
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Heterologous expression of the avirulence gene ACE1 from the fungal rice pathogen Magnaporthe oryzae†
Zhongshu
Song‡
a,
Walid
Bakeer‡
ab,
James W.
Marshall
a,
Ahmed A.
Yakasai
a,
Rozida Mohd
Khalid
a,
Jerome
Collemare
c,
Elizabeth
Skellam
d,
Didier
Tharreau
e,
Marc-Henri
Lebrun
fg,
Colin M.
Lazarus
h,
Andrew M.
Bailey
h,
Thomas J.
Simpson
a and
Russell J.
Cox
*ad
aSchool of Chemistry, University of Bristol, Cantock's Close, Bristol, BS8 1TS, UK
bMicrobiology Department, Faculty of Pharmacy, Beni Suef University, Egypt
cUMR1345, IRHS-INRA, 49071 Beaucouzé Cedex, France
dInstitute for Organic Chemistry, Leibniz University of Hannover, Schneiderberg 1B, 30167, Hannover, Germany. E-mail: russell.cox@oci.uni-hannover.de
eUMR BGPI, CIRAD, Campus International de Baillarguet, 34398 Montpellier Cedex 5, France
fUR 1290 BIOGER-CPP, INRA, Campus AgroParisTech, 78850 Thiverval-Grignon, France
gUMR 5240 MAP, CNRS, UCB, INSA, Bayer CropScience, 69263 Lyon Cedex 09, France
hSchool of Biological Sciences, University of Bristol, 24 Tyndall Avenue, Bristol BS8 1TQ, UK
First published on 1st June 2015
Abstract
The ACE1 and RAP1 genes from the avirulence signalling gene cluster of the rice blast fungus Magnaporthe oryzae were expressed in Aspergillus oryzae and M. oryzae itself. Expression of ACE1 alone produced a polyenyl pyrone (magnaporthepyrone), which is regioselectively epoxidised and hydrolysed to give different diols, 6 and 7, in the two host organisms. Analysis of the three introns present in ACE1 determined that A. oryzae does not process intron 2 correctly, while M. oryzae processes all introns correctly in both appressoria and mycelia. Co-expression of ACE1 and RAP1 in A. oryzae produced an amide 8 which is similar to the PKS-NRPS derived backbone of the cytochalasans. Biological testing on rice leaves showed that neither the diols 6 and 7, nor amide 8 was responsible for the observed ACE1 mediated avirulence, however, gene cluster analysis suggests that the true avirulence signalling compound may be a tyrosine-derived cytochalasan compound.
Introduction
The rice blast fungus Magnaporthe oryzae, which causes significant reductions in rice yields,1 is a well-known model for studying plant fungal interactions and pathogenicity.2 Rice resistance receptors (R) recognize M. oryzae secreted avirulence signals (AVR), allowing the rice plant to mount an effective defence response, killing or containing the fungus within the initial infected cell. Genetic and molecular studies have conclusively linked the avirulent phenotype of strain Guy11 on rice cultivars carrying the resistance gene Pi33, to the presence of the ACE1 gene (Avirulence Conferring Enzyme).3,4 Unlike the majority of known AVR genes which encode small secreted proteins acting directly on the host plant,5ACE1 encodes a cytoplasmic biosynthetic protein responsible for the production of a low molecular weight compound.4ACE1 belongs to an infection-specific secondary metabolite gene cluster (Scheme 1),6 expressed only during appressorium-mediated penetration, and not at any other stage of the M. oryzae life cycle.7 The 12.4 kb ACE1 gene encodes an enzyme consisting of a fungal highly-reducing polyketide synthase (hrPKS)8 fused to a single module of a nonribosomal peptide synthetase (NRPS).4 Similar fungal synthetases have been investigated genetically and biochemically and are known to produce acyl tetramic acids or pyrrolidones including pretenellin A 1,9 prefusarin C 2 (ref. 10) and preaspyridone A 3 (ref. 11 and 12) as well as other potent bioactive compounds including cytochalasin K 4 (ref. 13 and 14) and the important cholesterol biosynthesis inhibitor lovastatin 5 (ref. 15) (Scheme 1). It appears likely, therefore, that ACE1 encodes a biosynthetic protein which makes a small molecule consisting of a polyketide fused to an amino acid. Because ACE1 is under very tight temporal and cell type-specific control in M. oryzae, it has not yet been possible to identify or purify the avirulence compound. Knowledge of the ACE1 metabolite structure would be highly useful for investigating avirulence signalling and developing novel resistant cultivars. The research of our group and that of others has focused on the heterologous expression of other fungal PKS-NRPS genes in Aspergillus oryzae from gene clusters of known function.12,16–19 Here we set out to use heterologous expression of genes from a cryptic pathway to determine its unknown chemical product, since knowledge of the ACE1 compound will enhance efforts to design new compounds of high potential utility in crop protection.
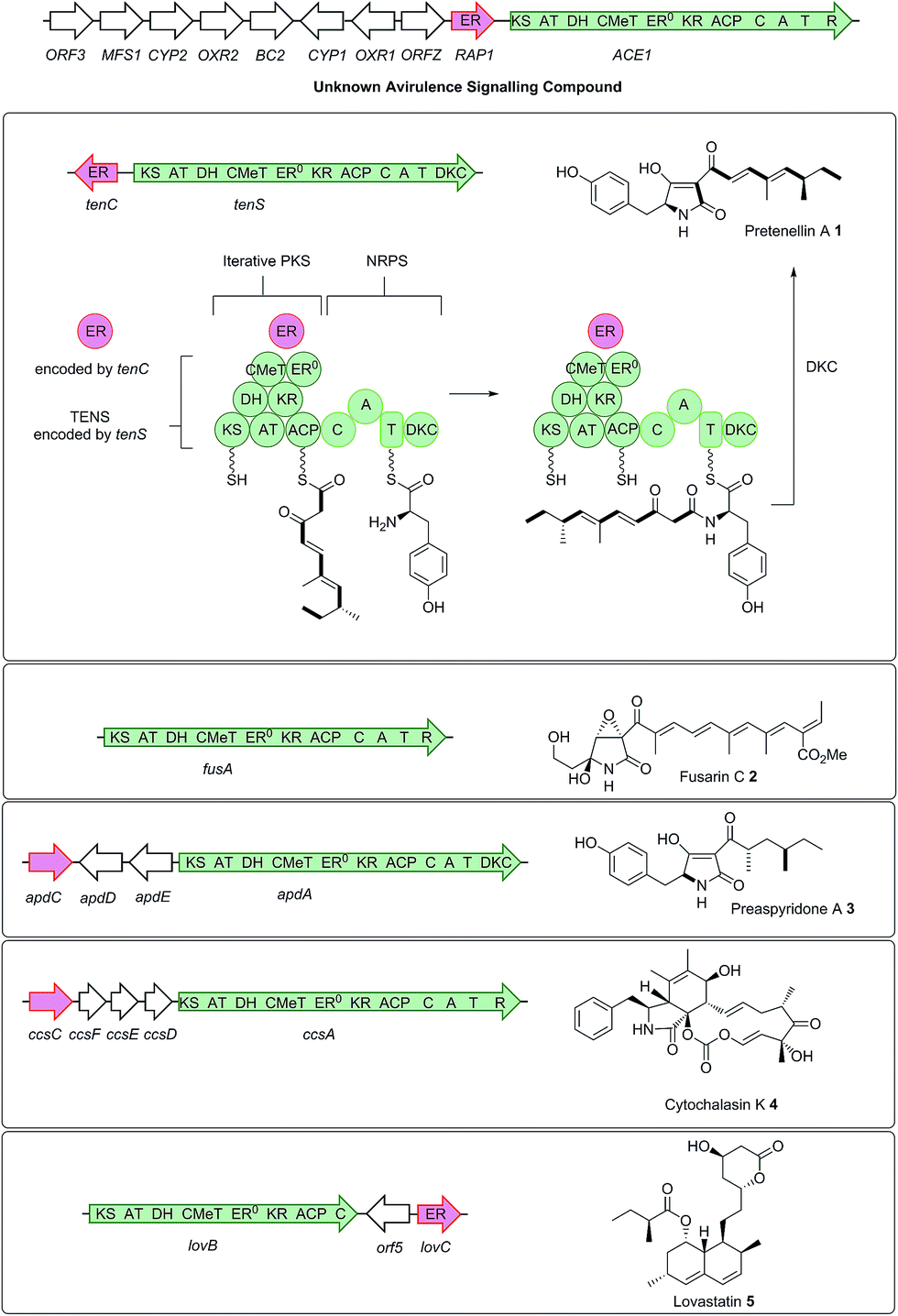 | ||
| Scheme 1 Relationship between biosynthetic gene clusters and the structures of known compounds in various fungi (only partial gene clusters are shown). The ACE1 gene cluster (MGG_08381 – MGG_08391, MGG_12447)6 is shown and the ACE1/RAP1 gene pair which show significant homology to other genes known to produce bioactive compounds are highlighted. The top panel shows key chemical steps catalysed by the fungal PKS-NRPS TENS, which synthesises pretenellin A 1 in partnership with the trans-acting ER TENC. KS, β-ketoacyl synthase; AT, acyl transferase, DH, dehydratase; ER, enoyl reductase; ERO, defective enoyl reductase; CMeT, C-methyl transferase; KR, β-ketoacyl reductase; ACP, acyl carrier protein; C, condensation; A, adenylation; T, thiolation; DKC, Dieckmann cyclase; R, thiolester reductase. | ||
Results
In initial experiments a construct known to express ACE1 in M. oryzae, containing ACE1 fused to eGFP at its 3′ terminus (pPACE1·ACE1eGFP·hyg),4 was used to construct the vector pPamyB·ACE1eGFP·argB (Fig. 1A) in which the ACE1-eGFP gene fusion was located downstream of PamyB.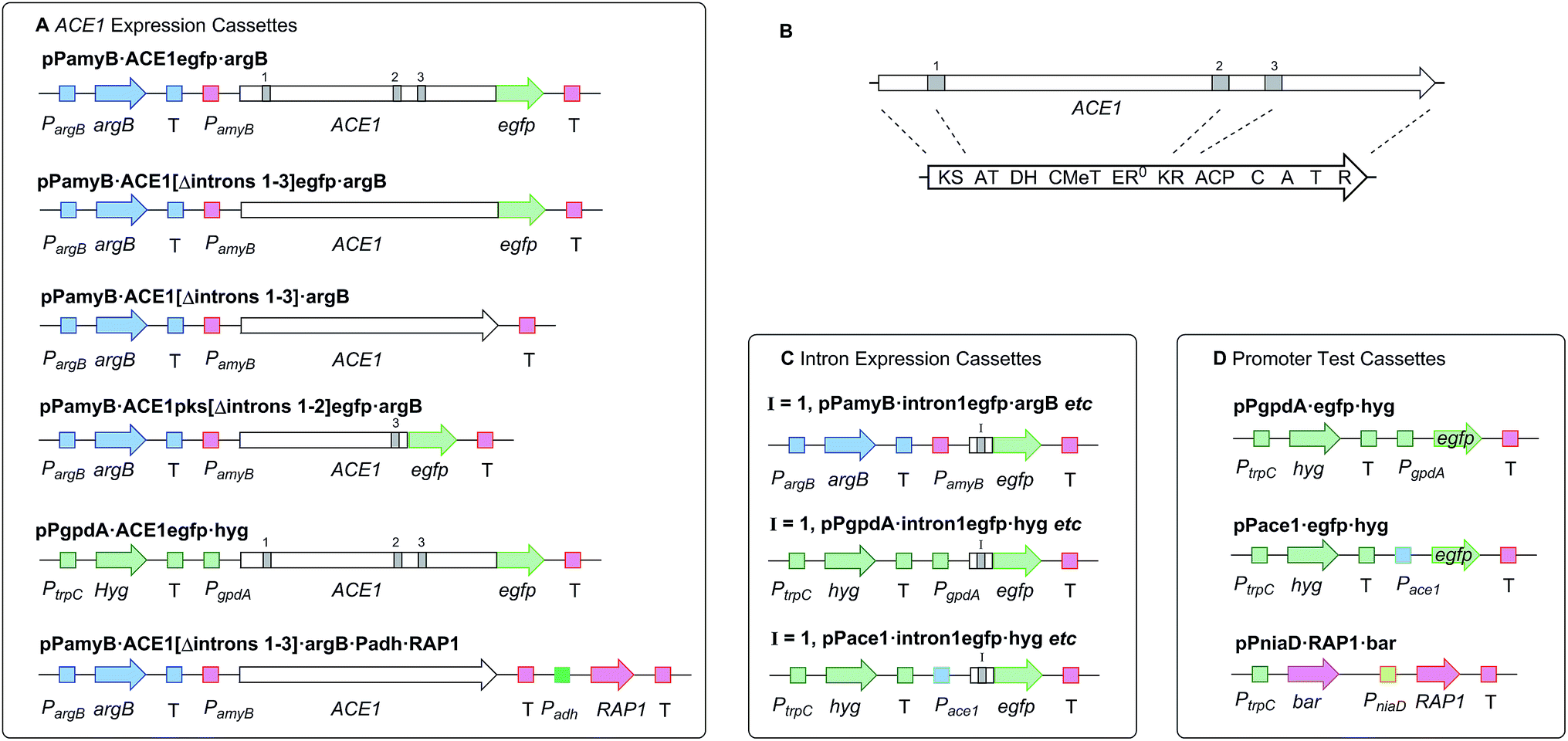 | ||
| Fig. 1 Diagrammatic representation of fungal expression systems used in this work (not to scale). Introns indicated by grey bars. PKS-NRPS functional domains as described in the legend to Scheme 1. P = promoter, T = terminator. (A) cassettes used for expression of ACE1 and RAP1; (B) relative positions of ACE1 introns in cDNA and mRNA; (C) cassettes used for testing intron splicing, I = 1/2/3; (D) cassettes used for promoter testing and coexpression of RAP1. | ||
This plasmid was then used to transform A. oryzae M-2-3. Transformants were selected on minimal medium and 100 colonies were selected and grown further on both minimal medium and minimal medium plus maltose. Four transformants showed weak fluorescence and these were picked together with 10 non-fluorescent transformants and grown in liquid medium (see ESI†). The resultant organic extracts were examined by LCMS but no significant chemical differences were observed between extracts from any of the transformants and the untransformed A. oryzae M-2-3 strain (data not shown).
Intron processing
Since A. oryzae is increasingly being used as a heterologous host for studying gene expression, we wanted to determine why our initial attempt at expressing ACE1 had failed. The ACE1 gene contains three introns located between exons corresponding to PKS domains (Fig. 1B). As there is a possibility that A. oryzae may not always splice heterologously expressed genes the same way as the host organism,18,20 we explored how the introns of ACE1 are processed by A. oryzae and M. oryzae.Each ACE1 intron was amplified with ca. 20 bp of flanking 5′ and 3′ exon sequences and cloned as in-frame fusions with eGFP downstream of either the native ACE1 promoter (PAce1), the strong constitutive promoter PgpdA from Aspergillus nidulans or the inducible promoter PamyB from A. oryzae. A total of nine plasmids were constructed (Fig. 1C). These constructs were transformed into either A. oryzae or M. oryzae, along with positive and negative controls, selected on the appropriate selection media and observed for fluorescence (Table 1 and Fig. 2). RT-PCR was performed for each intron construct using primers located either side of the introns and the products were sequenced (see ESI† for details).
| Construct name | Expression host | Conditions | Fluorescent transformants |
|---|---|---|---|
| pPamyB·intron1·eGFP·argB | A. oryzae | M | 80% |
| pPamyB·intron2·eGFP·argB | A. oryzae | M | 0% |
| pPamyB·intron3·eGFP·argB | A. oryzae | M | 90% (Fig. 2A) |
| Empty vector | A. oryzae | M | 0% |
| eGFP | A. oryzae | M | 87% |
| pPgpdA·intron1eGFP·hyg | M. oryzae | M, A | >70% |
| pPgpdA·intron2eGFP·hyg | M. oryzae | M, A | >70% (Fig. 2B) |
| pPgpdA·intron3eGFP·hyg | M. oryzae | M, A | >70% |
| Empty vector | M. oryzae | M, A | 0% |
| eGFP | M. oryzae | A | >70% |
| pPACE1·intron1eGFP·hyg | M. oryzae | A | >60% (Fig. 2C) |
| pPACE1·intron2eGFP·hyg | M. oryzae | A | >60% (Fig. 2D) |
| pPACE1·intron3eGFP·hyg | M. oryzae | A | >60% (Fig. 2E) |
| Empty vector | M. oryzae | M | 0% |
| eGFP | M. oryzae | M, A | >60% |
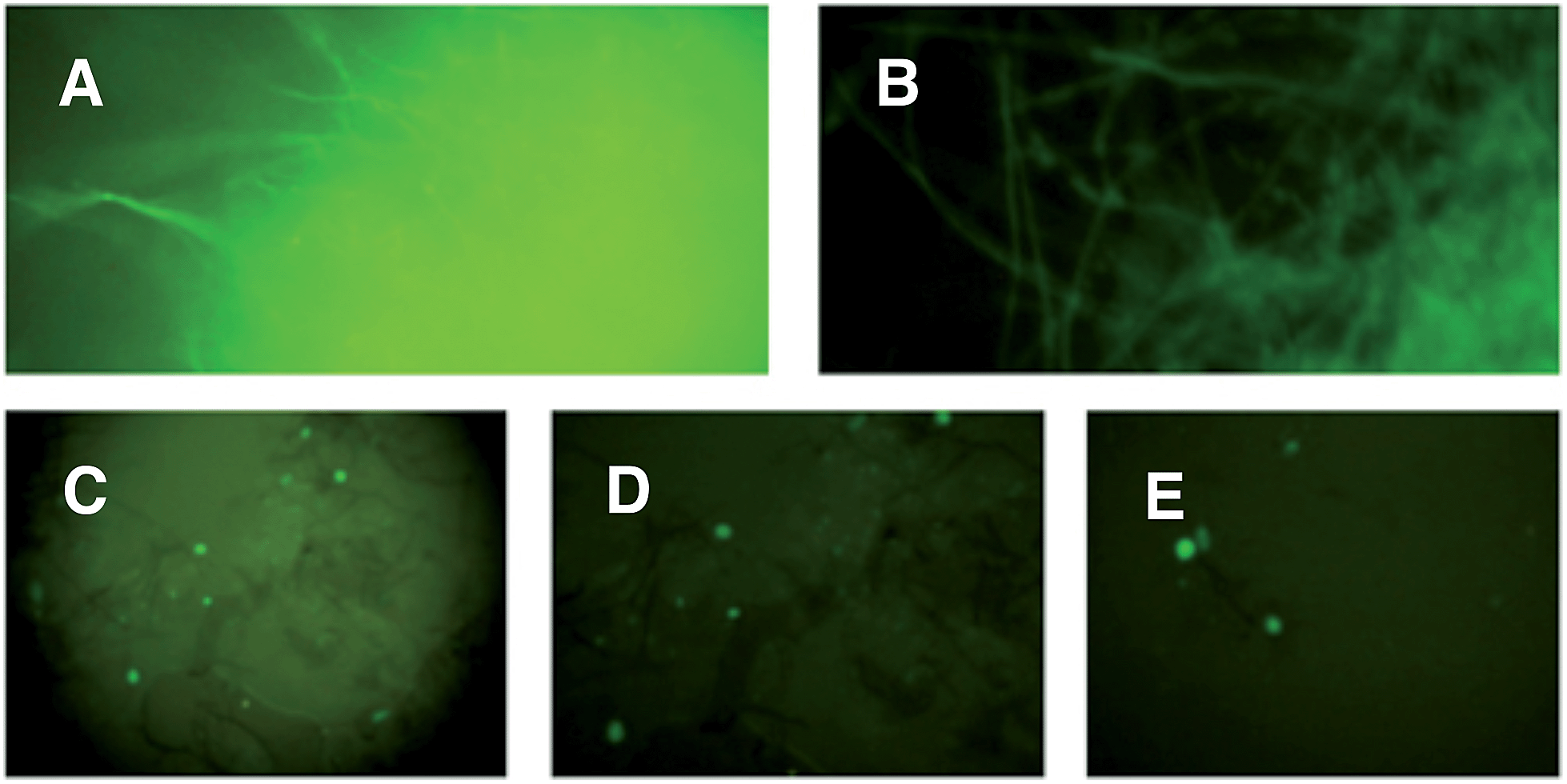 | ||
| Fig. 2 UV-stimulated green fluorescence of intron-eGFP clones. (A) A. oryzae M-2-3 pPamyB·intron3eGFP·argB; (B) M. oryzae Guy11 pPgpdA·intron2eGFP·hyg; (C) M. oryzae Guy11 pPACE1·intron1eGFP·hyg; (D) M. oryzae Guy11 pPACE1·intron2eGFP·hyg; (E) M. oryzae Guy11 pPACE1·intron3eGFP·hyg. A and B mycelia grown in liquid; C, D and E appressoria grown on cellophane. | ||
The results demonstrated that introns 1 and 3 are spliced identically in A. oryzae and M. oryzae whereas incorrect splicing of intron 2 by A. oryzae results in a frameshift and premature stop codon (Fig. S1†).
Heterologous expression of ACE1
Based on results from the intron processing experiments, we created two modified expression plasmids for further attempts to produce the ACE1 compound (Fig. 1A). The first consisted of the entire ACE1 gene lacking all introns fused to eGFP (pPamyB·ACE1[Δintrons1–3]eGFP·argB); the second consisted only of the PKS encoding portion of ACE1 – also fused to eGFP, and containing only intron-3 (pAmyB·ACE1pks[Δintrons1–2]eGFP·argB, see ESI† for construction details). Controls included empty vector and eGFP alone (pPgpdA·eGFP·hyg, Fig. 1D). These plasmids were used to transform A. oryzae M-2-3 and selection was achieved on minimal medium supplemented with hygromycin where appropriate. On malt extract agar (MEA) the transformants containing the ACE1 constructs produced a bright yellow colour, while the controls were colourless. Twenty colonies were selected for each construct, grown in liquid medium and extracted as described above. All transformants tested clearly produced a new peak compared to the controls (Fig. 3A) in the neutral organic extract, and in lesser amounts, in the acidic and mycelial extracts.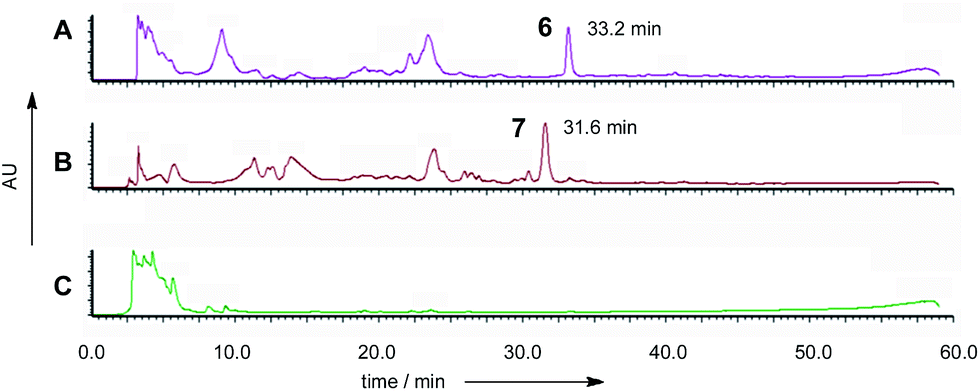 | ||
| Fig. 3 Diode array chromatograms of culture extracts from: (A) A. oryzae M-2-3 pPamyB·ACE1[Δintrons1–3]eGFP·argB; (B) M. oryzae pPgpdA·ACE1[Δintrons1–2]eGFP·hyg; (C) A. oryzae M-2-3 empty vector transformant. | ||
The new compounds appeared to be identical for the two ACE1 expression constructs (RT = 33.2 min; uv max = 214, 272, 361 nm; m/z = 315 [M]Na+, 293 [M]H+, 275 [M − H2O]H+). Both compounds were purified and shown to be identical by NMR and HRMS (m/z 291.1238 Da [M − H]−, consistent with a molecular formula of C16H20O5).
1H NMR analysis (see ESI†) showed that the sample consisted of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers each of which contained three methyl groups, a CHX, five contiguous olefinic protons, and two sharp doublets (J = 2.0 Hz) in the olefinic region. COSY correlations were used to determine that the olefinic protons were part of a distinct spin-system with an olefinic methyl group, connected by a vicinal coupling, visible only as a slight broadening of the terminal olefin doublet in the 1D 1H NMR. The COSY also showed that the CHX (which appeared as a complex multiplet) and one of the methyl groups (itself appearing as a pair of doublets) formed another distinct coupling system. An HMBC spectrum determined that the lone methyl group and olefins were all part of the same chain with a formula of C11H17O2, leaving C5H3O3 unaccounted for. The characteristic 13C NMR resonance of δ 163.6 ppm revealed that an ester group was present in the structure accounting for a further CO2, and leaving C4H3O unassigned.
:1 mixture of diastereomers each of which contained three methyl groups, a CHX, five contiguous olefinic protons, and two sharp doublets (J = 2.0 Hz) in the olefinic region. COSY correlations were used to determine that the olefinic protons were part of a distinct spin-system with an olefinic methyl group, connected by a vicinal coupling, visible only as a slight broadening of the terminal olefin doublet in the 1D 1H NMR. The COSY also showed that the CHX (which appeared as a complex multiplet) and one of the methyl groups (itself appearing as a pair of doublets) formed another distinct coupling system. An HMBC spectrum determined that the lone methyl group and olefins were all part of the same chain with a formula of C11H17O2, leaving C5H3O3 unaccounted for. The characteristic 13C NMR resonance of δ 163.6 ppm revealed that an ester group was present in the structure accounting for a further CO2, and leaving C4H3O unassigned.
The remaining two unassigned protons showed HMBC correlations to the ester carbonyl and two other quaternary carbons, the chemical shifts of which (δ 162.4 ppm and δ 162.6 ppm) were consistent with enolic carbons. Although these protons were coupled, they did not appear to be on adjacent carbons. An IR spectrum showed a characteristic absorbance at 1685 cm−1 suggesting that the ester group was present as part of a pyrone. Thus the structure was determined as the novel pyrone 6 to which we have assigned the name 12,13-dihydroxymagnaporthepyrone.
The polyketide origin of 6 was confirmed by feeding [1,2-13C2]-acetate and [methyl-13C]-methionine to A. oryzae pPamyB·ACE1[Δintrons1–3]eGFP·argB. Pyrone 6 isolated after the labelled acetate feed showed the presence of distinctive doublets in the 13C NMR indicative of the incorporation of intact 13C2 units (Fig. 4 and ESI†). Matching of the 2JCC values showed intact acetate incorporations consistent with the heptaketide origin of 6. The origin of both methyls from methionine was confirmed by the observation of a 9.4% incorporation of label at C-15 and C-16.
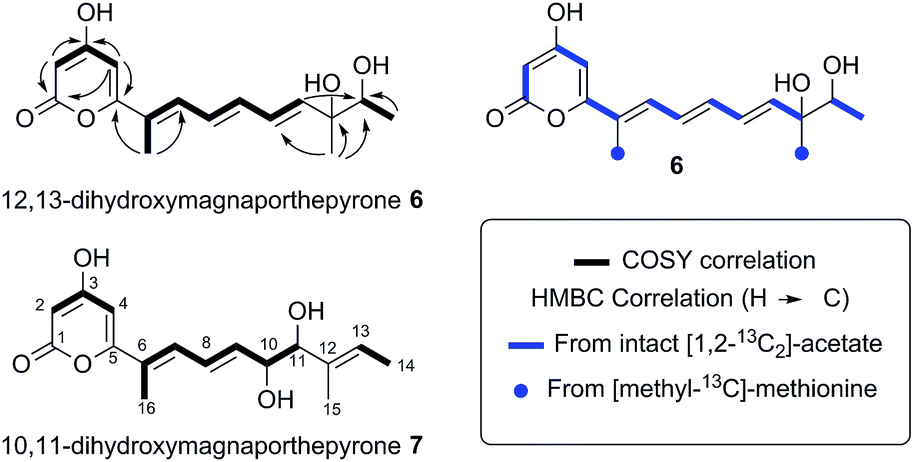 | ||
| Fig. 4 Structures and biosynthetic origin of the pyrones obtained by expressing ACE1 in A. oryzae M-2-3 and M. oryzae. | ||
Thus 6 consists entirely of a polyketide and does not contain the expected amino acid component. To rule out NRPS perturbance by the C-terminal eGFP fusion we modified the expression system to remove the 3′-eGFP gene to create pPamyB·ACE1[Δintrons1–3]·argB and transformed A. oryzae M-2-3. Selected transformants were grown in liquid medium and secondary metabolites detected using LCMS; 12,13-dihydroxymagnaporthepyrone 6 was the only new compound detected.
To overcome the possibility that A. oryzae lacks the specific amino acid required by the ACE1 NRPS in M. oryzae we constructed plasmids for homologous expression of ACE1 in M. oryzae Guy11 using the PgpdA constitutive promoter and hygromycin selection. We thus constructed an E. coli shuttle vector based on the pUC18 vector with a Gateway cassette downstream from PgpdA carrying a hygromycin resistance marker. The ACE1 eGFP construct was transferred into this vector by Gateway LR recombination in vitro to give pPgpdA·ACE1eGFP·hyg which was used to transform M. oryzae Guy11. The plasmid pPgpdA·eGFP·hyg was used as a control. Hygromyin resistant transformants were selected for each construct and grown on complete medium plates and examined for fluorescence. For pPgpdA·eGFP·hyg, 18/20 transformants were fluorescent while only 5/100 of the pPgpdA·ACE1eGFP·hyg transformants were fluorescent.
The five M. oryzae transformants expressing ACE1eGFP were then grown in complete liquid medium for 7 days then extracted and examined by LCMS as described in the ESI.† All five transformants produced a new compound which was not produced by the controls. This compound had a different retention time to 12,13 dihydroxymagnaporthepyrone 6 produced in A. oryzae (RT = 31.6 min, Fig. 3B) and a different UV spectrum (uvmax = 210 nm, 248 nm, 340 nm) although the low resolution ESI MS spectrum was identical to 6, as was the molecular formula predicted from the HRMS. Thus it appeared that this compound was isomeric to 12,13-dihydroxymagnaporthepyrone 6. 1H NMR showed it to consist of a single stereoisomer, but compared to 6 it had only three contiguous olefinic CH resonances, one of which was coupled to a CHO methine which was in turn coupled to another CHO methine. The methyl doublet of 6 was replaced by a methyl doublet coupling to an olefinic CH. These data, supported by 13C and 2D spectra showed this compound to be 10,11-dihydroxymagnaporthepyrone 7.
Co-expression with the RAP1 trans-ER protein
Bioinformatic analysis of the sequence of the ACE1 enoyl reductase (ER) domain suggests that it is inactive, similar to the tenellin and lovastatin nonaketide synthase (LNKS) ERs – such domains are designated ER0 to indicate their non-catalytic role. Many fungal hrPKS which contain an ER0 domain recruit a trans-acting ER which has a catalytic and programming role. For example in the cases of pretenellin A 1, preaspyridone 3 and lovastatin 5 biosynthesis trans-acting ERs encoded by tenC, apdC and lovC respectively interact with the PKS to provide programmed enoyl reduction and have the effect of reinforcing PKS programming including methylation and chain-length fidelity.15 RAP1 also encodes a trans-acting ER (e.g. RAP1 has a 48% similarity to TENC and 58% similarity to LOVC) and thus it may play a similar role in concert with ACE1, possibly controlling fidelity as well as enoyl reduction. We therefore constructed plasmids to coexpress RAP1 with ACE1 in both A. oryzae and M. oryzae.For A. oryzae we created a plasmid carrying both the ACE1 and RAP1 genes in which ACE1 lacking all introns was expressed under the control of PamyB, and RAP1 was expressed from the strong constitutive alcohol dehydrogenase promoter Padh from A. oryzae (pPamyB·ACE1[Δintrons 1–3]·argB·Padh·RAP1, Fig. 1A). A. oryzae M-2-3 protoplasts were transformed with pPamyB·ACE1[Δintrons 1–3]·argB·Padh·RAP1 and transformants were selected on minimal medium.
Twenty seven of the resulting A. oryzae transformants were selected and transferred to new agar plates. Ten of these transformants were grown in liquid medium containing starch for seven days, then extracted in the usual way. LCMS analysis of the extracts showed that of the A. oryzae transformants, five produced a new peak compared to the controls in the neutral organic extract, and in lesser amounts, in the acidic and mycelial extracts. The new compound had different retention times to compounds 6 and 7 (RT = 22.5 min, uv max = 270, 280 nm, m/z 481.4 [M − H]− (Fig. 5)).
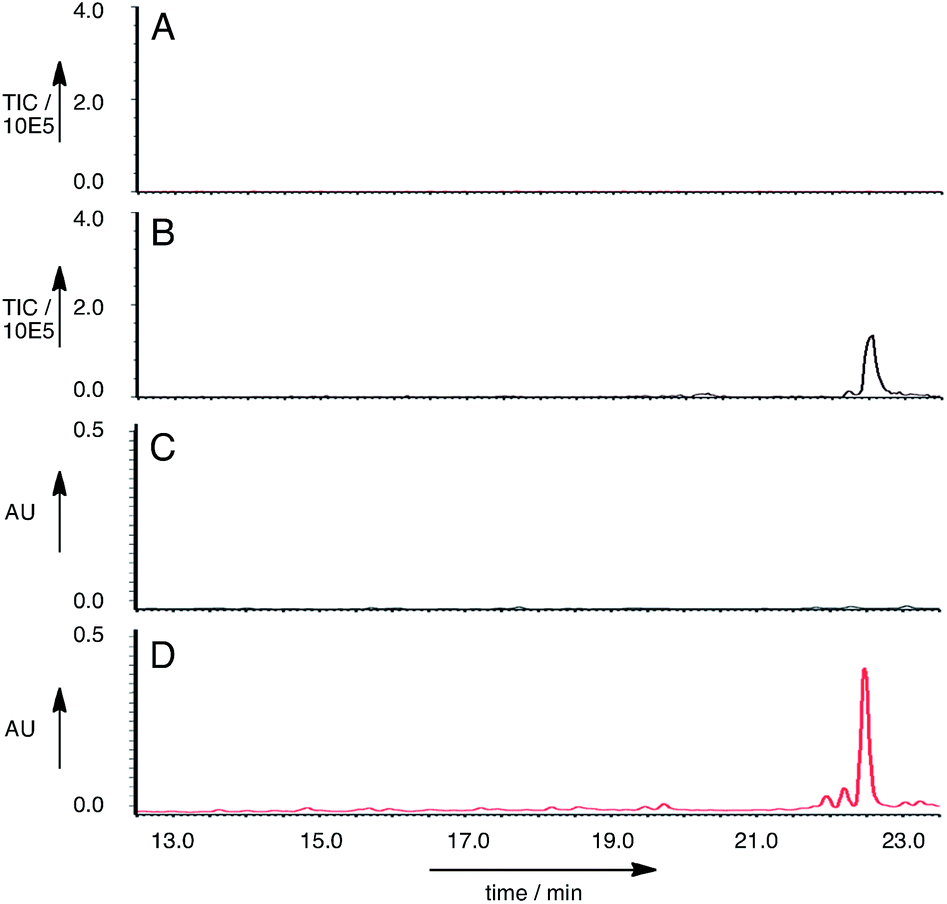 | ||
| Fig. 5 LCMS analysis of ACE1 + RAP1 expression: A, EIC (ES−) m/z 480.4 for A. oryzae M-2-3; B, EIC (ES−) m/z 480.4 for A. oryzae M-2-3 + ACE1 + RAP1; C, uv chromatogram (280 nm) for A. oryzae M-2-3; D, uv chromatogram (280 nm) for A. oryzae M-2-3 + ACE1 + RAP1. | ||
A single A. oryzae transformant was grown at large scale (800 mL) to yield 65.5 mg of crude extract. The new compound was purified by HPLC and analysed by NMR and HRMS (m/z 482.3265 Da [M]H+, consistent with a molecular formula of C30H43NO4). 1H NMR analysis showed that the compound contained four methyl groups, six olefinic protons and an aromatic group indicative of tyrosine. 13C NMR, COSY, HMBC and HSQC determined the structure as 8 (Fig. 6, ESI†).
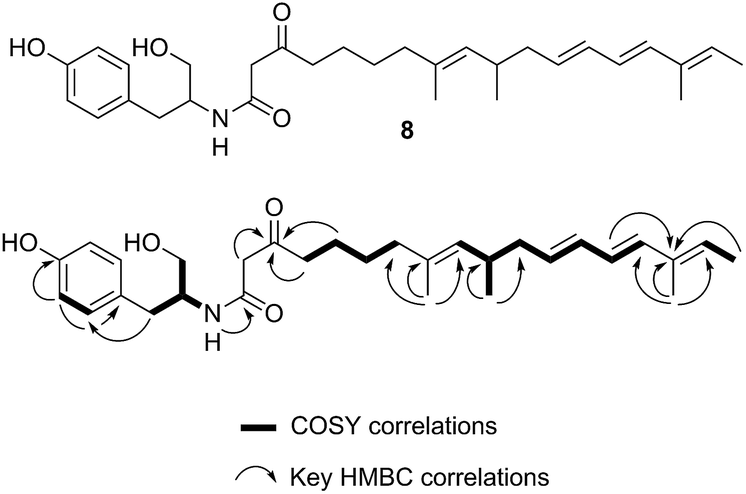 | ||
| Fig. 6 Structure and NMR data of 8 from ACE1 + RAP1 expression transformants. | ||
Biological testing
Purified magnaporthepyrones 6 and 7 were tested for biological activity on susceptible, (Maratelli, Sariceltik CO39) and resistant rice cultivars carrying Pi33 (IR64 and C101LAC).21 Compounds were deposited onto normal or wounded rice leaves. Both compounds induced localised brown necrosis on all cultivars, irrespective of host genotype (Fig. 7). These results suggest that magnaporthepyrones do not induce a phenotype specifically on rice cultivars carrying Pi33. Addition of these compounds to spore suspensions of an isolate virulent on Pi33 rice cultivars did not induce an AVR reaction, but slightly reduced pathogenicity on both susceptible and resistant cultivars. Compound 8 had no biological activity: no induction of leaf symptoms, no changes of a virulent interaction into AVR, no enhancement nor reduction in pathogenicity. Therefore compounds, 6, 7 and 8 do not appear to be the avirulence signal compound produced by ACE1/M. oryzae Guy11 during appressorium-mediated penetration.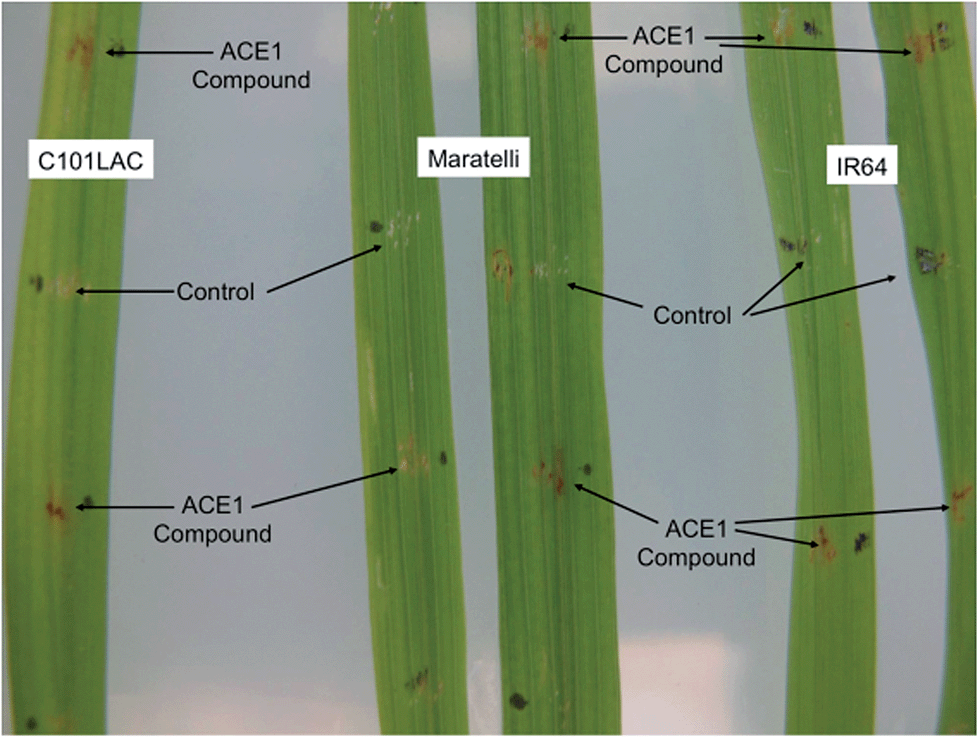 | ||
| Fig. 7 Biological testing of 12,13-dihydroxymagnaporthepyrone 6 on both resistant (IR64 and C101LAC) and susceptible (Maratelli) rice cultivars. Testing of 7 gave similar results. 8 Had no effect. | ||
Discussion
Initial attempts to express ACE1 in A. oryzae produced no observable new metabolites. This was surprising as all of our previous efforts to express foreign fungal genes in this organism had been successful.Further investigation using RT-PCR and subsequent sequence analysis clearly showed that ACE1 intron-2 is not spliced correctly in A. oryzae, resulting in an mRNA with a frame-shift and premature stop codon. Intron-2 is located centrally in the PKS portion of ACE1, upstream of the sequence encoding the key acyl carrier protein (ACP) domain. Any truncated ACE1 protein resulting from the translation of this incorrectly spliced mRNA would not be capable of producing a polyketide product.
Removal of the ACE1 introns resulted in transcripts which were translated correctly in A. oryzae. Indeed more than 75% of transformants produced full-length ACE1-eGFP protein fusions as evidenced by strong fluorescence of transformed hyphae, and the production of novel metabolites. Expression of both full-length and NRPS-deleted ACE1 intronless constructs resulted in the production of the same compound, 12,13-dihydroxymagnaporthepyrone 6 which is exclusively of polyketide origin as shown by isotopic labelling. Expression of the same clones in M. oryzae resulted in the production of the closely related 10,11-dihydroxymagnaporthepyrone 7. Similar compounds were observed by Vederas and coworkers when the lovasatin nonaketide synthase (LNKS) was expressed without its cognate ER encoded by lovC.15
These results can be interpreted if the PKS portion of ACE1 produces the polyunsaturated pyrone 9 (which we name magnaporthepyrone, Scheme 2). This compound is then epoxidised by different monooxygenases in either A. oryzae or M. oryzae and spontaneous hydrolysis of the epoxides results in the observed diols 6 and 7. Support for this hypothesis comes from the expression of tenS in A. oryzae where we observed the presence of prototenellin C 10 (ref. 22) which displays exactly the same chemical motif as 12,13-dihydroxymagnaporthepyrone 6 – thus A. oryzae must possess a monooxidase selective for the terminal methylbutenyl motif of such precursors. Likewise, M. oryzae is known to produce diol compounds such as pyriculol 11 (ref. 23) which must also derive from epoxidation of a polyunsaturated polyketide followed by hydrolysis. Therefore, M. oryzae must have a monooxygenase with selectivity for epoxidation of mid-chain polyenes.
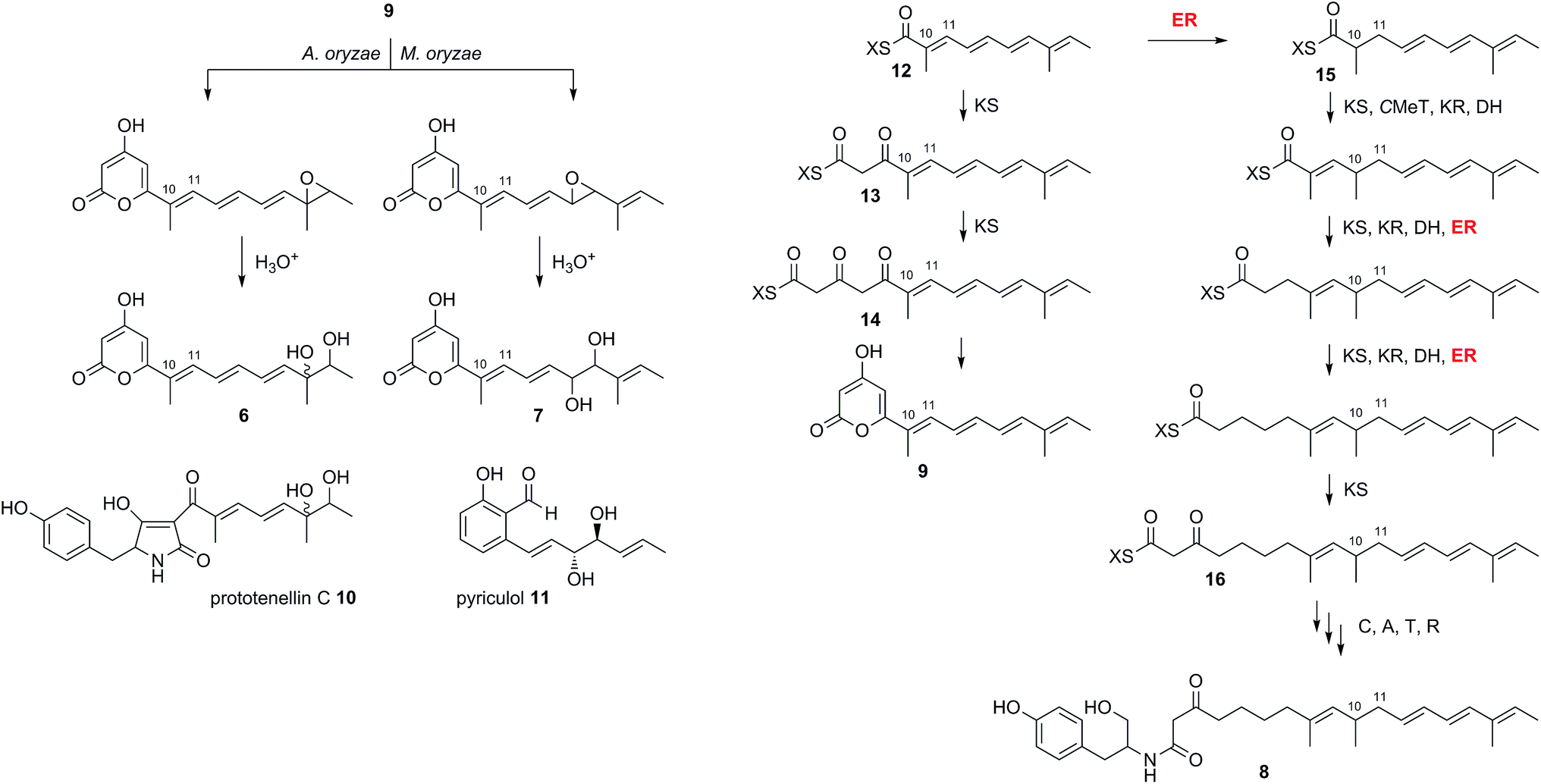 | ||
| Scheme 2 Deduced biosynthesis of ACE1 products in the presence and absence of RAP1. X = ACE1 ACP domain. | ||
The biosynthesis of 6 and 7 do not require an enoyl reductase step, consistent with the observation that the ACE1 ER domain is predicted to be inactive. Coexpression of ACE1 with RAP1, which encodes a trans-acting ER, changed the biosynthetic pathway. In the heterologous host A. oryzae, where we constructed a single vector to carry both genes, the amide 8 was produced and compounds derived from 9 were no longer observed.
Amide 8 is closely related to pyrone 9 and its biosynthesis can be rationalised by the intervention of the RAP1 ER at the pentaketide stage of biosynthesis (Scheme 2). Pentaketide intermediate 12 is recognised by the ACE1 KS domain and extended to 13, but in the absence of prior enoyl reduction the next required CMeT step is prevented. Continued chain extension by an unselective KS then gives the tricarbonyl 14 which can spontaneously offload itself from the synthase as the pyrone 9. Alternatively, in the presence of the RAP1 ER, pentaketide 12 is reduced giving a substrate which can be extended by the KS and then correctly methylated to give 15 before following three more extension and processing cycles to give the fully extended nonaketide 16. Thus the ACE1 CMeT domain appears to display an additional level of substrate selectivity in blocking correct chain processing in the absence of 10,11 reduction. Crucially, however, the KS is unselective and continues to extend the chain to the tricarbonyl 14 which can be off-loaded as the pyrone 9. Thus the lack of selectivity by the KS effectively removes incorrectly processed chains from the PKS, preventing its blockage.
In many other investigated PKS-NRPS systems the NRPS then attaches the amino acid to the fully extended polyketide and either reductively cleaves it,24 or performs a non-reductive Dieckmann cyclisation reaction to release an acyl tetramic acid.9 In this case it appears that tyrosine is used as the amino acid and attached to the polyketide in the same way as observed in the cases of pretenellin A 1 and preaspyridone 3. After amide formation, however, the fully elaborated product is probably reductively released. This could be via 2 electron reduction to form an aldehyde (as in the classic case of fungal lysine biosynthesis25) which could be further reduced to a primary alcohol by an adventitious A. oryzae enzyme, or via double (i.e. 4 electron) reduction catalysed by the NRPS R-domain itself as observed during the biosynthesis of myxochelin.26
Consistent with this hypothesis is the observation that the ACE1 terminal-domain has an intact conserved NADPH binding motif (GXSXXG) and the catalytic triad Ser-Tyr-Lys more closely resembles ‘reducing’ R-domains than DKC cyclising domains (see ESI†).24 Qiao et al. predicted a reductive release for ccsA, the PKS-NRPS gene involved in cytochalasin K 4 biosynthesis in Aspergillus clavatus,27 based on the same rationale, and the co-expression of ccsA with ccsC by Oikawa and co-workers led to the production of the amide 17,14 also at the alcohol oxidation state.
The Oikawa compound 17 differs from 8 in being an octaketide rather than a nonaketide and being evidently constructed from phenylalanine rather than tyrosine (Scheme 3). A further difference is that 8 is unsaturated between carbons 8 and 9 of the polyketide, whereas 17 is fully saturated at the corresponding position. Genetic experiments have already shown that ccsA and ccsC are involved in cytochalasin K 4 biosynthesis, most likely via ketocytochalasin 18.27 Although 17 is unlikely to be a direct precursor of 4, it is probably a shunt metabolite from the corresponding aldehyde. Vederas, Tang and coworkers have also recently elucidated that ccsB from the cytochalasin K biosynthetic pathway is an FAD-dependent oxygenase which catalyses (among other reactions) the creation of the distinctive carbonate moiety of 4.28
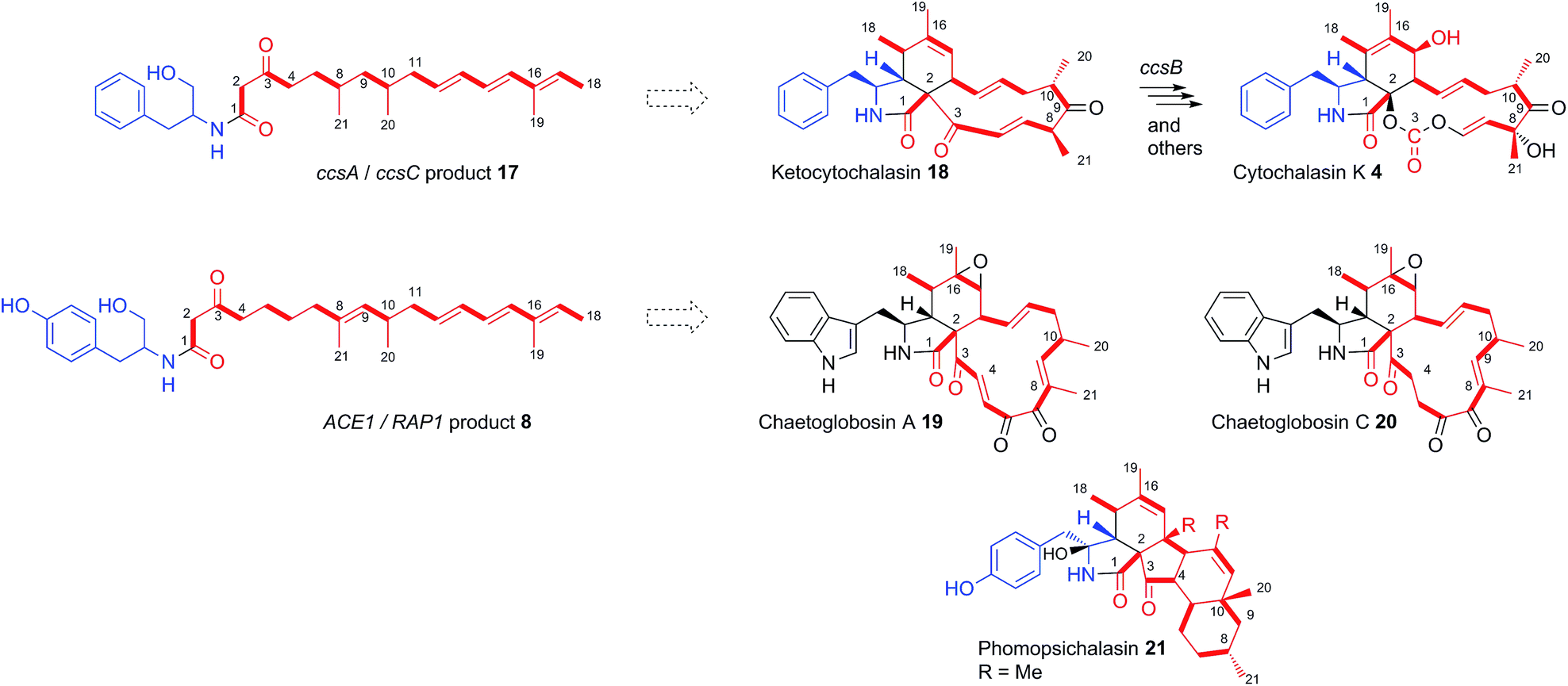 | ||
| Scheme 3 Structural relationships between isolated chemical products of fungal PKS/NRPS and trans-ER proteins and known natural products and biosynthetic pathways. Although cytochalasans are usually grouped according to the specific incorporated amino acid, the nature of the polyketide chain (shown in red) also contributes to their chemical diversity. Numbering for illustrative purposes only. Dashed arrow indicates biosynthetic relationship and does not indicate a direct precursor/product relationship. | ||
The structural similarities between the ccsA/C product 17 and the ACE1/RAP1 product 8 strongly suggest that 8 is also a shunt metabolite of a cytochalasan-like biosynthetic pathway.13 This is supported by the observation that 8 does not show the expected biological activity as an avirulence signalling compound.
To our knowledge, only two other gene clusters have been correlated with cytochalasan biosynthesis in fungi. These are the gene clusters for chaetoglobosin A 19 biosynthesis in Penicillium expansum and Chaetomium globosum.29,30 Chaetoglobosin A 19 is biosynthesised from tryptophan and a nonaketide likely to be almost identical to that of amide 8, except for the unsaturation between carbons 4 and 5. However, several chaetoglobosins are also known which are fully saturated at this position (e.g. chaetoglobosin C 20)31 indicating that amide 8 could include the correct polyketide precursor. Analysis of the tailoring genes in the ACE1 cluster show that they have more similarity to the chaetoglobosin biosynthetic genes than those in the ccs cytochalasin gene cluster (Table S2†).32
The nonaketide nature of the amide 8 and the fact that tyrosine is incorporated, distinguishes the RAP1/ACE1 product from the ccsA/ccsC compound and mean that the likely ultimate product of the ACE1 pathway will be a member of the tyrosine-derived cytochalasans, an example of which is phomopsichalasin (also known as diaporthichalasin) 21.33,34 In addition, the differences between the ACE1 and ccs gene clusters, such as the lack of a ccsB homolog, plus additional oxidoreductases similar to those found in the chaetoglobosin A 19 gene cluster, indicate that the tailoring of the polyketide backbone is likely to be more similar to 19, which could arise from a polyketide identical or very similar to that of 8.
Thus we have shown that heterologous expression is an effective tool to probe cryptic gene clusters in fungi, which enables important metabolic relationships to be unravelled. It is now clear that avirulence signalling between the rice blast fungus Magnaporthe oryzae and rice is mediated by a natural product bearing structural similarities to the cytochalasans. Current and future work in our group is focused on exploring the enzymatic activities of the proteins encoded by other genes from the ACE1 biosynthetic cluster to identify the bioactive compound responsible for avirulence signalling.
Experimental
For construction of vectors, medium conditions, transformation protocols, LCMS methods, NMR data, isotope feeding experiments and details on biological testing see the ESI.†Characterisation of new natural products
:1 mixture of two diastereoisomers (A and B) by preparative HPLC. 1H NMR (500 MHz acetone-d6): δ 1.09 ppm (1.5H, d, 3JHH 6.7, H-14A), 1.11 ppm (1.5H, d, 3JHH 6.3, H-14B), 1.255 (1.5H, s, H-15A), 1.260 (1.5H, s, H-15B), 2.02 (1.5H, s, H-16A), 2.03 (1.5H, s, H-16B), 3.60 (0.5H, q, 3JHH 6.7, H-13A), 3.61 (0.5H, q, 3JHH 6.3, H-13B), 5.39 (1H, d, 3JHH 2.0, H-2A + H-2B), 6.07 (0.5H, d, 3JHH 15.3, H-11A), 6.08 (0.5H, d, 3JHH 15.3, H-11B), 6.20 (1H, d, 3JHH 2.0, H-4A + H-4B), 6.55 (1H, m, H-10AB), 6.69 (2H, m, H-8AB + H-9AB), 7.09 (1H, d, 3JHH 9.6, H-7AB); 13C NMR (151 MHz acetone-d6): δ 12.7 ppm (C-15), 18.1 & 18.3 (C-14), 24.5 & 23.7 (C-16), 74.2 (C-13), 75.8 (C-12), 90.1 (C-2), 99.0 (C-4), 126.8 (C-6), 128.2 (C-8), 129.5 & 129.8 (C-10), 132.5 (C-7), 139.6 & 139.6 (C-9), 142.8 & 143.2 (C-11), 162.4 (C-3/5), 162.6 (C-5), 163.6 (C-1), 170.7 (C-3); UV/vis λmax (H2O/MeOH) 273, 362 nm; IR νmax (neat) 3375 brs, 2926, 1685 br s, 1530 cm−1; MS (ESI+) m/z (%): 293 (100) [M]H+, 315 (50) [M]Na+; MS (ESI−) m/z (%): 291 (100) [M − H]−; HRMS (ESI+) calculated [M − H2O]H+ 275.1277 found 275.0922; HRMS (ESI−) calculated [M − H]− 291.1033 found 291.1238.
Acknowledgements
Walid Bakeer was funded by a Government of Egypt Scholarship. James Marshall was funded by The School of Chemistry at the University of Bristol and the Mark Evans Scholarship. Ahmed Yakasai was funded by Kano State Government Nigeria, The MacArthur Foundation, Bayero University, and the Nigerian Petroleum Technology Fund. Rozida Mohd Khalid was funded by a Malaysian Govenment Scholarship. Dr Z. Song and LCMS equipment were funded by EPSRC (EP/F066104/1).References
- P. Skamnioti and S. J. Gurr, Trends Biotechnol., 2009, 27, 141–150 CrossRef CAS PubMed
.
- E. Perez-Nadales, M. F. Nogueira, C. Baldin, S. Castanheira, M. El Ghalid, E. Grund, K. Lengeler, E. Marchegiani, P. V. Mehrotra, M. Moretti, V. Naik, M. Oses-Ruiz, T. Oskarsson, K. Schäfer, L. Wasserstrom, A. A. Brakhage, N. A. Gow, R. Kahmann, M.-H. Lebrun, J. Perez-Martin, A. Di Pietro, N. J. Talbot, V. Toquin, A. Walther and J. Wendland, Fungal Genet. Biol., 2014, 70, 42–67 CrossRef CAS PubMed
- R. Berruyer, H. Adreit, J. Milazzo, S. Gaillard, A. Berger, W. Dioh, M.-H. Lebrun and D. Tharreau, Theor. Appl. Genet., 2003, 107, 1139–1147 CrossRef CAS PubMed
- H. U. Bohnert, I. Fudal, W. Dioh, D. Tharreau, J.-L. Notteghem and M.-H. Lebrun, Plant Cell, 2004, 16, 2499–2513 CrossRef PubMed
- I. Stergiopoulos and P. J. G. M. de Wit, Annu. Rev. Phytopathol., 2009, 47, 233–263 CrossRef CAS PubMed
- J. Collemare, M. Pianfetti, A.-E. Houlee, D. Morin, L. Camborde, M.-J. Gagey, C. Barbisan, I. Fudal, M.-H. Lebrun and H. U. Böhnert, New Phytol., 2008, 179, 196–208 CrossRef CAS PubMed
- I. Fudal, J. Collemare, H. U. Bohnert, D. Melayah and M.-H. Lebrun, Eukaryotic Cell, 2007, 6, 546–554 CrossRef CAS PubMed
- R. J. Cox, Org. Biomol. Chem., 2007, 5, 2010–2026 CAS
- L. M. Halo, M. N. Heneghan, A. A. Yakasai, Z. Song, K. Williams, A. M. Bailey, R. J. Cox, C. M. Lazarus and T. J. Simpson, J. Am. Chem. Soc., 2008, 130, 17988–17996 CrossRef CAS PubMed
- Z. Song, R. J. Cox, C. M. Lazarus and T. J. Simpson, ChemBioChem, 2004, 5, 1196–1203 CrossRef CAS PubMed
- S. Bergmann, J. Schümann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef CAS PubMed
- Z. Wasil, K. A. K. Pahirulzaman, C. P. Butts, T. J. Simpson, C. M. Lazarus and R. J. Cox, Chem. Sci., 2013, 4, 3845–3856 RSC
- K. Scherlach, D. Boettger, N. Remme and C. Hertweck, Nat. Prod. Rep., 2010, 27, 869–886 RSC
- R. Fujii, A. Minami, K. Gomi and H. Oikawa, Tetrahedron Lett., 2013, 54, 2999–3002 CrossRef CAS PubMed
- J. Kennedy, K. Auclair, S. Kendrew, C. Park, J. Vederas and C. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS
- T. B. Kakule, Z. Lin and E. W. Schmidt, J. Am. Chem. Soc., 2014, 136, 17882–17890 CrossRef CAS PubMed
- M. N. Heneghan, A. A. Yakasai, L. M. Halo, Z. Song, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, ChemBioChem, 2010, 11, 1508–1512 CrossRef CAS PubMed
- K. M. Fisch, E. Skellam, D. Ivison, R. J. Cox, A. M. Bailey, C. M. Lazarus and T. J. Simpson, Chem. Commun., 2010, 46, 5331–5333 RSC
- I. Fujii, Y. Mori, A. Watanabe, Y. Kubo, G. Tsuji and Y. Ebizuka, Biosci., Biotechnol., Biochem., 1999, 63, 1445–1452 CrossRef CAS PubMed
- A. M. Bailey, R. J. Cox, K. Harley, C. M. Lazarus, T. J. Simpson and E. Skellam, Chem. Commun., 2007, 4053–4055 RSC
- R. Berruyer, H. Adreit, J. Milazzo, S. Gaillard, A. Berger, W. Dioh, M. H. Lebrun and D. Tharreau, Theor. Appl. Genet., 2003, 107, 1139–1147 CrossRef CAS PubMed
- M. N. Heneghan, A. A. Yakasai, K. Williams, K. A. Kadir, Z. Wasil, W. Bakeer, K. M. Fisch, A. M. Bailey, T. J. Simpson, R. J. Cox and C. M. Lazarus, Chem. Sci., 2010, 2, 972 RSC
- A. N. Rao and S. Suryanarayanan, Curr. Sci., 1974, 43, 348–349 Search PubMed
- X. Liu and C. T. Walsh, Biochemistry, 2009, 48, 8746–8757 CrossRef CAS PubMed
- D. Ehmann, A. Gehring and C. T. Walsh, Biochemistry, 1999, 38, 6171–6177 CrossRef CAS PubMed
- Y. Li, K. J. Weissman and R. Müller, J. Am. Chem. Soc., 2008, 130, 7554–7555 CrossRef CAS PubMed
- K. Qiao, Y.-H. Chooi and Y. Tang, Metab. Eng., 2011, 13, 723–732 CrossRef CAS PubMed
- Y. Hu, D. Dietrich, W. Xu, A. Patel, J. A. J. Thuss, J. Wang, W.-B. Yin, K. Qiao, K. N. Houk, J. C. Vederas and Y. Tang, Nat. Chem. Biol., 2014, 10, 552–554 CrossRef CAS PubMed
- J. Schümann and C. Hertweck, J. Am. Chem. Soc., 2007, 129, 9564–9565 CrossRef PubMed
- K. Ishiuchi, T. Nakazawa, F. Yagishita, T. Mino, H. Noguchi, K. Hotta and K. Watanabe, J. Am. Chem. Soc., 2013, 135, 7371–7377 CrossRef CAS PubMed
- J. P. Springer, J. Clardy, J. M. Wells, R. J. Cole, J. W. Kirksey, R. D. Macfarlane and D. F. Torgerson, Tetrahedron Lett., 1976, 17, 1355–1358 CrossRef
- N. Khaldi, J. Collemare, M.-H. Lebrun and K. H. Wolfe, Genome Biol., 2008, 9, R18 CrossRef PubMed
- W. S. Horn, M. S. J. Simmonds, R. E. Schwartz and W. M. Blaney, Tetrahedron, 1995, 51(14), 3969–3978 CrossRef CAS
- S. Pornpakakul, S. Roengsumran, S. Deechangvipart, A. Petsom, N. Muangsin, N. Ngamrojnavanich, N. Sriubolmas, N. Chaichit and T. Ohta, Tetrahedron Lett., 2007, 48, 651–655 CrossRef CAS PubMed
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03707c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |