
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In vitro and in vivo comparative and competitive activity-based protein profiling of GH29 α-L-fucosidases†
Jianbing
Jiang‡
a,
Wouter W.
Kallemeijn‡
b,
Daniel W.
Wright
c,
Adrianus M. C. H.
van den Nieuwendijk
a,
Veronica Coco
Rohde
a,
Elisa Colomina
Folch
a,
Hans
van den Elst
a,
Bogdan I.
Florea
a,
Saskia
Scheij
b,
Wilma E.
Donker-Koopman
b,
Marri
Verhoek
b,
Nan
Li
a,
Martin
Schürmann
d,
Daniel
Mink
d,
Rolf G.
Boot
b,
Jeroen D. C.
Codée
a,
Gijsbert A.
van der Marel
a,
Gideon J.
Davies
c,
Johannes M. F. G.
Aerts
*ab and
Herman S.
Overkleeft
*a
aLeiden Institute of Chemistry, Leiden University, P. O. Box 9502, 2300 RA Leiden, The Netherlands. E-mail: h.s.overkleeft@chem.leidenuniv.nl; j.m.aerts@amc.uva.nl
bDepartment of Medical Biochemistry, Academic Medical Center, Meibergdreef 15, 1105 AZ Amsterdam, The Netherlands
cDepartment of Chemistry, University of York, Heslington, York, YO10 5DD, UK
dDSM Innovative Synthesis, Urmonderbaan 22, NL-6167 RD Geleen, The Netherlands
First published on 9th February 2015
Abstract
GH29 α-L-fucosidases catalyze the hydrolysis of α-L-fucosidic linkages. Deficiency in human lysosomal α-L-fucosidase (FUCA1) leads to the recessively inherited disorder, fucosidosis. Herein we describe the development of fucopyranose-configured cyclophellitol aziridines as activity-based probes (ABPs) for selective in vitro and in vivo labeling of GH29 α-L-fucosidases from bacteria, mice and man. Crystallographic analysis on bacterial α-L-fucosidase confirms that the ABPs act by covalent modification of the active site nucleophile. Competitive activity-based protein profiling identified L-fuconojirimycin as the single GH29 α-L-fucosidase inhibitor from eight configurational isomers.
Introduction
GH29 α-L-fucosidases catalyze the hydrolysis of terminal α-L-fucosidic linkages.1 The GH29 (ref. 2) glycoside hydrolase family of retaining α-L-fucosidases contains members from various kingdoms of life including the eukaryota3 and bacteria (for example, Bacteroides thetaiotaomicron,4Sulfolobus solfataricus,5 and Thermotoga maritima6). GH29 α-L-fucosidases process their substrate with overall retention of configuration at the anomeric center of the cleaved fucopyranose and do so through a double-displacement mechanism. In this mechanism, first proposed by Koshland,7 (Fig. 1A), SN2 displacement of the aglycon (activated through protonation by the general acid/base residue) by nucleophilic attack of the catalytic nucleophile yields a fucosyl-enzyme intermediate, which is subsequently hydrolyzed to yield α-L-fucopyranose together with the released aglycon.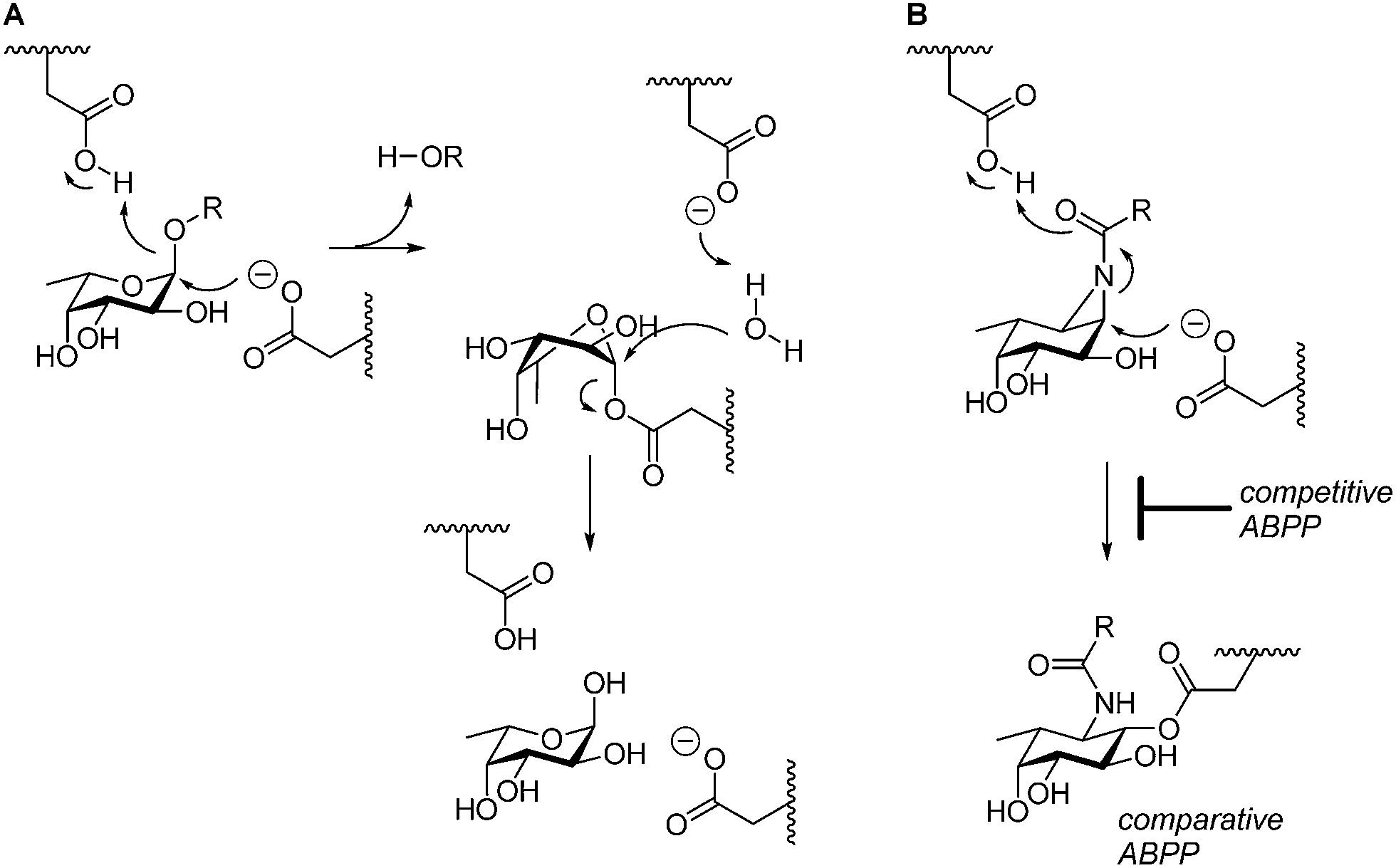 | ||
| Fig. 1 (A) Double-displacement mechanism of retaining α-L-fucosidases. (B) Comparative and competitive activity-based profiling of GH29 α-L-fucosidases presented here. | ||
Two GH29 α-L-fucosidases are expressed in man. Of these, FUCA1 is found in lysosomes whereas FUCA2 is secreted into the plasma.8 Deficiency of FUCA1 α-L-fucosidase activity causes fucosidosis,9 a rare autosomal recessive lysosomal storage disorder. Next to its role in lysosomal turnover of fucosylated substrates, FUCA1 is also involved in sperm transport and sperm–egg interactions.10 FUCA1 activity levels are considered to be a biomarker for cellular senescence11 as well as for the diagnosis of hepatocellular cancers.12 Deficiency of human FUCA2 has been shown to protect against Helicobacter pylori adhesion to gastric cancer cells.8
The biological and biomedical relevance of GH29 retaining α-L-fucosidases warrant the development of efficient methods to monitor their functional state and activity in vitro, in situ and in vivo. In this respect, activity-based probes (ABPs) have shown their merit as tools to detect active enzyme molecules in their native environment (Fig. 1B).13 We have previously reported the use of cyclophellitol aziridines as scaffolds for the design of in situ and in vivo active ABPs directed at GH1 retaining β-glucosidases14 and GH27 retaining α-galactosidases.15 The specificity of these probes appeared due to their configuration, with the β-glucopyranose configured cyclitol aziridine being highly selective towards retaining β-glucosidases and their α-galacto-configured counterparts selective towards α-galactosidases. Here we describe the development of retaining GH29 α-L-fucosidase ABPs. The ABPs are based on the cyclophellitol aziridine structure having α-L-fucoside configuration and are equipped with a green (1, JJB256) or red (2, JJB244) BODIPY fluorophore and biotin tag (3, JJB243) (Fig. 2). We reveal that these probes are highly sensitive and selective and can be used for in situ and in vivo monitoring of mammalian and bacterial GH29 retaining α-L-fucosidases.
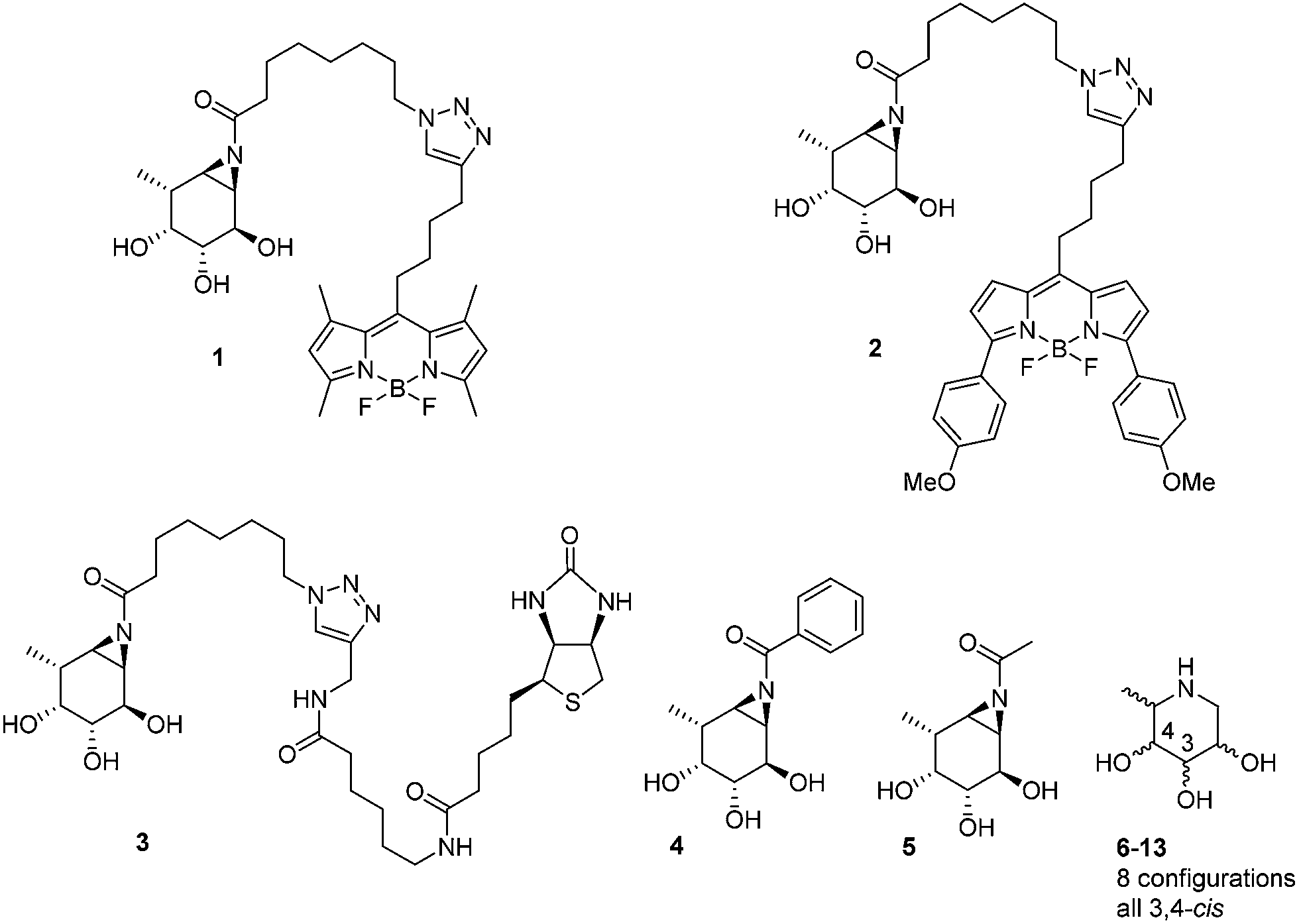 | ||
| Fig. 2 Structures of inhibitors and ABPs presented in this study. | ||
We also demonstrate that ABPs 1 and 2 can be used in a competitive activity-based protein profiling (ABPP) assay16 to identify rapidly retaining α-L-fucosidase inhibitors from a library of eight configurational isomers of deoxy-L-fuconojirimycin (6–13); a library we prepared specifically for this purpose. Finally we unambiguously establish the validity of the cyclophellitol aziridine design platform for ABP development of retaining glycosidases by solving the crystal structure of retaining α-L-fucosidase from Bacteroides thetaiotaomicron 2970, covalently bound to N-acyl cyclophellitol aziridines 4 and 5.
Results and discussion
The synthesis of α-L-fucopyranose-configured cyclophellitol aziridine-based target compounds started with aldol condensation of aldehyde 15 and chiral acrylamide 14, following the procedure reported17 by Llebaria and co-workers for the enantiomer of 16 (Scheme 1). Reductive removal of the Evans template in 16 followed by ring-closing metathesis yielded according to the Llebaria procedure17 partially protected L-galactopyranose-configured cyclohexene 17 in good yield. Tosylation of the primary alcohol in 17 was followed by hydride displacement of the tosylate to afford L-fucopyranose-configured cyclohexene 19. The benzyl groups in 19 were reduced under Birch conditions, after which the cis-diol was protected as the isopropylidene acetal to give 20. The secondary alcohol in 20 was transformed into the corresponding trichloroacetimidate after which iodocyclisation yielded in a stereospecific fashion intermediate 21 analogous to the procedure we reported for the synthesis of retaining β-glucosidase ABPs.18 Acidic hydrolysis of both acetal and iminal in 21 was followed by base-induced intramolecular nucleophilic substitution of the iodine to yield aziridine 22. Acetylation or benzoylation of the aziridine in 22 under the agency of 2-ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ) following conditions we had developed previously18 yielded compounds 4 and 5, respectively. EEDQ-induced acylation of 22 with 8-azidooctanoic acid1924 provided azide 23, which was transformed into target ABPs 1, 2 and 3 by conjugation to BODIPY-alkynes 25, 26 and biotin-alkyne 27, respectively, via copper(I)-catalyzed Huisgen [2 + 3] cycloaddition. The final compounds were purified by reverse phase HPLC.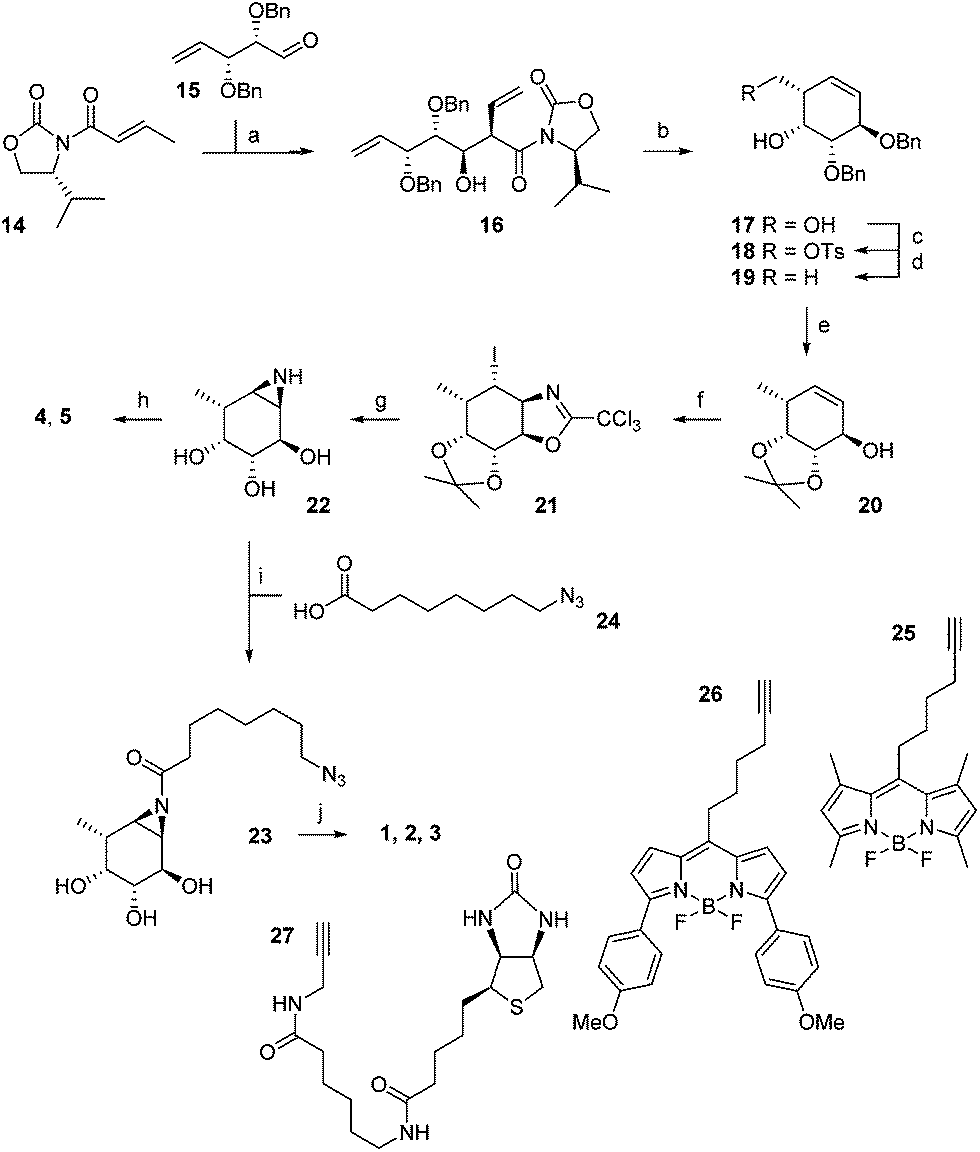 | ||
| Scheme 1 Synthesis of aziridine ABPs 1, 2, 3 and inhibitors 4, 5. Reagents and conditions: (a) DBBT, Et3N, CH2Cl2, −78 °C, 71%; (b) (i) LiBH4, THF, 83%; (ii) Grubbs 2nd generation, CH2Cl2, 95%; (c) p-TsCl, Et3N, CH2Cl2, 87%; (d) LiAlH4, THF, 0 °C to rt, 87%; (e) (i) Li, NH3 (l), THF, −60 °C, 73%, (ii) 2,2-dimethoxypropane, CSA, rt, 60%; (f) (i) CCl3CN, DBU, CH2Cl2, rt; (ii) NaHCO3, I2, H2O, rt, 46%; (g) (i) 37% HCl (aq.), MeOH, 60 °C; (ii) NaHCO3, MeOH, rt, 65%; (h) EEDQ, benzoic acid or acetic acid, DMF, 0 °C, 7% 4, 28% 5; (i) EEDQ, 24, DMF, 0 °C 25%; (j) CuSO4, sodium ascorbate, 25, 26 or 27, rt, 19% 1, 12% 2, 13% 3. | ||
