quantification of the released amino acid by HPLC.
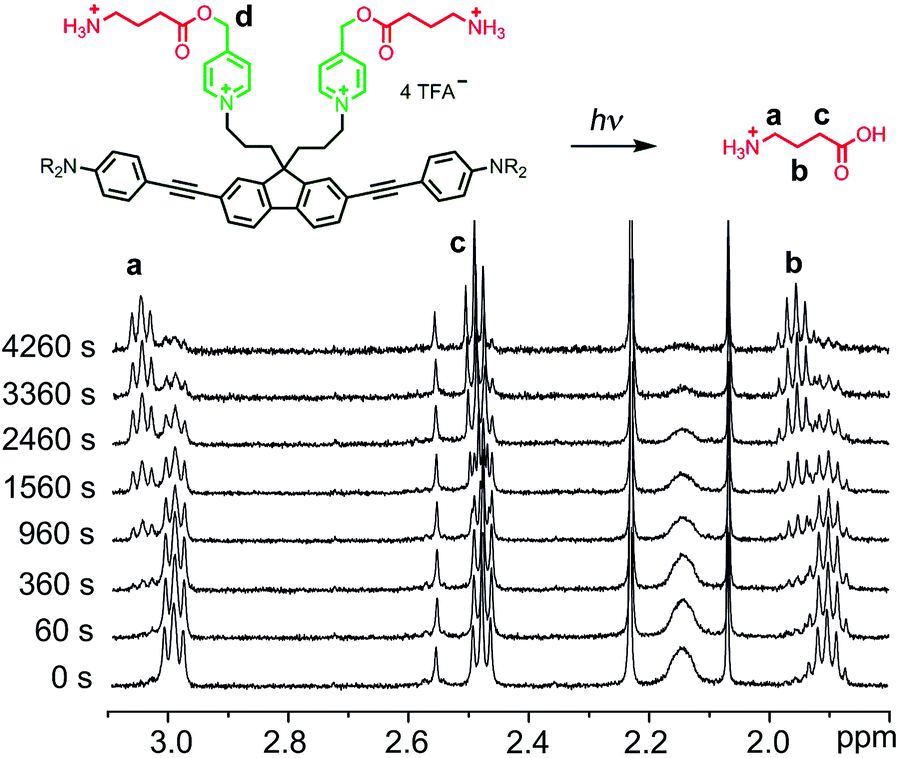
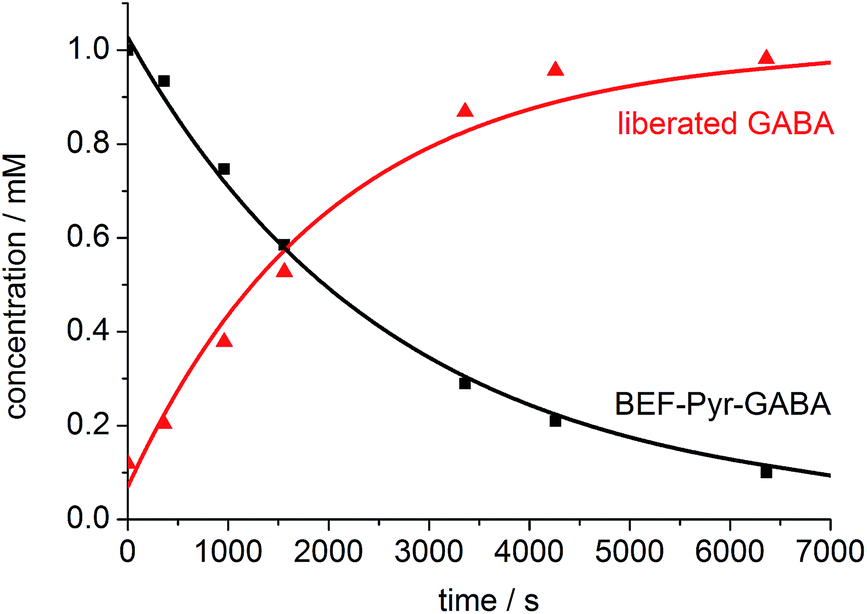
One-photon uncaging of the pyridinium-based protecting group was initially investigated by irradiating a 1 mM D2O solution of BEF-Pyr-GABA with a broad UV-A source (300–400 nm, peak 350 nm) in an NMR tube. At this concentration, the transmittance of the solution is negligible across the entire wavelength range of the light source. 1H NMR spectroscopy (with t-butanol as an internal reference) demonstrated the release of GABA with a chemical yield of >95% (Fig. 7 and 8). Two GABA molecules are released from each molecule of BEF-Pyr-GABA, which demonstrates that the BEF chromophore is able to undergo two cycles of photoreduction. We were unable to identify the chemical products generated by photolysis of the caging group.
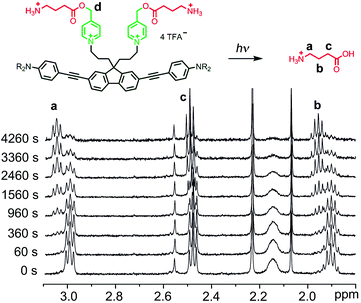 |
| | Fig. 7
1H-NMR spectra from a representative uncaging experiment, which shows the disappearance of caged-GABA signals with a concurrent increase of a new set of multiplets later found to be free GABA (signals “a-c”). Decrease in intensity of signal “d” (5.29 ppm) is not shown. | |
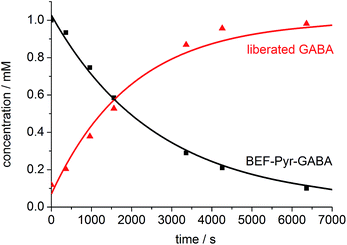 |
| | Fig. 8 The change in concentration of BEF-Pyr-GABA (black squares) and free GABA (red triangles) over time. Concentration of free GABA and BEF-Pyr-GABA were determined by integration of signal “c” and “d” respectively relative to t-butanol. The presence of free GABA was confirmed by doping an irradiated solution with an authentic sample, which resulted in no new signals and an increase in the intensity of the suspected GABA signals (Fig. S30†). | |
The photolysis of BEF-Pyr-Trp (20 μM in water) with 300–400 nm light (350 nm peak) was monitored by HPLC. Complete consumption of the starting material resulted in release of tryptophan with 83% chemical yield. No photochemically generated byproducts were detected but decomposition of the chromophore unit was observed after the uncaging events.
The quantum yield of uncaging of BEF-Pyr-GABA was determined by comparison with the commercially available DPNI-GABA, which has a known ϕu of 0.085.26 Solutions of BEF-Pyr-GABA and DPNI-GABA (1 mM, D2O) were irradiated simultaneously (300–400 nm, peak 350 nm), and their respective rates of uncaging (ku) were determined by 1H-NMR (see ESI†). Under these concentrated conditions, the rate of uncaging depends only on the light intensity and the quantum yield, but not on the molar absorption coefficient. The uncaging quantum yield of BEF-Pyr-GABA was calculated using eqn (4) to give ϕu = 0.009 ± 0.003.
| | 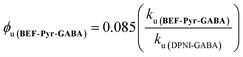 | (4) |
This determination of the uncaging quantum yield of BEF-Pyr-GABA was verified using ferric oxalate actinometry,27 which gave an uncaging quantum yield of ϕu = 0.088 ± 0.004 for DPNI-GABA (in close accord with the published value) and an uncaging quantum yield of ϕu = 0.009 ± 0.004 for BEF-Pyr-GABA.
The fluorescence quenching experiments showed that 98% of absorption events lead to charge transfer, while the overall uncaging quantum yield is only about 1%, which suggests that decay of the excited state is dominated by back electron transfer to the ground state. Nevertheless, due to the high value of the TPA cross section (1100 GM at 700 nm), the calculated two-photon uncaging cross section for BEF-Pyr-GABA is δu = δaϕu = 10 ± 3 GM at 700 nm, which is comparable to the highest reported value (11 GM for the 2-(o-nitrophenyl)propyl caged GABA).11a
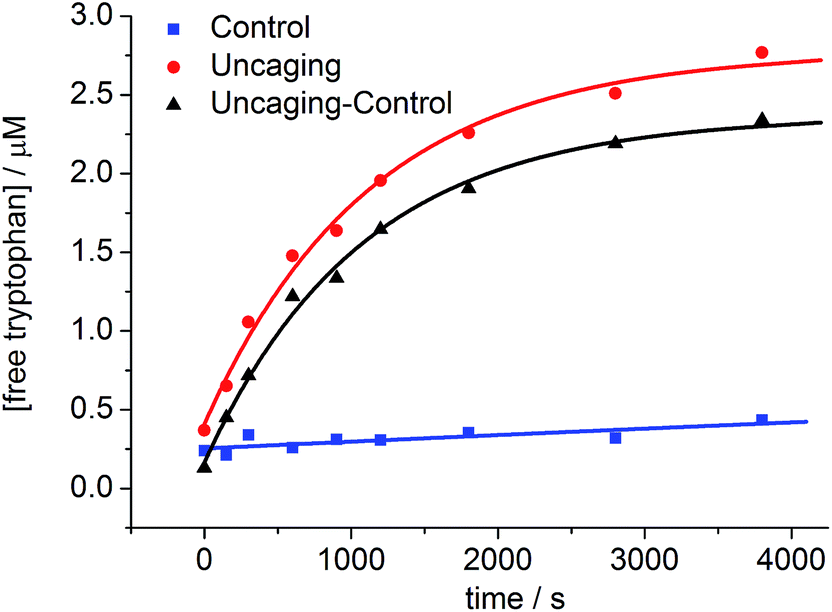
The uncaging quantum yield for BEF-Pyr-Trp was also determined by ferric oxalate actinometry. A solution of BEF-Pyr-Trp (∼1.5 μM, pH 3.0) was irradiated at 360 nm with a fluorimeter and photorelease of tryptophan was monitored by HPLC (Fig. 9), using a protocol designed to take account of competing background hydrolysis (see ESI†). The quantum yield of uncaging was measured for BEF-Pyr-Trp as ϕu = 0.0025 (at 360 nm), so the calculated two-photon uncaging cross-section for BEF-Pyr-Trp at 720 nm is only δu = 2.5 GM. The reasons for the difference in uncaging quantum yield between BEF-Pyr-Trp and BEF-Pyr-GABA are unclear and will require further investigation.
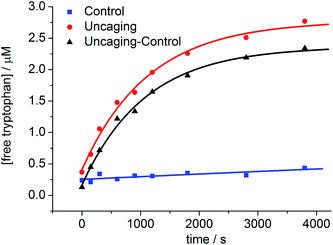 |
| | Fig. 9 The change in concentration of tryptophan upon photolysis of BEF-Pyr-Trp at pH 3.0 and 360 nm; red circles – photolyzed sample, blue squares – control samples stored in the dark, black triangles – net concentration of tryptophan released upon uncaging. The concentration of tryptophan was determined by HPLC. | |
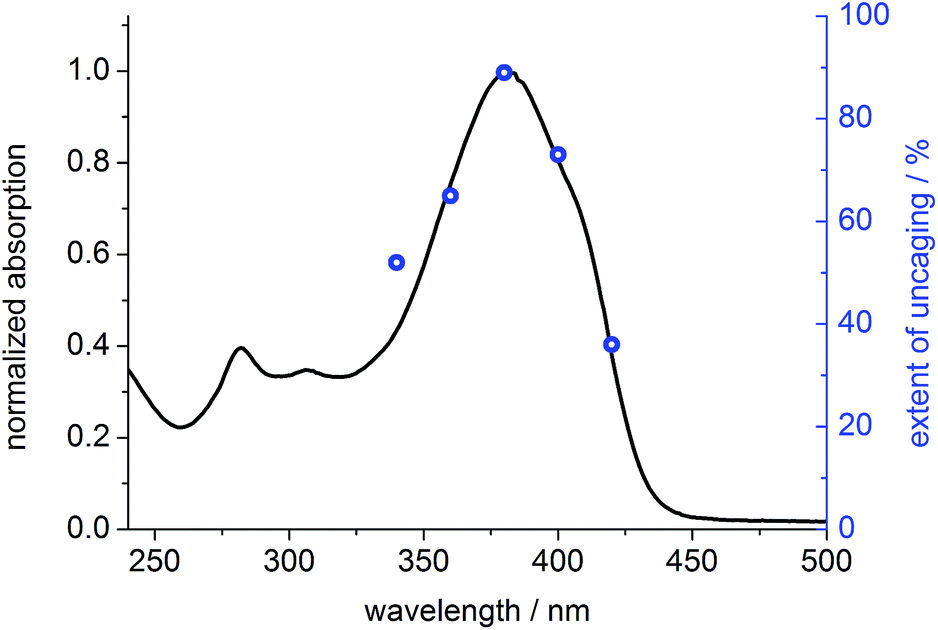
To prove that photo-cleavage of our new protecting group is sensitized by the absorption of the fluorene-based dye, rather than by direct excitation of pyridinium unit, we investigated the efficiency of uncaging as a function of irradiation wavelength. For this purpose, solutions of BEF-Pyr-Trp (1.5 μM, pH 3.0) were irradiated at 340, 360, 380, 400 and 420 nm for 3800 s, and dark control experiments were carried out, to account for background hydrolysis (Fig. 9). The extent of uncaging correlates closely with the absorption spectrum of the BEF chromophore (Fig. 10), confirming the active role of the fluorene-dye in uncaging and PeT mediated release of tryptophan from BEF-Pyr-Trp.
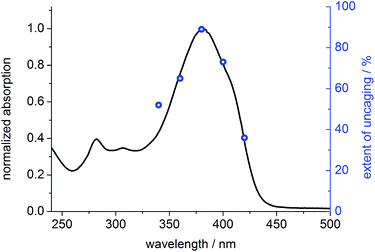 |
| | Fig. 10 Normalized absorption spectrum of BEF-Pyr-Trp in citric acid/sodium citrate buffer (pH 3.0) (black line) plotted in the scale with extent of photolysis, expressed as yield of released tryptophan upon 3800 s of irradiation of BEF-Pyr-Trp at 340–420 nm (blue circles). | |
Preliminary experiments were carried out to test the two-photon excited release of GABA in the proximity of cultured neurons using BEF-Pyr-GABA. Upon two-photon excitation at 720 nm (300 fs, Ti:sapphire laser; 2–5 ms duration), we observed changes in membrane potential with kinetics consistent with activation of GABA-A receptors, indicating GABA release. Further experiments are needed to test the effectiveness of this caged compound, and to quantify side-effects, such as the biological activity of the caged drug.
Non-pyridinium designs
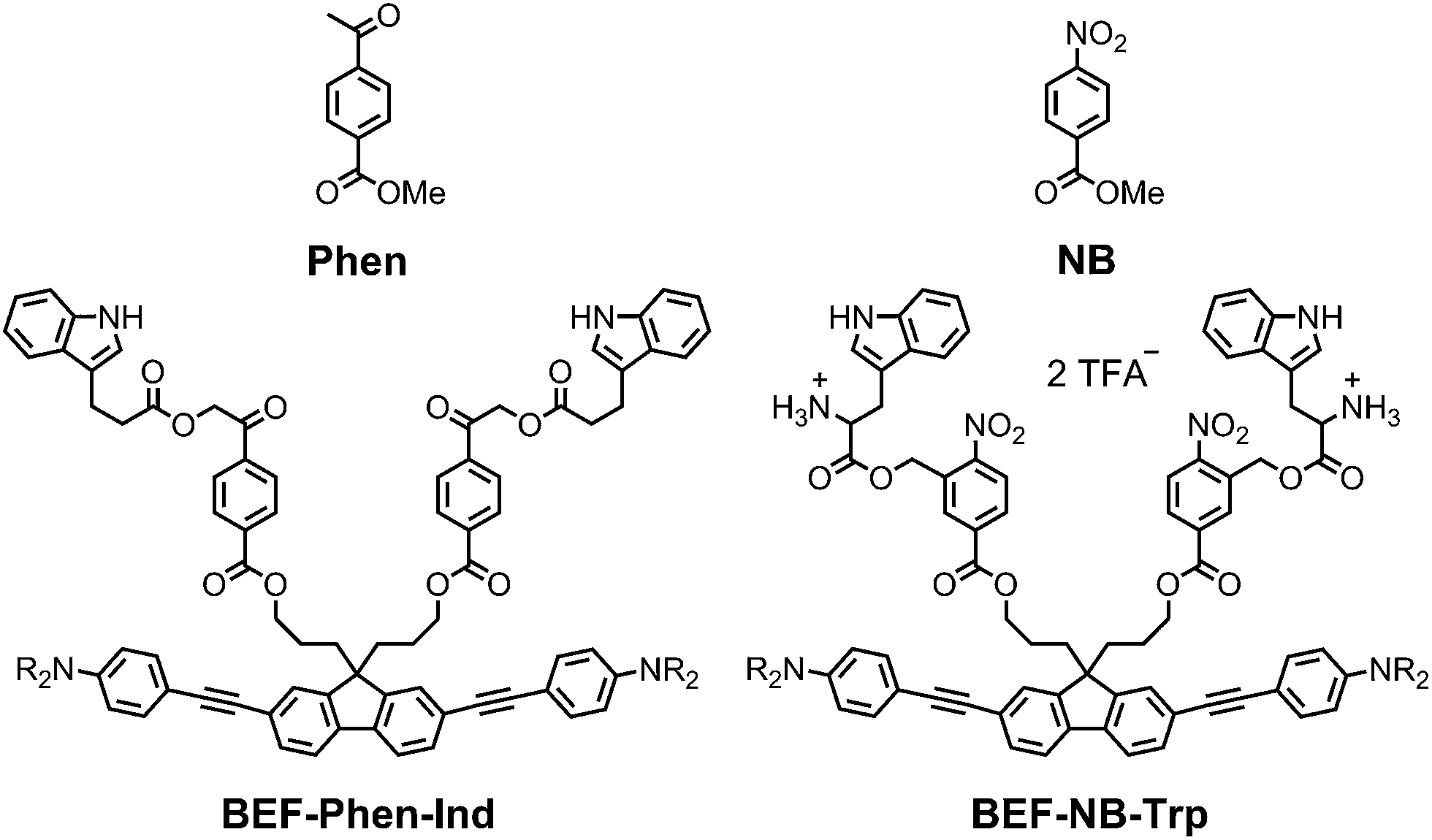
The pyridinium group is an excellent electron-acceptor, and the results presented above show that it gives very efficient PeT, however the efficiency of cleavage of the charge-separated state is disappointing (ϕu ≈ 1%). The pyridinium group may also exhibit undesirable reactivity towards nucleophiles, so we decided to explore other electron acceptors. We chose to investigate phenacyl, for which uncaging via PeT has been previously reported13b and nitrobenzyl esters (Fig. 11). The reduction potentials of both methyl 4-acetylbenzoate (Phen) and methyl 4-nitrobenzoate (NB) were measured by cyclic and square wave voltammetry, giving ERED of Phen: −2.18 V; ERED of NB: −1.47 V (vs. Fc/Fc+ in THF with 0.1 M Bu4PF6). We estimated the values of the Coulombic term, eqn (2), in BEF-Phen-Ind and BEF-NB-Trp from the distance of photoinduced charge separation (a) using molecular mechanics calculations (see ESI†). This gave w(D+˙A−˙) = −0.21 eV for both compounds. Calculation of Gibbs energy of the photoinduced electron transfer (ΔGET), according to eqn (1), revealed that it is energetically favorable in both systems, giving ΔGET of −0.54 eV and −1.25 eV for Phen and NB respectively. To evaluate the photo-release properties of phenacyl and nitrobenzyl derived groups, we synthesized their tryptophan analogues for HPLC-monitored uncaging experiments. In the case of the phenacyl group, attack of the free amino group of tryptophan on the ketone group to form a 6-membered ring posed a limitation for protection of α-amino acids. This issue was overcome by use of the alternative structure: 3-indolepropionic acid (Ind), which possesses an indole chromophore but lacks the amino group, allowing us to preserve the absorption properties of tryptophan while avoiding cyclization. Structures of nitrobenzyl tryptophan (BEF-NB-Trp) and phenacyl protected 3-indolepropionic acid (BEF-Phen-Ind) are shown in Fig. 11 (for synthesis see ESI†).
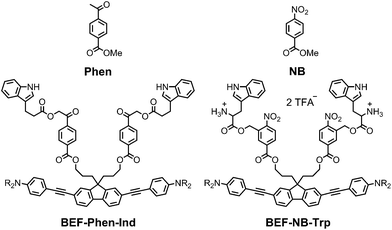 |
| | Fig. 11 The structures of alternative electron acceptor units: methyl 4-acetylbenzoate (Phen) and methyl 4-nitrobenzoate (NB) and corresponding protecting groups based on the banana-shaped fluorene dye BEF-Phen-Ind and BEF-NB-Trp; R = (CH2CH2O)7CH3. | |
Photolysis of BEF-Phen-Ind was investigated by irradiating a solution in water (17 μM) with a broad UV-A light source (300–400 nm; peak: 350 nm), monitoring the progress of uncaging by HPLC. 3-Indolepropionic acid was liberated with a modest 20% chemical yield, despite full consumption of the starting material. HPLC analysis did not reveal formation of any other products of photolysis and we were unable to define the fate of the remaining 80% of starting material. The quantum yield of uncaging was determined by use of ferric oxalate actinometry, giving ϕu = 0.0022, corresponding to a two-photon uncaging cross section of δu = 2.4 GM at 700 nm. The limited scope of substrates that can be caged (due to the reactivity of the ketone group), the low chemical yield of photorelease and the susceptibility to hydrolysis under physiological conditions make ester-linked phenacyl platforms unattractive release units.
Photolysis of BEF-NB-Trp was tested in a range of solvents (5 μM concentration in water, NaHCO3-aCSF, acetonitrile, methanol and THF) using a broad UV-A light source (300–400 nm; peak: 350 nm), but no release of tryptophan was observed. Photochemical decomposition of BEF-NB-Trp occurred, but it did not result information of free tryptophan. The same result was observed when irradiation was carried out at 280 nm. Tryptophan was liberated cleanly in 75% yield by hydrolysis of BEF-NB-Trp in the dark over 40 h in NaHCO3-based aCSF buffer.
Conclusions
In this study, we have investigated an approach to the rational design of two-photon photo-labile protecting groups. A protecting group has been developed which operates by PeT between an electron-rich fluorene-based dye and a pyridinium electron-acceptor. The fluorescence of the dye is quenched by electron transfer to the pyridinium; charge-transfer leads to bond scission, to liberate a carboxylic acid. Our protecting group has been demonstrated to release the neurotransmitter GABA and amino acid L-tryptophan upon irradiation with light of wavelength 340–420 nm, in aqueous solution in nearly quantitative chemical yields. This group exhibits a high TPA cross-section (1100 GM at 700 nm) and highly efficient charge-transfer between the electron donor and acceptor was observed (98%). The fast back electron transfer from the charge-shifted state reduces the overall quantum efficiency of uncaging to around 1% which, when combined with the TPA cross-section, results in a two-photon uncaging cross-section of approximately δu = 10 GM (700 nm) for BEF-Pyr-GABA. Wavelength-dependent uncaging experiments confirmed electron-transfer mediated release of caged tryptophan with efficiency of release proportional to the extinction coefficient of the fluorene dye within 340–420 nm.
A key objective for future research will be to apply the modular design strategy demonstrated in this study to create an electron donor–acceptor pair for which the back electron transfer is suppressed, so that bond-scission becomes the main decay pathway. The susceptibility of pyridinium esters towards hydrolysis can lead to practical difficulties for uncaging studies in aqueous media, and it would be useful to extend these systems to non-ester linking unit that is are more stable to aqueous hydrolysis, such as carbamates.
Acknowledgements
We thank Prof. Seth R. Marder for valuable discussion, Prof. Ole Paulsen, Dr Yu Zhang and Dr Golnaz Borghei for carrying out preliminary electrophysiology experiments with BEF-Pyr-GABA. This work was funded by the European Commission Seventh Framework Programme (TOPBIO ITN, project-grant agreement no. 264362). We thank the EPSRC Mass Spectrometry Service (Swansea) for mass spectra.
Notes and references
- J. H. Kaplan, B. Forbush III and J. F. Hoffman, Biochemistry, 1978, 17, 1929 CrossRef CAS PubMed.
-
(a) A. P. Pelliccioli and J. Wirz, Photochem. Photobiol. Sci., 2002, 1, 441 RSC;
(b) C. G. Bochet, J. Chem. Soc., Perkin Trans. 1, 2002, 125 CAS;
(c) P. Klán, T. Šolomek, C. G. Bochet, A. Blanc, R. Givens, M. Rubina, V. Popik, A. Kostikov and J. Wirz, Chem. Rev., 2013, 113, 119 CrossRef PubMed;
(d) H. Yu, J. Li, D. Wu, Z. Qiua and Y. Zhang, Chem. Soc. Rev., 2010, 39, 464 RSC.
- G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2006, 45, 4900 CrossRef CAS PubMed.
- M. Pawlicki, H. A. Collins, R. G. Denning and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 3244 CrossRef CAS PubMed.
- W. Denk, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 6629 CrossRef CAS.
-
(a) G. Bort, T. Gallavardin, D. Ogden and P. I. Dalko, Angew. Chem., Int. Ed., 2013, 52, 4526 CrossRef CAS PubMed;
(b) A. Specht, F. Bolze, Z. Omran, J.-F. Nicoud and M. Goeldner, HFSP J., 2009, 3, 255 CrossRef CAS PubMed;
(c) C. Brieke, F. Rohrbach, A. Gottschalk, G. Mayer and A. Heckel, Angew. Chem., Int. Ed., 2012, 51, 8446 CrossRef CAS PubMed;
(d) D. Warther, S. Gug, A. Specht, F. Bolze, J.-F. Nicoud, A. Mourot and M. Goeldner, Bioorg. Med. Chem., 2010, 18, 7753 CrossRef CAS PubMed.
-
(a) O. D. Fedoryak, J.-Y. Sul, P. G. Haydon and G. C. R. Ellis-Davies, Chem. Commun., 2005, 3664 RSC;
(b) G. C. R. Ellis-Davies, M. Matsuzaki, M. Paukert, H. Kasai and D. E. Bergles, J. Neurosci., 2007, 27, 6601 CrossRef CAS PubMed;
(c) N. Gagey, P. Neveu and L. Jullien, Angew. Chem., Int. Ed., 2007, 46, 2467 CrossRef CAS PubMed;
(d) N. Gagey, P. Neveu, C. Benbrahim, B. Goetz, I. Aujard, J.-B. Baudin and L. Jullien, J. Am. Chem. Soc., 2007, 129, 9986 CrossRef CAS PubMed;
(e) T. Furuta, S. S. Wang, J. L. Dantzker, T. M Dore, J. W. Bybee, E. M. Callaway, W. Denk and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1193 CrossRef CAS PubMed;
(f) V. Nikolenko, R. Yuste, L. Zayat, L. M. Baraldo and R. Etchenique, Chem. Commun., 2005, 1752 RSC;
(g) M. Salierno, E. Marceca, D. S. Peterka, R. Yuste and R. Etchenique, J. Inorg. Biochem., 2010, 104, 418 CrossRef CAS PubMed;
(h) E. Fino, R. Araya, D. S. Peterka, M. Salierno, R. Etchenique and R. Yuste, Front. Neural Circuits, 2009, 3, 1 Search PubMed;
(i) A. Specht, J.-S. Thomann, K. Alarcon, W. Wittayanan, D. Ogden, T. Furuta, Y. Kurakawa and M. Goeldner, ChemBioChem, 2006, 7, 1690 CrossRef CAS PubMed;
(j) Y. Zhu, C. M. Pavlos, J. P. Toscano and T. M. Dore, J. Am. Chem. Soc., 2006, 128, 4267 CrossRef CAS PubMed;
(k) M. J. Davis, C. H. Kragor, K. G. Reddie, H. C. Wilson, Y. Zhu and T. M. Dore, J. Org. Chem., 2009, 74, 1721 CrossRef CAS PubMed;
(l) M. Petit, C. Tran, T. Roger, T. Gallavardin, H. Dhimane, F. Palma-Cerda, M. Blanchard-Desce, F. C. Acher, D. Ogden and P. I. Dalko, Org. Lett., 2012, 14, 6366 CrossRef CAS PubMed.
- N. I. Kiskin, R. Chillingworth, J. A. McCray, D. Piston and D. Ogden, Eur. Biophys. J., 2002, 30, 588 CrossRef CAS PubMed.
-
(a) J. P. Olson, H.-B. Kwon, K. T. Takasaki, C. Q. Chiu, M. J. Higley, B. L. Sabatini and G. C. R. Ellis-Davies, J. Am. Chem. Soc., 2013, 135, 5954 CrossRef CAS PubMed;
(b) J. M. Amatrudo, J. P. Olson, G. Lur, C. Q. Chiu, M. J. Higley and G. C. R. Ellis-Davies, ACS Chem. Neurosci., 2014, 5, 64 CrossRef CAS PubMed;
(c) Y. Sakamoto, S. Boinapally, C. Katan and M. Abe, Tetrahedron Lett., 2013, 54, 7171 CrossRef CAS;
(d) C. Bao, G. Fan, Q. Lin, B. Li, S. Cheng, Q. Huang and L. Zhu, Org. Lett., 2012, 14, 572 CrossRef CAS PubMed.
-
(a) I. Aujard, C. Benbrahim, M. Gouget, O. Ruel, J.-B. Baudin, P. Neveu and L. Jullien, Chem.–Eur. J., 2006, 12, 6865 CrossRef CAS PubMed;
(b) S. Boinapally, B. Huang, M. Abe, C. Katan, J. Noguchi, S. Watanabe, H. Kasai, B. Xue and T. Kobayashi, J. Org. Chem., 2014, 79, 7822 CrossRef CAS PubMed.
-
(a) L. Donato, A. Mourot, C. M. Davenport, C. Herbivo, D. Warther, J. Léonard, F. Bolze, J.-F. Nicoud, R. H. Kramer, M. Goeldner and A. Specht, Angew. Chem., Int. Ed., 2012, 51, 1840 CrossRef CAS PubMed;
(b) S. Gug, F. Bolze, A. Specht, C. Bourgogne, M. Goeldner and J.-F. Nicoud, Angew. Chem., Int. Ed., 2008, 47, 9525 CrossRef CAS PubMed;
(c) A. Specht, F. Bolze, L. Donato, C. Herbivo, S. Charon, D. Warther, S. Gug, J.-F. Nicoud and M. Goeldner, Photochem. Photobiol. Sci., 2012, 11, 578 RSC;
(d) S. Gug, S. Charon, A. Specht, K. Alarcon, D. Ogden, B. Zietz, J. Léonard, S. Haacke, F. Bolze, J.-F. Nicoud and M. Goeldner, ChemBioChem, 2008, 9, 1303 CrossRef CAS PubMed;
(e) F. Bolze, J.-F. Nicoud, C. Bourgogne, S. Gug, X. H. Sun, M. Goeldner, A. Specht, L. Donato, D. Warther, G. F. Turi and A. Losonczy, Opt. Mater., 2012, 34, 1664 CrossRef CAS.
- S. Picard, E. Genin, G. Clermont, V. Hugues, O. Mongin and M. Blanchard-Desce, New J. Chem., 2013, 37, 3899 RSC.
-
(a) D. E. Falvey and C. Sundararajan, Photochem. Photobiol. Sci., 2004, 3, 831 RSC;
(b) K. Lee and D. E. Falvey, J. Am. Chem. Soc., 2000, 122, 9361 CrossRef CAS;
(c) C. Sundararajan and D. E. Falvey, Org. Lett., 2005, 7, 2631 CrossRef CAS PubMed.
-
(a) D. Wöll, J. Smirnova, W. Pfleiderer and U. E. Steiner, Angew. Chem., Int. Ed., 2006, 45, 2975 CrossRef PubMed;
(b) G. Papageorgiou, D. Ogden and J. E. T. Corrie, J. Org. Chem., 2004, 69, 7228 CrossRef CAS PubMed;
(c) G. Papageorgiou, D. Ogden and J. E. T. Corrie, Photochem. Photobiol. Sci., 2008, 7, 423 RSC.
-
(a) S. Picard, E. J. Cueto-Diaz, E. Genin, G. Clermont, F. Acher, D. Ogden and M. Blanchard-Desce, Chem. Commun., 2013, 49, 10805 RSC;
(b) E. J. Cueto Díaz, S. Picard, V. Chevasson, J. Daniel, V. Hugues, O. Mongin, E. Genin and M. Blanchard-Desce, Org. Lett., 2015, 17, 102 CrossRef PubMed.
- M. C. Pirrung, T. M. Dore, Y. Zhu and V. S. Rana, Chem. Commun., 2010, 46, 5313 RSC.
- Related studies were reported in a PhD thesis by Jing Wang, University of Arizona, 2007; thesis advisor: S. R. Marder.
- O. Mongin, L. Porrès, M. Charlot, C. Katan and M. Blanchard-Desce, Chem.–Eur. J., 2007, 13, 1481 CrossRef CAS PubMed.
-
(a) C. Sundararajan and D. E. Falvey, J. Org. Chem., 2004, 69, 5547 CrossRef CAS PubMed;
(b) C. Sundararajan and D. E. Falvey, J. Am. Chem. Soc., 2005, 127, 8000 CrossRef CAS PubMed;
(c) C. Sundararajan and D. E. Falvey, Photochem. Photobiol. Sci., 2006, 5, 116 RSC.
- C. Monnereau, E. Blart and F. Odobel, Tetrahedron Lett., 2005, 46, 5421 CrossRef CAS.
- F. Terenziani, A. Painelli, C. Katan, M. Charlot and M. Blanchard-Desce, J. Am. Chem. Soc., 2006, 128, 15742 CrossRef CAS PubMed.
- C. Katan, M. Charlot, O. Mongin, C. Le Droumaguet, V. Jouikov, F. Terenziani, E. Badaeva, S. Tretiak and M. Blanchard-Desce, J. Phys. Chem. B, 2010, 114, 3152 CrossRef CAS PubMed.
- IUPAC. Compendium of Chemical Terminology, 2nd ed. (the “Gold Book”). Compiled by A. D. McNaught and A. Wilkinson. Blackwell Scientific Publications, Oxford (1997). XML on-line corrected version: http://www.goldbook.iupac.org (2006-) created by M. Nic, J. Jirat, B. Kosata; updates compiled by A. Jenkins. ISBN 0-9678550-9-8.
- A. I. Ciuciu, K. A. Korzycka, W. J. Lewis, P. M. Bennett, H. L. Anderson and L. Flamigni, Phys. Chem. Chem. Phys., 2015, 17 RSC.
-
(a) I. M. Kovach, I. H. Pitman and T. Higuchi, J. Pharm. Sci., 1981, 70, 881 CrossRef CAS PubMed;
(b) R. W. Hay and L. Main, Aust. J. Chem., 1968, 21, 155 CrossRef CAS.
- G. Papageorgiou and J. E. T. Corrie, Tetrahedron, 2007, 63, 9668 CrossRef CAS.
-
(a) J. Goedhart and T. W. J. Gadella, Biochemistry, 2004, 43, 4263 CrossRef CAS PubMed;
(b) H. J. Kuhn, S. E. Braslavsky and R. Schmidt, Pure Appl. Chem., 1989, 61, 187 CrossRef CAS;
(c)
M. Montalti, A. Credi, L. Prodi and M. T. Gandolfi, Handbook of Photochemistry, Taylor & Francis Group, Florida, 2006, pp. 601–604 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: One- and two-photon absorption spectra, emission spectra, synthetic procedures and characterization, square-wave and cyclic voltammetry, photolysis protocols. See DOI: 10.1039/c4sc03775h |
| ‡ K.A.K. and P.M.B. made equal contribution to this work. |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a