The configurational fuconojirimycin isomers 6–13 (Fig. 3) were synthesized following the strategy exemplified for 1-deoxy-L-fuconojirimycin 6 (Scheme 2). Key steps in the synthetic scheme include a DIBAL-H reduction-transimination-sodium borohydride reduction cascade of reactions involving enantiomerically pure cyanohydrin 28, prepared employing (S)-hydroxynitrile lyase (S-HbHNL), from Hevea brasiliensis rubber tree,20 and the allylic amine 29 prepared by reported strategy,21 to give secondary amine 30. N-Boc protection (30 to 31), ring-closing metathesis (31 to 32) and Upjohn dihydroxylation afforded a mixture of syn-diols, which were acetylated and separated by silica gel purification to yield diastereomers 33 and 34 in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ratio.
:3 ratio.
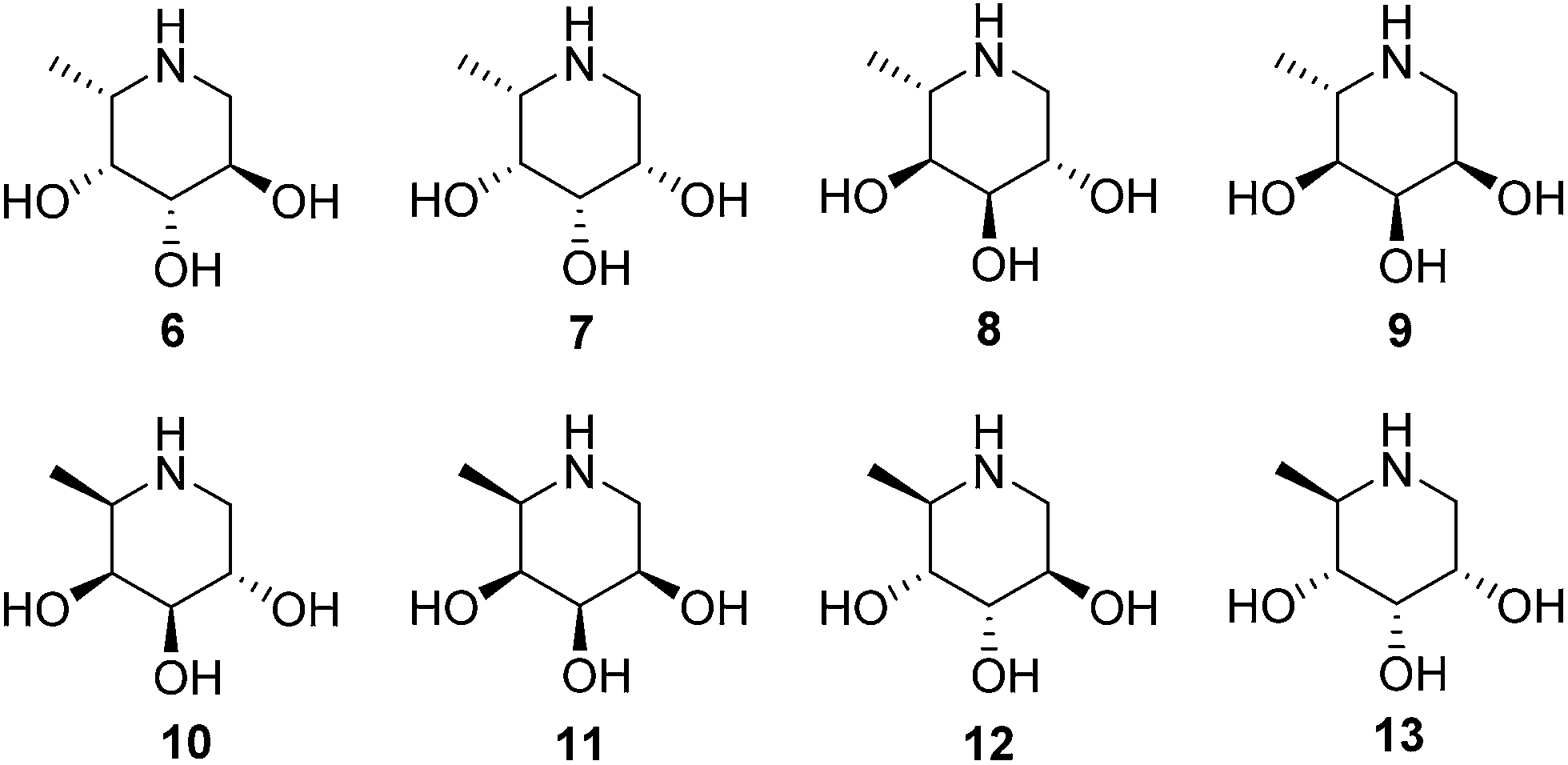 | ||
| Fig. 3 Structures of 1-deoxy-L-fuconojirimycin 6 and configurational isomers 7–13. | ||
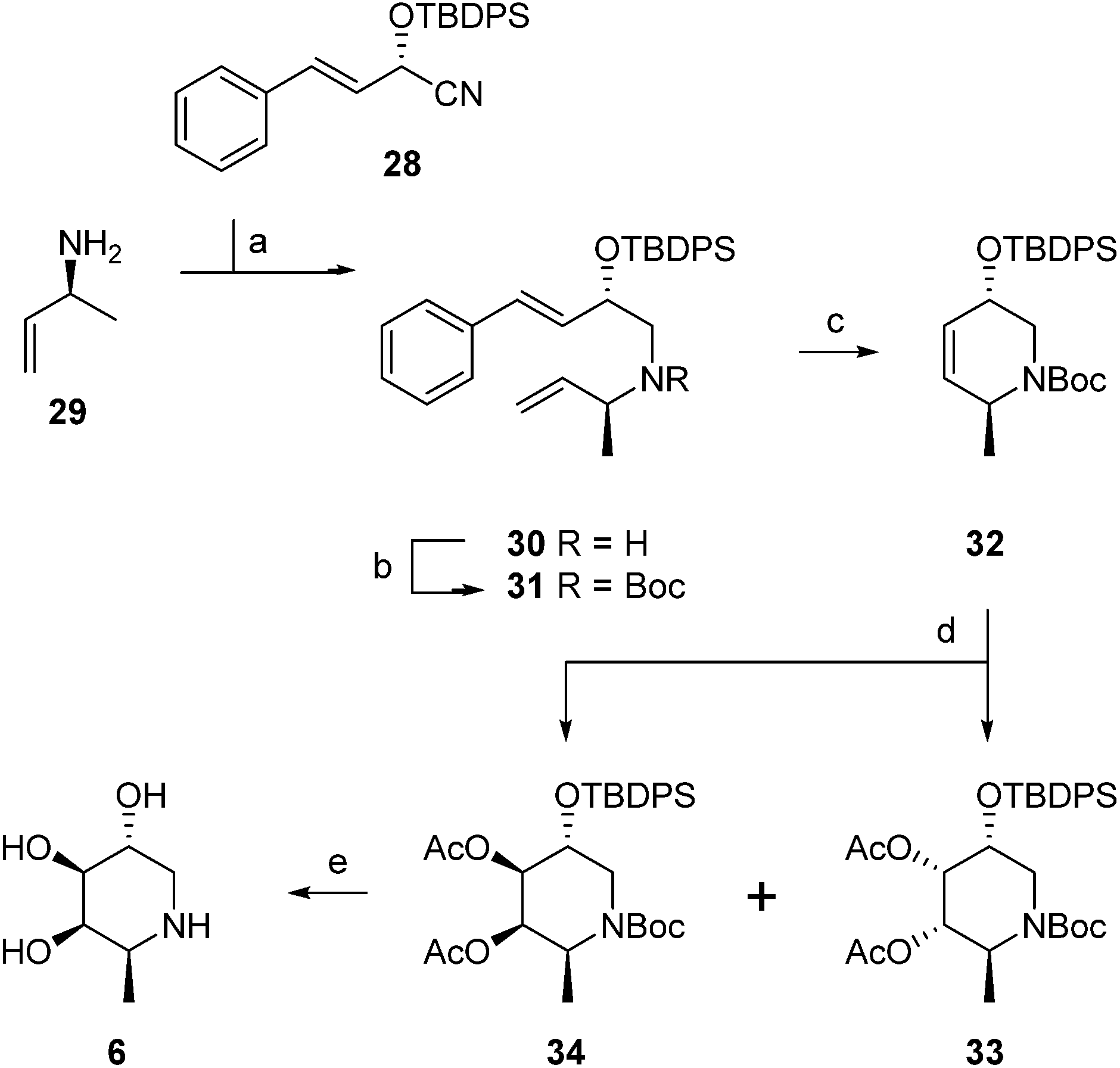 | ||
| Scheme 2 Synthesis of 1-deoxy-L-fuconojirimycin 6. Reagents and conditions: (a) (i) Et2O, DIBAL-H, −80 °C, (ii) MeOH, −90 °C, (iii) amine 29, NaOMe, (iv) NaBH4, −15 °C to rt, 88%; (b) Boc2O, 50 °C, 100%; (c) Grubbs 1st generation, CH2Cl2, 88%; (d) (i) K2OSO4·2H2O, NMO, acetone–H2O (1:1), −10 °C, (ii) Ac2O, pyridine, DMAP, 0 °C, 88% (33:34 = 1:3); (e) (i) K2CO3, MeOH, (ii) TBAF, THF, (iii) 6M HCl, MeOH, 71%. | ||
Global deprotection of 34 afforded 1-deoxy-L-fuconoijirimycin 6, the analytical and spectroscopical data of which were in full agreement with those reported in the literature.22 The seven configurational isomers 7–13 were prepared by alteration of the building blocks and/or the chemical transformations. It should be noted that four out of the eight isomers of fuconojirimycin are derived from 28. To enable the synthesis of the four enantiomers we needed access to the enantiomer of cyanohydrin 28, which we prepared using almond (R)-hydroxynitrile lyase (R-PaHNL).23
In vitro GH29 α-L-fucosidase activity assays
Having the cyclophellitol aziridine inhibitors and probes in hand, we determined their inhibitory potency towards the human lysosomal α-L-fucosidase, FUCA1. Inhibition potency was determined by measuring the residual enzyme activity using the fluorogenic substrate 4-methylumbelliferyl-α-L-fucopyranoside after pre-incubation of lysates of COS-7 cells over-expressing recombinant human FUCA1, with varying concentrations of the non-fluorescent, irreversible cyclophellitol aziridine inhibitors 4, 5 and 23; the ABPs 1 (JJB256), 2 (JJB244), 3 (JJB243), 1-deoxy-L-fuconojirimycin 6, and the seven fuconojirimycin isomers 7–13. All N-acyl aziridines inhibited human FUCA1 activity with nanomolar IC50 activities (Table 1).| In vitro IC50 | In situ IC50 | |
|---|---|---|
| a Values are given in nanomolar. ND: not determined. It should be noted that IC50 values on competitive inhibitors do not compare well to those obtained for mechanism-based inhibitors. The values given above allow for a comparison of inhibitory potency within the two classes of compounds studied. | ||
| Aziridine | ||
| 1 (JJB256) | 6.9 ± 0.8 | 28.9 ± 4.4 |
| 2 (JJB244) | 8.7 ± 1.2 | 157.8 ± 22 |
| 3 (JJB243) | 7.2 ± 1.1 | 25512 ± 2278 |
| 4 | 371.6 ± 21.2 | 515.4 ± 72.9 |
| 5 | 46.8 ± 3.4 | 77.9 ± 9.1 |
| 23 | 8.7 ± 1.1 | 65.4 ± 7.5 |
|
||
| Iminosugars | ||
| 6 | 3979 ± 257 | ND |
| 7–13 | >100000 |
ND |
Perusal of the inhibitory data does reveal some trends that allow us to draw some tentative conclusions. N-Benzoyl aziridine 4, bearing a bulky aromatic N-benzoate group, is about eight-fold less active compared to its N-acetylated counterpart, 5. In contrast, comparing 5 with 23 reveals that a bulky aliphatic N-acyl substituent is in fact favored for enzyme inhibition (5-fold increase, Table 1). ABPs 1 and 2, bearing a BODIPY fluorophore attached to the alkyl chain, inhibit FUCA1 in the same order as their azide precursor 23. In situ inhibition of FUCA1 in living fibroblasts by ABP 1 and 2 occurs with similar efficacy as 23 and ABP 3, the latter carrying a biotin attached to the alkyl chain. Exposure of cells to ABP 3 revealed in a 25-fold decreased inhibitory potency, suggesting a reduced ability to penetrate into cells to reach the lysosomal FUCA1. The known competitive fucosidase inhibitor, 1-deoxy-L-fuconojirimycin 6 inhibits FUCA1 with an IC50 of 3.9 μM, in accordance with the literature values.24 The seven configurational isomers 7–13 do not significantly inhibit FUCA1 activity up to 100 μM, a result that corroborates previous findings on some of the configurational analogues, which were reported as poor fucosidase inhibitors.25
As the next research objective, we examined activity-based profiling of GH29 α-L-fucosidases from varying sources with green-fluorescent aziridine ABP 1, in the presence or absence of excess concentrations of, either the mechanism-based inhibitor 4 or the competitive inhibitor 6. As is shown in Fig. 4A, ABP 1 efficiently labels purified α-L-fucosidase from Bacteroides thetaiotaomicron. In lysates from an E. coli culture overexpressing recombinant α-L-fucosidase from Bacteroides thetaiotaomicron gene 2970, several fluorescent protein bands are visible upon labeling with 1 (ESI Fig. S1†), with the most prominent band at around 50 kDa, corresponding to the predicted molecular weight of the enzyme. Labeling of the major band at 50 kD could moreover be blocked following pre-incubation with either 100 μM 4 or with 5 mM 6 (ESI Fig. S1†). Red fluorescent ABP 2 labels α-(1-2,3,4) and α-(1-6)-fucosidases from various bacterial sources in a similar fashion (Fig. 4B).
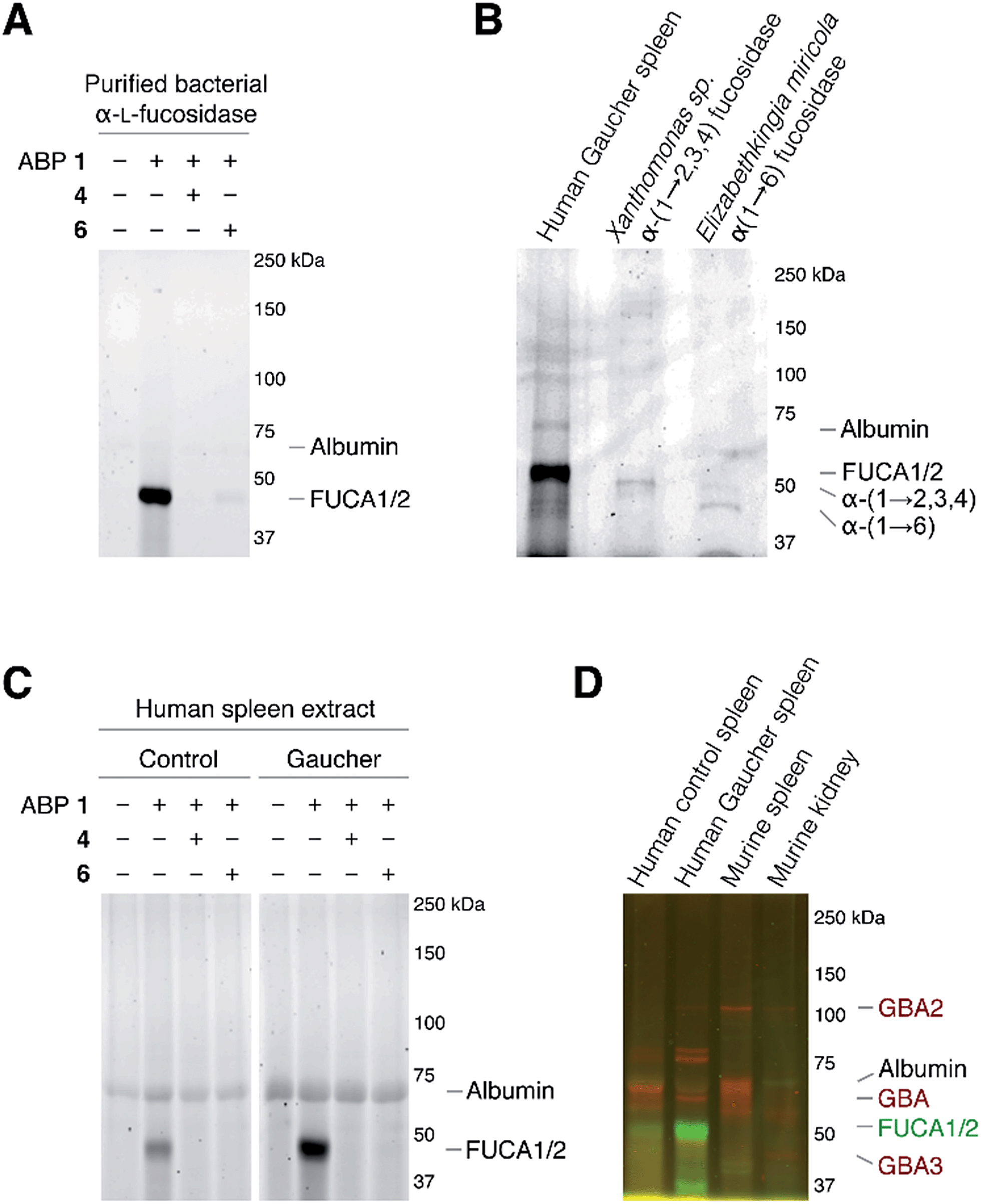 | ||
| Fig. 4 In vitro activity-based protein profiling of GH29 α-L-fucosidases. (A) Labeling with ABP 1 of recombinant α-L-fucosidase from Bacteroides thetaiotaomicron 2970. (B) In vitro labeling of lysate of spleen from a Gaucher disease patient, α-(1-2,3,4)-fucosidase from Xanthomonas sp. and α-(1-6)-fucosidase from Elizabethkingia miricola with ABP 2. (C) In vitro labeling of human healthy and Gaucher disease spleen. (D) Direct labeling of GH29 α-L-fucosidases with green-fluorescent ABP 1 and retaining β-glucosidases GBA, GBA2 and GBA3 with red-fluorescent JJB75.18 The location of albumin autofluorescence is designated on each gel. | ||
Subsequently, we exposed lysates of spleens from a healthy human individual and a patient suffering from Gaucher disease to ABP 1. As can be seen in Fig. 4C, a single fluorescent protein migrating slightly below 50 kDa was fluorescently labeled. The labeled band from human Gaucher spleen lysate is considerably more intense than the corresponding band in healthy human spleen, reflecting elevated α-L-fucosidase activity in the former. This result corroborates earlier observations that mRNA encoding lysosomal glycosidases are upregulated in Gaucher tissue.26 As before, labeling with ABP 1 was suppressed after pre-treatment with either N-benzoyl aziridine 4 or L-fuconojirimycin 6. To further ascertain that ABP 2 labels mammalian GH29 retaining α-L-fucosidases we performed a number of control experiments (ESI Fig. S2a†). Compound 1 labeled both FUCA1 and FUCA2 that were overexpressed in COS cells. Treatment of murine spleen lysates with 1 yielded a result essentially as observed for healthy human tissue (ESI Fig. S2b†).
Finally, we evaluated the simultaneous labeling of retaining β-glucosidases and retaining α-L-fucosidase. To this end we incubated tissue lysates with 1 and our previously18 reported broad-spectrum activity-based retaining β-glucosidase probe, JJB75 (see for its structure, ESI Fig. S3†). As can be seen (Fig. 4D) the applied ABPs label a distinct set of proteins. Since they bear complementary fluorophores they can be used jointly to profile both retaining glycosidase families in a single experiment.
The FUCA1 enzymatic activity is maximal at around pH 4.5 (see ESI Fig. S4†), consistent with the acidic pH of its natural lysosomal environment. Labeling efficiency with 1 largely reflects the pH dependence of FUCA1 at pH below 7. Of note, the pH dependence of labeling of FUCA1 does not follow that of its enzymatic activity. At alkaline conditions, where enzymatic activity is low, labeling still proceeds. We made similar observations in the past for ABPP of retaining β-exoglucosidases using cyclophellitol β-aziridine ABPs.14 This result reveals the high reactivity of the aziridine probes in the initial displacement step employed by retaining glycosidases, a feature which, in conjunction with their selectivity in binding and relative stability in physiological environment, explains their efficacy as activity-based glycosidase probes.27
Assessment of fluorescent labeling kinetics by employing 10 nM 1 labeling at stoichiometric concentrations of rhFUCA1 at 4 °C and 37 °C revealed that, during time-course experiments, examined on SDS-PAGE gels, labeling is near complete within the first minute at 4 °C (ESI Fig. S5a†), impairing accurate determination of kinetic constants. This rapid reaction with both 1 and 2 also precluded a classic determination of kinetic constants following a time-dependent decrease in detectable 4-methylumbelliferone (ESI Fig. S5b†).
In vivo GH29 α-L-fucosidase assays
The ability of 1 to label α-L-fucosidase in living mice was investigated next. Four wild-type C57Bl/6J male mice were injected with 100 μL vehicle (PBS) or PBS containing 10, 100 or 1000 pmol 1. After two hours, the mice were anesthetized, perfused with PBS and then brain, spleen, liver and kidney tissues were isolated. Tissue homogenates were prepared and each lysate was labeled prior to gel electrophoresis with red-fluorescent cyclophellitol β-aziridine JJB75, which labels β-exoglucosidases as loading control (ESI Fig. S6†). Furthermore, tissue homogenates of vehicle-treated animals were labeled with excess 1 to visualize the maximal α-L-fucosidase labeling achievable in each tissue.After treatment of mice with ABP 1, a dose-dependent labeling of retaining α-L-fucosidases is observed in spleen, liver and kidney (Fig. 5). Injection of 1000 pmol 1 results in substantial labeling of α-fucosidase in spleen, liver and kidney. Detected fluorescence levels are comparable to that in matching samples from vehicle-treated mice incubated in vitro with excess 1. In contrast, no in vivo brain FUCA1 labeling was observed after any of the administered doses of ABP 1, and we conclude that ABP 1, just as its β-glucose and α-galactose congeners,14,15 does not penetrate the brain.
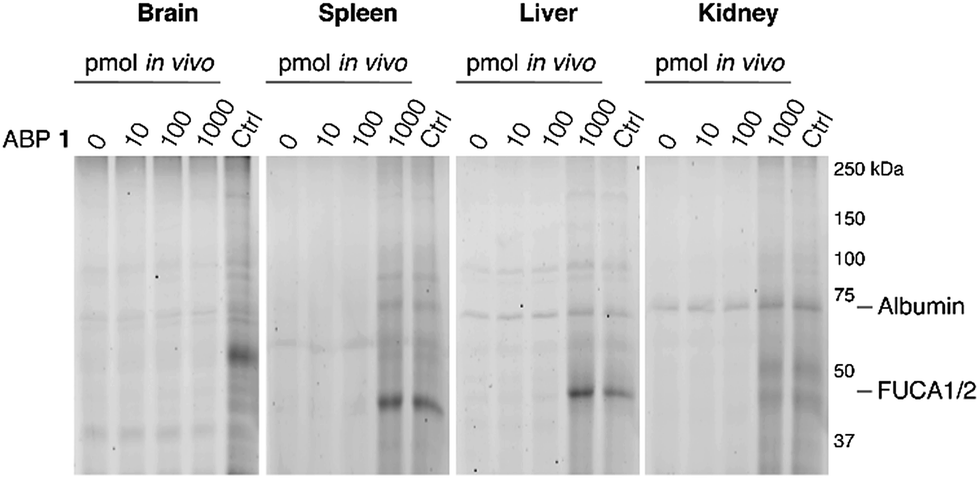 | ||
| Fig. 5 In vivo labeling of α-L-fucosidases in mice with various concentrations of ABP 1 during 2 hours. In vivo labeling compared to maximal labeling with excess ABP 1 of matched homogenates (Ctrl). The location of albumin autofluorescence is designated on each gel. | ||
Competitive activity-based GH29 α-L-fucosidase profiling
Having established the efficacy of ABP 2 to selectively label GH29 retaining α-L-fucosidases from various sources in an activity-based manner, we determined the inhibition potential of deoxyfuconojirimycin 6 and its 7 stereoisomers 7–13 in a competitive ABPP format (Fig. 6). In contrast to 6, none of the seven configurational isomers 7–13 were capable of blocking ABP 1 labeling of α-L-fucosidase, a result that matches with the data on inhibition of recombinant FUCA1 in the fluorogenic activity assay (Table 1).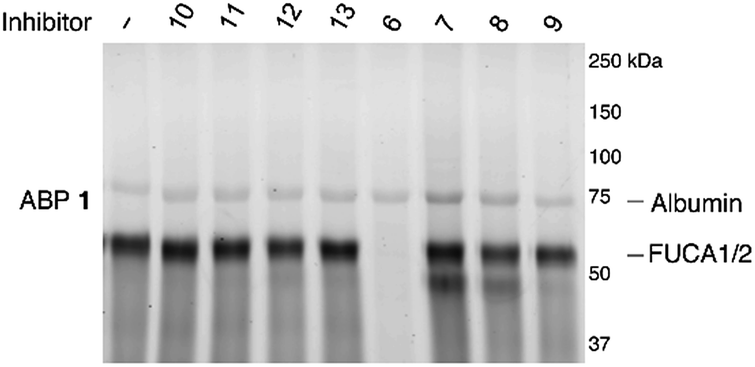 | ||
| Fig. 6 Competitive ABPP on recombinant FUCA1 with deoxyfuconojirimycin 6 and configurational analogues 7–13 towards α-L-fucosidases, with ABP 1 labeling as readout. | ||
Identification of biotin labeled GH29 α-L-fucosidases
To further determine the specificity of the developed ABPs, we have analyzed the ABP-labeled proteins in a complex tissue homogenate. For this purpose, we incubated Gaucher spleen lysate with biotinylated ABP 3 or with DMSO only (control) or competitive ABP 3-labeling by first incubating with fluorescent ABP 1. Glycosylated proteins were then enriched via ConA, followed by affinity purification with streptavidin coated paramagnetic beads. The identity of biotinylated and ABP 3-labeled proteins was determined by on-bead digestion with trypsin, peptide analysis by LC-MS/MS and matching against the human UniProt database, using the Mascot search engine as previously reported.14 FUCA1 was identified after ABP 3 pull-down as one of the top identified proteins (Table S1A) in the ESI data, but was not found in the competitive (Table S1B) or untreated control (Table S1C†). Proteins with higher scores were background proteins such as abundant endogenously biotinylated PCCA and PC, and keratin contaminations. FUCA1 was selectively found in the ABP 3-pull down experiment only. ESI Table S2† shows the analysis parameters of the identified peptides from FUCA1, with accuracy below 5 ppm, and Mascot ion scores above 40 (indicating reliable MS/MS fragment annotation and match) and manually curated fragmentation patterns. These results show FUCA1 can be undisputedly affinity purified and identified via biotinylated ABP 3. Moreover the binding of ABP 3 can be completely blocked by pre-incubation with ABP 1, which may indicate that both ABPs bind at the same site of the enzyme.3-D crystal structure analysis of bacterial FUCA1 complexed to 4 and 5
In order to obtain experimental evidence for the formation of a covalent adduct between α-L-fucosidase and ABPs 1, 2 and 3, a crystal structure of BtFuc2970 (often used as a surrogate for the mammalian enzyme) with mechanism-based inhibitor 4 was obtained (PDB code 4WSK). While the resulting crystal structure clearly demonstrates the formation of an enzyme–4 complex, electron density around the aryl group of the inhibitor “aglycon” moiety was close to the side-chain of the catalytic acid/base of the enzyme, residue E288 (ESI Fig. S7†) resulting in disorder. This likely reflects steric clashing and considerable conformational flexibility in the 4 aglycon when bound to BtFuc2970. “Aglycon” here reflects the aziridine N-acyl moiety to which the BODIPY tags are grafted in ABPs 1 and 2 and which is designed to occupy the space normally occupied by the substrate fucoside.In our design of aziridine-based retaining exo-glycosidase ABPs we made the assumption that such aglycon-like moieties would not interfere with enzyme binding (this also based on the numerous fluorogenic substrates that are in use to study exoglycosidases and in which the aglycon moiety can take on made shapes and sizes). The 3-D fold of BtFuc2970, however, appears not to provide sufficient space to accommodate an extended aryl aglycon pendant to the atom, which would equate to the ring oxygen in fucose. In order to minimize steric clashes, BtFuc2970 was subsequently incubated with 5. The crystal structure of the resulting complex (PDB code 4WSJ) revealed unambiguous electron density for the presence of a covalent enzyme–5 complex (Fig. 7). The complex has C–O bond lengths between the BtFuc2970 catalytic nucleophile and 5 of ca. 1.43 Å as would be expected for a C–O ester bond. As expected from the reduction in aglycon size, 5 is better ordered than 4 when bound to the bacterial enzyme and provides a clearer definition of the resulting conformation and interactions. Upon trans-diaxial opening of the acylaziridine the covalently bound and substituted cyclohexane adopts a slightly distorted 3H4 conformation (between 3H4 and 3S1); consistent with the expected catalytic itinerary.28
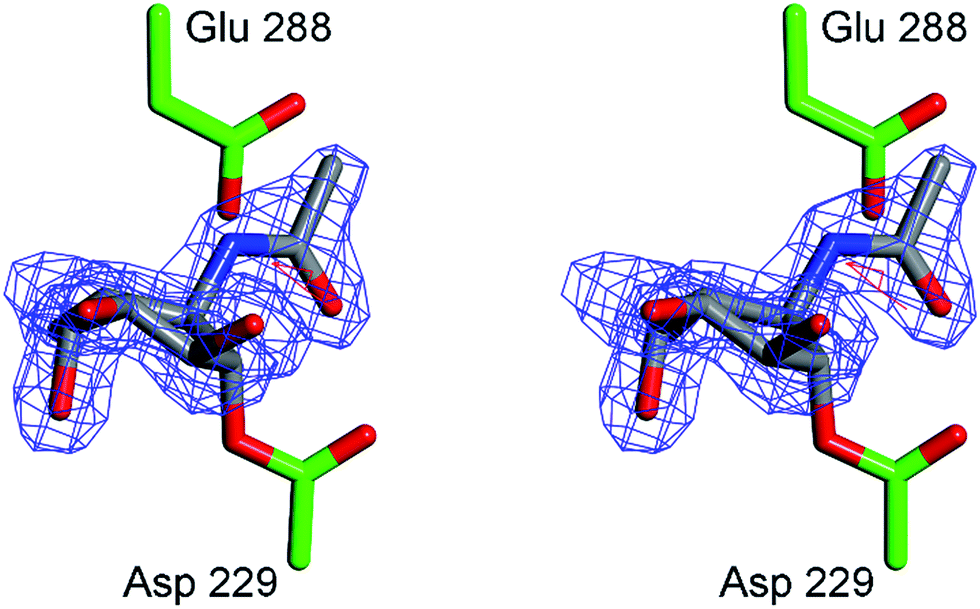 | ||
| Fig. 7 Crystal structure of α-L-fucosidase from Bacteroides thetaiotaomicron in complex with 5. Catalytic residues are annotated: Asp 229 (nucleophile) and Glu 288 (acid/base). Electron density displayed is Fo–Fc density from phases calculated prior to the inclusion of 5 in refinement, contoured at 3σ. Figure was prepared using CCP4MG.29 | ||
Conclusions
We have developed the potent and selective aziridine-based ABPs 1, 2 and 3 for selective profiling of active GH29 α-L-fucosidases in cell extracts from bacteria, mice and man as well as in vivo in mice. Labeling of GH29 retaining α-L-fucosidases with our L-fucopyranose-configured, cyclophellitol aziridine-based ABPs proceeds with good efficiency and high selectively both in vitro and in vivo with the single caveat that the probes do not penetrate brain tissue in mice. The covalent irreversible aziridine inhibitors proved much more potent than their iminosugar counterparts, of which L-fuconojirimycin 6 appeared to be the single compound from this set of configurational isomers that is able to inhibit FUCA1 in a competitive ABPP setting. The crystal structures of α-L-fucosidase from Bacteroides thetaiotaomicron in complex with compound 4 and 5 provide strong evidence for the covalent binding of cyclophellitol aziridine to active α-L-fucosidases and by this virtue the validity of the cyclophellitol aziridine design for activity-based profiling of retaining glycosidases that employ the Koshland double displacement mechanism. Whereas aziridines and epoxides that are annulated to cyclohexane rings preferably open in a trans-diaxial fashion through a chair-like transition state, reaction with the α-L-fucosidase nucleophile takes place at the aziridine carbon corresponding to the anomeric center of a substrate α-L-fucoside. This corresponds to ring opening to yield a skew boat, as is observed in the trapped enzyme active site in the co-crystal.In conclusion, we offer ABPs 1, 2 and 3 as reagents for the discovery and annotation of new members of the GH29 family of α-L-fucosidase in comparative ABPP experiments, for monitoring retaining α-L-fucosidase activities in health and disease, and for the discovery of inhibitors able to interfere with specific α-L-fucosidases in competitive ABPP experiments. ABPs 1, 2 and 3 add to our growing series of in situ and in vivo active retaining glycosidase ABPs and moreover their design hold promise for the design of ABPs targeting retaining glycosidases recognizing and processing differently configured and substituted carbohydrates.
Experimental section
Synthesis
The synthesis, analytical and spectroscopical analysis of L-fuconojirimycin configured cyclophellitol aziridines 1–5 and L-fuconojirimycin isomers 6–13 is described in the ESI.†Biological assays
000 g, supernatant collected carefully, protein concentration determined and 200 mg total protein was then incubated with either 0.1% (v/v) DMSO, ABP 1 or ABP 3, or firstly with 10 μM ABP 1 followed by 10 μM of ABP 3, each step taking 30 min at 37 °C, in a total volume of 1 mL McIlvaine buffer, pH 4.5. Glycosylated biomolecules were enriched by using 1 mL ConA-Sepharose per sample, according to manufacturer's instructions (Amersham Pharmacia Biotech AB, Sweden) and subsequently denatured by the presence of 2% (w/v) SDS and boiling for 5 min at 100 °C. From here on, samples were prepared for MS as published earlier.14 After desalting on StageTips, peptides were analyzed with a 2 h gradient of 5–25% ACN on nano-LC, hyphenated to an LTQ-Orbitrap and identified via the Mascot protein search engine.14
:1 ratio. After ca. 1 h soaking with ligands, crystals were fished into cryo-protectant solutions (mother liquor supplemented with 20% v/v glycerol) and cryo-cooled in liquid nitrogen. Diffraction data were collected at Diamond Light Source. Diffraction images were indexed and integrated using MOSFLM29 (4) or XDS31 (5) and scaled and merged using AIMLESS.32 Crystals grew in an almost isomorphous space group to PDB entry 4JFV, and coordinates from this entry were used directly to obtain a starting model for refinement. Iterative stages of model-building (COOT33) and maximum-likelihood refinement (REFMAC5 (ref. 34)) were conducted to yield final models. Maximum-likelihood restraints for compounds 4 and 5 were created using the PRODRG online server35 and link restraints generated using JLIGAND.36 X-ray crystallographic data statistics are available in ESI Table S3.†
Acknowledgements
The work in Leiden and Amsterdam was supported by the China Research Council (PhD grant, to JBJ), the Netherlands Organization for Scientific Research (NWO, ChemThem grant to JMA and HSO) and the European Research Council (ERC AdG ‘ChemBioSphing’, to HSO and JMA). The work in York was supported by the Biotechnology and Biological Sciences Research Council (BBSRC) through a Ph.D. studentship to D. W. W. ABPP work in York is further supported through the ERC (ERC-2012-AdG-322942). The authors thank ChemAxon for the academic licensed software.Notes and references
- J. Intra, M.-E. Perotti, G. Pavesi and D. Horner, Gene, 2007, 392, 34–46 CrossRef CAS PubMed.
- V. Lombard, H. Golaconda Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, Nucleic Acids Res., 2014, 42, D490–D495 CrossRef CAS PubMed.
- S. W. Liu, C. S. Chen, S. S. Chang, K. K. T. Mong, C. H. Lin, C. W. Chang, C. Y. Tang and Y. K. Li, Biochemistry, 2009, 48, 110–120 CrossRef CAS PubMed.
- L. Guillotin, P. Lafite and R. Daniellou, Biochemistry, 2014, 53, 1447–1455 CrossRef CAS PubMed.
- B. Cobucci-Ponzano, M. Mazzone, M. Rossi and M. Moracci, Biochemistry, 2005, 44, 6331–6342 CrossRef CAS PubMed.
- F. A. Shaikh, A. L. van Bueren, G. J. Davies and S. G. Withers, Biochemistry, 2013, 52, 5857–5864 CrossRef CAS PubMed.
- D. E. Koshland, Biol. Rev., 1953, 28, 416–436 CrossRef CAS PubMed.
- T. W. Liu, C. W. Ho, H. H. Huang, S. M. Chang, S. D. Popat, Y. T. Wang, M. S. Wu, Y. J. Chen and C. H. Lin, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14581–14586 CrossRef CAS PubMed.
- J. S. O'Brien, P. J. Willems, H. Fukushima, J. R. de Wet, J. K. Darby, R. D. Cioccio, M. L. Fowler and T. B. Shows, Enzyme, 1987, 38, 45–53 Search PubMed.
- K. Phopin, W. Nimlamool, L. J. Lowe-Krentz, E. W. Douglass, J. N. Taroni and B. S. Bean, Mol. Reprod. Dev., 2013, 80, 273–285 CrossRef CAS PubMed.
- D. G. Hildebrand, S. Lehle, A. Borst, S. Haferkamp, F. Essmann and K. Schulze-Osthoff, Cell Cycle, 2013, 12, 1922–1927 CrossRef CAS PubMed.
- M. G. Giardina, M. Matarazzo, R. Morante, A. Lucariello, A. Varriale, V. Guardasole and G. De Marco, Cancer, 1998, 83, 2468–2474 CrossRef CAS.
- B. F. Cravatt, A. T. Wright and J. W. Kozarich, Annu. Rev. Biochem., 2008, 77, 383–414 CrossRef CAS PubMed.
- W. W. Kallemeijn, K. Y. Li, M. D. Witte, A. R. Marques, J. Aten, S. Scheij, J. Jiang, L. I. Willems, T. M. Voorn-Brouwer, C. P. van Roomen, R. Ottenhoff, R. G. Boot, H. van den Elst, M. T. Walvoort, B. I. Florea, J. D. Codee, G. A. van der Marel, J. M. Aerts and H. S. Overkleeft, Angew. Chem., Int. Ed. Engl., 2012, 51, 12529–12533 CrossRef CAS PubMed.
- L. I. Willems, T. J. M. Beenakker, B. Murray, S. Scheij, W. W. Kallemeijn, R. G. Boot, M. Verhoek, W. E. Donker-Koopman, M. J. Ferraz, E. R. van Rijssel, B. I. Florea, J. D. C. Codée, G. A. van der Marel, J. M. F. G. Aerts and H. S. Overkleeft, J. Am. Chem. Soc., 2014, 136, 11622–11625 CrossRef CAS PubMed.
- M. J. Niphakis and B. F. Cravatt, Annu. Rev. Biochem., 2014, 83, 341–377 CrossRef CAS PubMed.
- Y. Harrak, C. M. Barra, A. Delgado, A. R. Castano and A. Llebaria, J. Am. Chem. Soc., 2011, 133, 12079–12084 CrossRef CAS PubMed.
- K.-Y. Li, J. Jiang, M. D. Witte, W. W. Kallemeijn, H. van den Elst, C.-S. Wong, S. D. Chander, S. Hoogendoorn, T. J. M. Beenakker, J. D. C. Codée, J. M. F. G. Aerts, G. A. van der Marel and H. S. Overkleeft, Eur. J. Org. Chem., 2014, 2014, 6030–6043 CrossRef CAS.
- O. David, W. J. N. Meester, H. Bieräugel, H. E. Schoemaker, H. Hiemstra and J. H. van Maarseveen, Angew. Chem., Int. Ed., 2003, 42, 4373–4375 CrossRef CAS PubMed.
- H. Griengl, N. Klempier, P. Pöchlauer, M. Schmidt, N. Shi and A. A. Zabelinskaja-Mackova, Tetrahedron, 1998, 54, 14477–14486 CrossRef CAS.
- A. D. Campbell, T. M. Raynham and R. J. K. Taylor, Synthesis, 1998, 1707–1709 CrossRef CAS PubMed.
- G. W. J. Fleet, A. N. Shaw, S. V. Evans and L. E. Fellows, J. Chem. Soc., Chem. Commun., 1985, 841–842 RSC.
- E. G. J. C. Warmerdam, A. M. C. H. van den Nieuwendijk, J. Brussee, A. van der Gen and C. G. Kruse, Recl. Trav. Chim. Pays-Bas, 1996, 115, 20–24 CrossRef CAS.
- E. Moreno-Clavijo, A. T. Carmona, A. J. Moreno-Vargas, L. Molina and I. Robina, Curr. Org. Synth., 2011, 8, 102–133 CrossRef CAS.
- (a) Y. Bleriot, D. Gretzke, T. M. Krulle, T. D. Butters, R. A. Dwek, R. J. Nash, N. Asano and G. W. Fleet, Carbohydr. Res., 2005, 340, 2713–2718 CrossRef CAS PubMed; (b) A. Bordier, P. Compain, O. R. Martin, K. Ikeda and N. Asano, Tetrahedron: Asymmetry, 2003, 14, 47–51 CrossRef CAS; (c) P. Jakobsen, J. M. Undbeck, M. Kristiansen, J. Breinholt, H. Demuth, J. Pawlas, M. P. Torres Candela, B. Andersen, N. Westergaard, K. Lundgren and N. Asano, Bioorg. Med. Chem., 2001, 9, 733–744 CrossRef CAS; (d) J. Streith, A. Boiron, J.-L. Paillaud, E.-M. Rodriguez-Perez, C. Strehler, T. Tschamber and M. Zehnder, Helv. Chim. Acta, 1995, 78, 61–72 CrossRef CAS.
- M. T. Moran, J. P. Schofield, A. R. Hayman, G. P. Shi, E. Young and T. M. Cox, Blood, 2000, 96, 1969–1978 CAS.
- L. I. Willems, J. Jiang, K.-Y. Li, M. D. Witte, W. W. Kallemeijn, T. J. N. Beenakker, S. P. Schröder, J. M. F. G. Aerts, G. A. van der Marel, J. D. C. Codée and H. S. Overkleeft, Chem.–Eur. J., 2014, 20, 10864–10872 CrossRef CAS PubMed.
- A. Lammerts van Bueren, A. Ardèvol, J. Fayers-Kerr, B. Luo, Y. Zhang, M. Sollogoub, Y. Blériot, C. Rovira and G. J. Davies, J. Am. Chem. Soc., 2010, 132, 1804–1806 CrossRef CAS PubMed.
- S. Mcnicholas, E. Potterton, K. S. Wilson and M. E. M. Noble, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 386–394 CrossRef CAS PubMed.
- I. M. Aerts and S. van Weely, Hum. Mutat., 1997, 10, 348–358 CrossRef.
- A. G. W. Leslie, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 48–57 CrossRef PubMed.
- W. Kabsch, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 125–132 CrossRef CAS PubMed.
- P. Emsley, B. Lohkamp, W. G. Scott and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 486–501 CrossRef CAS PubMed.
- G. N. Murshudov, P. Skubak, A. A. Lebedev, N. S. Pannu, R. A. Steiner, R. A. Nicholls, M. D. Winn, F. Long and A. A. Vagin, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2011, 67, 355–367 CrossRef CAS PubMed.
- A. W. Schuttelkopf and D. M. F. van Aalten, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 1355–1363 CrossRef PubMed.
- A. A. Lebedev, P. Young, M. N. Isupov, O. V. Moroz, A. A. Vagin and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2012, 68, 431–440 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental part including synthesis procedures, characterization data, copies of NMR spectra of new compounds, supporting biological figures and crystallographic information. See DOI: 10.1039/c4sc03739a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |