
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting parameter space in MOFs: a 20-fold enhancement of phosphate-ester hydrolysis with UiO-66-NH2†
Michael J.
Katz
a,
Su-Young
Moon
a,
Joseph E.
Mondloch
a,
M. Hassan
Beyzavi
a,
Casey J.
Stephenson
a,
Joseph T.
Hupp
*ab and
Omar K.
Farha
*ac
aDepartment of Chemistry, Northwestern University, 2145 Sheridan Road, Evanston, Illinois 60208, USA. E-mail: j-hupp@northwestern.edu; o-farha@northwestern.edu
bChemical Science and Engineering Division, Argonne National Laboratory, 9700 S. Cass Avenue, Argonne, Illinois 60439, USA
cDepartment of Chemistry, Faculty of Science, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 24th February 2015
Abstract
The hydrolysis of nerve agents is of primary concern due to the severe toxicity of these agents. Using a MOF-based catalyst (UiO-66), we have previously demonstrated that the hydrolysis can occur with relatively fast half-lives of 50 minutes. However, these rates are still prohibitively slow to be efficiently utilized for some practical applications (e.g., decontamination wipes used to clean exposed clothing/skin/vehicles). We thus turned our attention to derivatives of UiO-66 in order to probe the importance of functional groups on the hydrolysis rate. Three UiO-66 derivatives were explored; UiO-66-NO2 and UiO-66-(OH)2 showed little to no change in hydrolysis rate. However, UiO-66-NH2 showed a 20 fold increase in hydrolysis rate over the parent UiO-66 MOF. Half-lives of 1 minute were observed with this MOF. In order to probe the role of the amino moiety, we turned our attention to UiO-67, UiO-67-NMe2 and UiO-67-NH2. In these MOFs, the amino moiety is in close proximity to the zirconium node. We observed that UiO-67-NH2 is a faster catalyst than UiO-67 and UiO-67-NMe2. We conclude that the role of the amino moiety is to act as a proton-transfer agent during the catalytic cycle and not to hydrogen bond or to form a phosphorane intermediate.
Introduction
Phosphate-based compounds are key ingredients in biological systems. The cleavage and formation of P–O bonds are responsible for converting ATP to ADP and vice versa.1,2 It is thus not surprising that organophosphorous compounds such as the ones shown in Fig. 1 are capable of disrupting biological functions.2–5 Sarin, for example, is a phosphate-based chemical warfare agent that has unfortunately shown resurgence in recent times.4,6,7 The P–F bond is easily cleaved followed by the formation of a strong acetylcholinesterase inhibitor due to the favorable P–O bond formed; this often leads to asphyxiation.6,8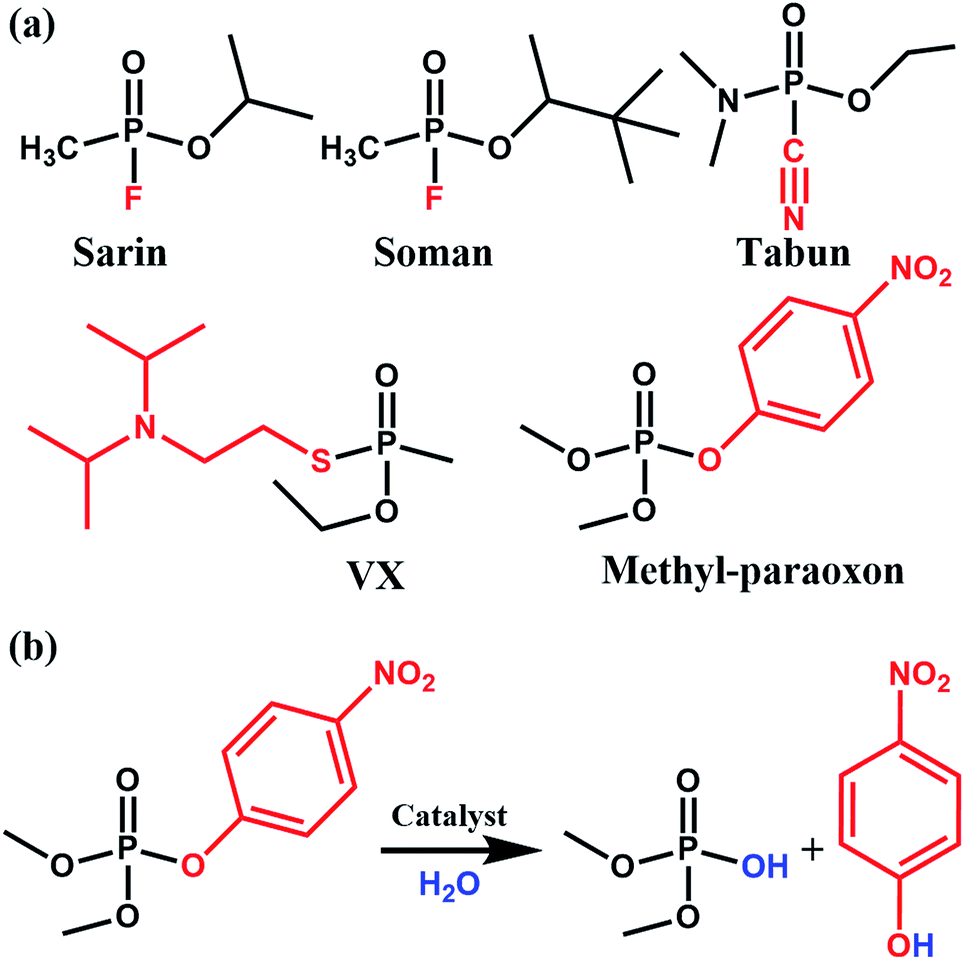 | ||
| Fig. 1 (a) Molecular structure of various nerve agents as well as the pesticide methyl-paraoxon. The fragment shown in red is the desired leaving group during hydrolysis. (b) Catalytic hydrolysis reaction for the hydrolysis of methyl-paraoxon. | ||
Other organophosphorous containing compounds, such as methyl-paraoxon, are phosphate-based pesticides. Methyl paraoxon's toxicity is due to the ease in which the P–O group of the nitrophenol can undergo transesterification under physiological conditions. Interestingly, due to the high-use of these pesticides, enzymes have evolved as a response to these pesticides. Ultimately, this has led to enzymes, both natural and synthetic, which have a high specificity9 and efficacy towards this family of nerve agents. Thus, given the importance of phosphate esters in biological systems, it is not surprising that there has been much work done in studying enzymes, proteins, and small molecules which are capable of performing phosphate-ester hydrolysis on nitrophenol-containing pesticides/nerve-agent simulants.
Transition-metal, or rather Lewis-acid, hydrolysis is ubiquitous in the field of phosphate-hydrolysis.10–16 Taking a lesson from nature, many transition-metal complexes employ hydroxide bridged dimers as catalysts.10,11,17–20 For example, the family of phosphoesterase enzymes utilize a ZnII–OH–ZnII active site which is often further supported by a bridging carboxylate.21 Given that early row transition metals are often found in biological materials due to bioavailability, researchers such as Brown have utilized coordination complexes with more Lewis-acidic centres such as LaIII and ZrIV as catalysts for phosphate-ester hydrolysis.11,14,22
Our group has also been investigating the role of Lewis-acidic centres such AlIII and ZnII as catalysts for the degradation of phosphates such as methyl-paraoxon.23–26 We observed that AlIII–porphyrin dimers24 and tetramers26 significantly outperformed their ZnII counterparts; half-lives of 10 hours were observed for the methanolysis of methyl- and phenyl-paraoxon by these AlIII-complexes. In-line with these materials, we began looking at porous-organic polymers (POPs) as potential catalysts.23,25 These POPs offered the combined advantage of homogeneous catalysis (e.g., well defined catalysts such as AlIII porphyrins23 and LaII catecholates25) and heterogeneous catalysis (e.g., insoluble catalyst that can be easily removed).
We observed two key design features in the aforementioned systems: (i) strong Lewis-acidic metals were key for fast turn over, and (ii) the use of simple synthesis, such as those used in the formation of POPs, are attractive heterogeneous catalysts. Thus, we turned our attention to another class of porous materials, namely, metal–organic frameworks (MOFs).
MOFs are porous materials formed from inorganic cationic nodes (e.g., Zr6O4(OH)412+) and anionic bridging ligands (e.g., 1,4-benzenedicarboxylate).27 In addition to our two design principles above, MOFs are ordered/crystalline materials which are needed for rapid screening and hypothesis driven research. Due to their high porosity28 as well as thermal and chemical stability,29–32 MOFs are promising materials in applications such as gas-storage,33–35 chemical separations,36–41 sensing,42,43 catalysis,44–47 and light harvesting.48–51 In keeping with our aforementioned design principle, we have shown that UiO-66 (Fig. 2), a MOF containing highly Lewis-acidic ZrIV ions, in the form of ZrIV6O4(OH)412+ clusters, which can be easily formed in high yield using a variety of different synthetic procedures,52 had a half-life of 50 minutes for the hydrolysis of methyl-paraoxon at room temperature.53 This half-life is among the fastest, non-enzymatic, half-lives reported to date. Hatton et al.54 have also demonstrated that the Lewis-acidic CrIII centre in Cr-MIL-101 is also capable of hydrolysing phosphate esters with a half-life of nearly 3 hours.
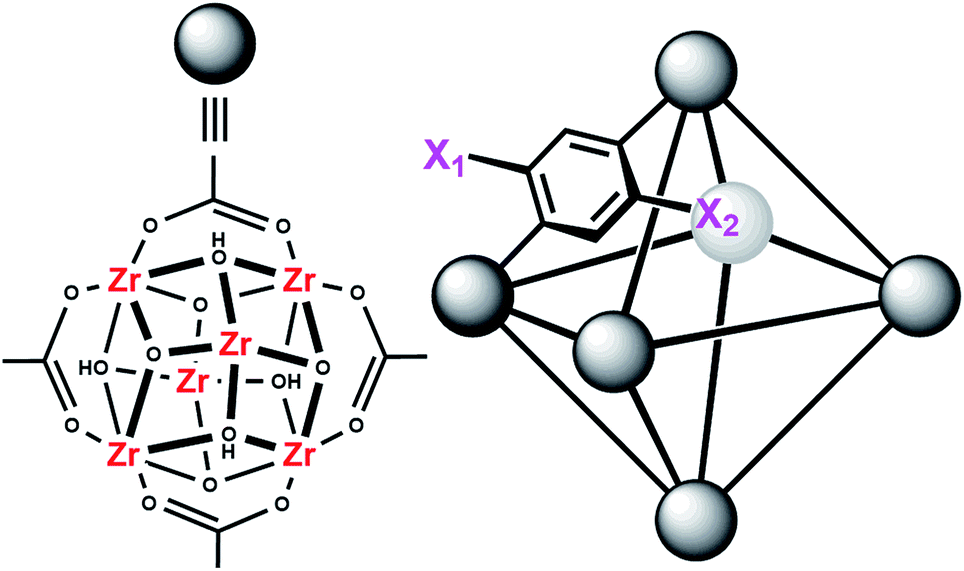 | ||
| Fig. 2 Molecular structure of the node of UiO-66 showing four of the twelve bound carboxylates (left) and the connectivity of the octahedral pore in UiO-66 (right). X1 = X2 = H for UiO-66, X1 = NO2 and X2 = H for UiO-66-NO2, X1 = X2 = OH for UiO-66-(OH)2, and X1 = NH2 and X2 = H for UiO-66-NH2. | ||
Our initial success with UiO-66 was based on the observation that enzymatic systems use bimetallic Lewis-acidic metal centres bridge by a hydroxide. Along the same line, we continue with the biomimetic approach by looking at the necessary components of the mechanism.16,21,55–59 Raushel, for example, has used a combination of experimental and computational chemistry to probe the mechanism (Fig. 3). They, and others, have shown the importance of a proximal aspartate and histidine.60 Both moieties act as a proximal base, transferring protons to and from these moieties at key portions of the catalytic cycle. In the absence of these moieties, the hydrolysis rates were diminished.
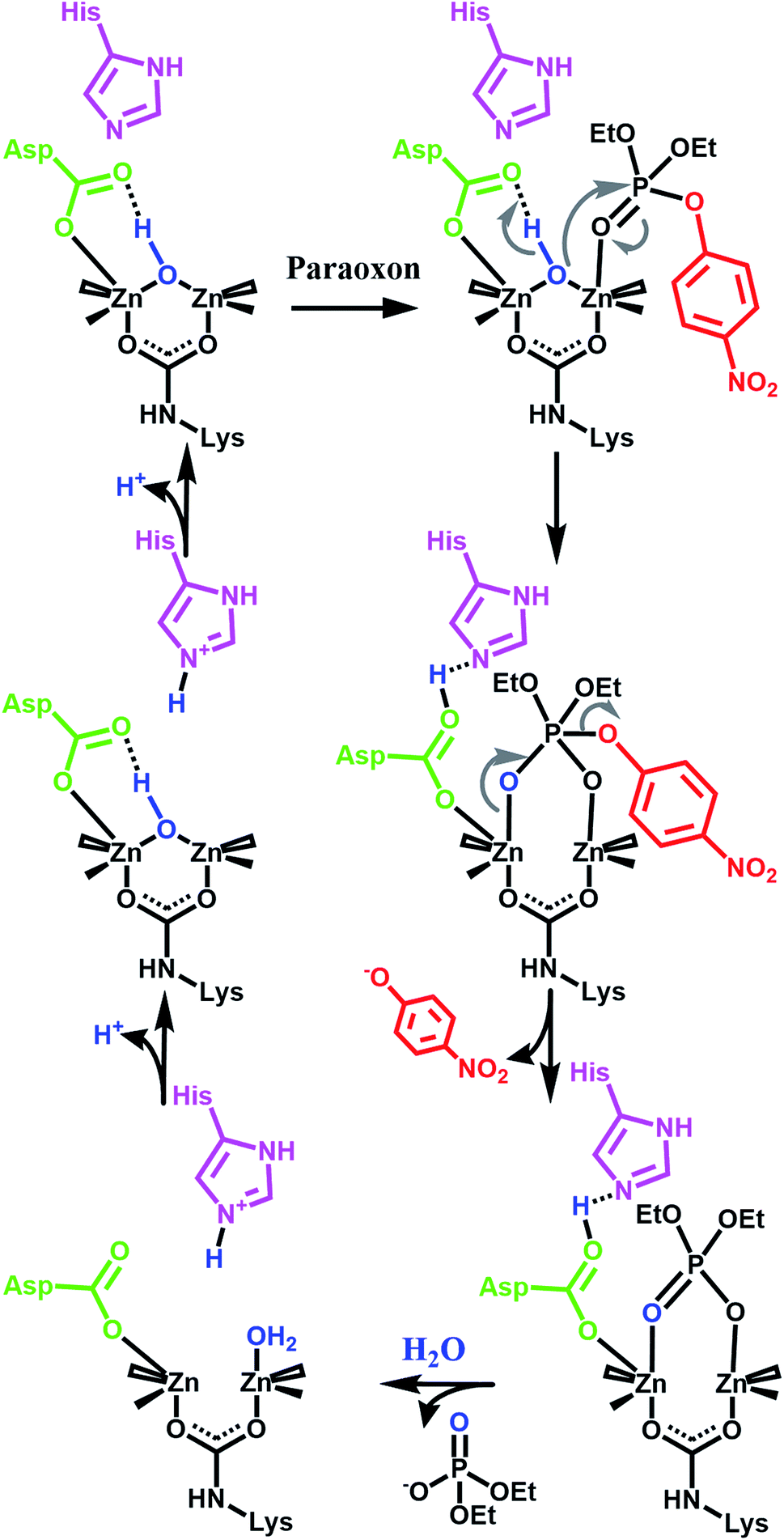 | ||
| Fig. 3 One proposed enzymatic mechanism by Raushel for the hydrolysis of paraoxon.21 Note the presence of proximal bases such as aspartate carboxylate and a histidine imidazole. | ||
With this mechanistic insights in-mind, we hypothesized that UiO-66-NH2 would be a faster catalyst than UiO-66. To that end, we investigate the role of UiO-66-NH2 for its ability to enhance the reaction rate of our catalysis. As shown below, UiO-66-NH2 is capable of decreasing the half-life by roughly 20 fold.
Experimental
All reagents were purchased from commercial sources and used without further purification. UiO-66, UiO-66-NH2, UiO-66-(OH)2, UiO-66-NO2, UiO-67, UiO-67-NMe2, UiO-67-NH2, and methyl-paraoxon were synthesized according to literature procedures (see the ESI† for powder X-ray diffractograms and N2 isotherms).52,61 UiO-67, UiO-67-NMe2, and UiO-67-NH2 were de-solvated by supercritical CO2.26,52Hydrolysis experiments were carried out at room temperature as described previously. Briefly, to a solid sample of UiO-66 (2.5 mg, 6 mol%, 0.0015 mmol; 0.045 mol% of active surface sites) in an Eppendorf tube was added an aqueous solution of N-ethyl-morpholine (0.45 M); the N-ethyl-morpholine is used as a buffer to limit pH changes. The resulting mixture was stirred for 30 minutes to finely disperse the MOF particles. To the suspension was then added methyl-paraoxon (6.2 mg, 0.025 mmol). Periodic monitoring was carried out by removing a 20 μL aliquot from the reaction mixture and diluting with an aqueous solution of N-ethyl-morpholine (10 mL, 0.45 M) prior to UV-Vis measurements (Varian Cari 5000) (Fig. 4a). Progress of the reaction was monitored by following the p-nitrophenoxide absorbance at 407 nm to avoid overlapping absorptions with other species. No spectral evidence for the p-nitrophenol was observed at this pH (10.2). All background reactions were carried out under identical reaction conditions without the MOF catalyst.
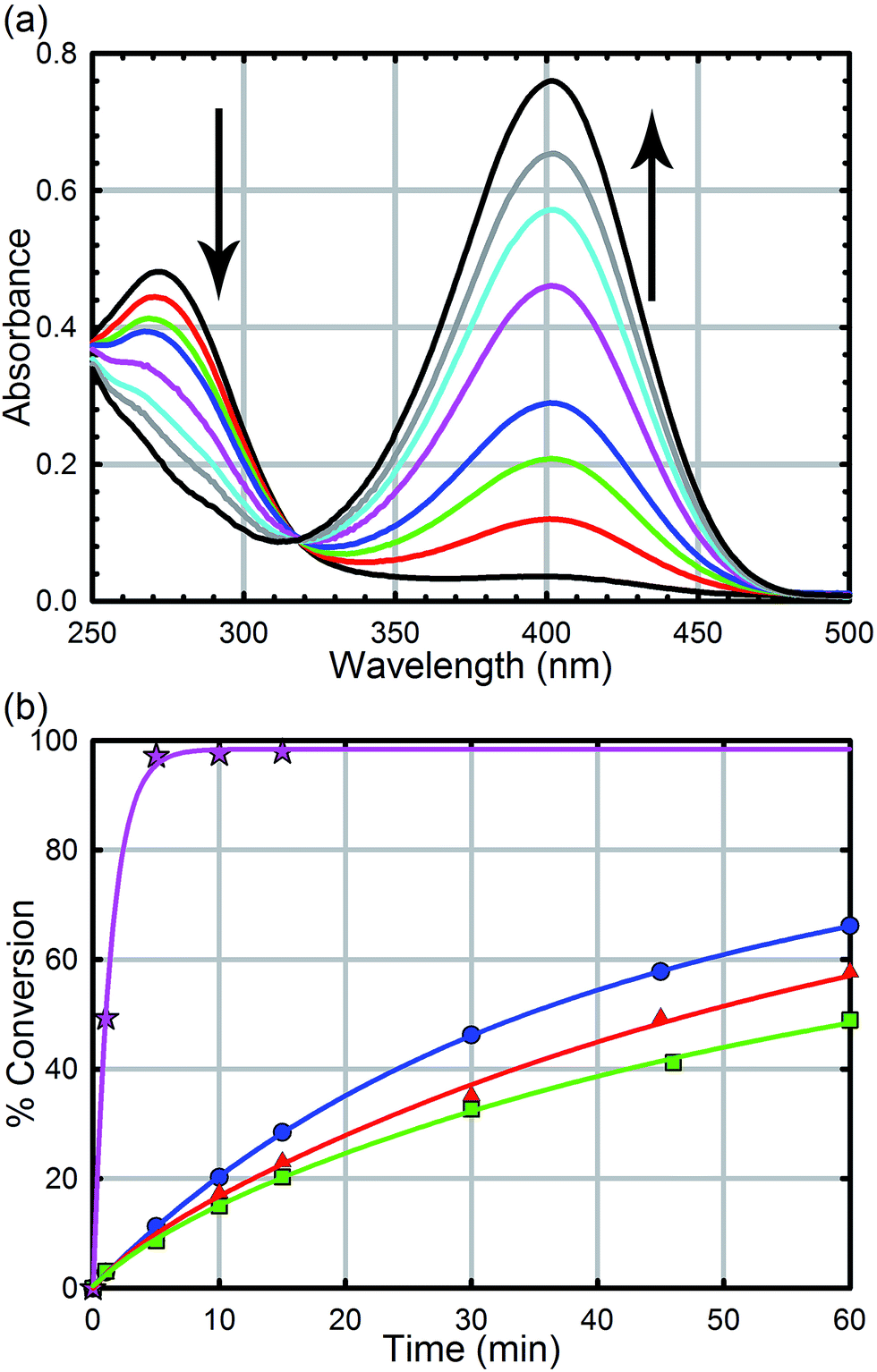 | ||
| Fig. 4 (a) UV-Vis trace of the hydrolysis of methyl-paraoxon as a function of time using UiO-66. The arrows denote the disappearance of the starting material at 275 nm and the appearance of the product at 407 nm. (b) Hydrolysis rate of UiO-66 (blue circles), UiO-66-NO2 (red triangles), UiO-66-(OH)2 (green squares), and UiO-66-NH2 (pink stars). | ||
Initial rates were determined using the method of initial rates.62 Polynomial fits of order 3–5 were used with the lowest observed correlation coefficient of 0.98.
Results and discussion
As we have previously demonstrated, the hydrolysis of methyl-paraoxon with UiO-66 is the fastest MOF-based catalysts reported to date.53 The ease in which alternative linkers can be incorporated into the framework29,31,52,63–72 makes UiO-66 an attractive platform for generating a large parameter space which can be used to easily probe a mechanism. As illustrated in Fig. 4b and Table 1, just as the enzymatic hydrolysis is enhanced by the presence of a proximal anchored base, the hydrolysis rate of UiO-66-NH2 is 20 times greater than that of UiO-66.73 In order to compare the amino moiety in our system with the aspartate and histidine moieties in the enzymatic system, we also synthesized UiO-66-NO2 and UiO-66-(OH)2. These two MOFs are capable of hydrogen bond donating and receiving, similar to UiO-66-NH2, but they cannot act as a Brønsted base like the amino moiety. As illustrated in in Fig. 4 and Table 1, there is no significant difference between the initial rates of UiO-66-(OH)2 and UiO-66-NO2; the initial rates for the catalytic hydrolysis by UiO-66-(OH)2 and UiO-66-NO2 (0.0063–0.0070 mmol s−1 respectively) are slightly slower than that of UiO-66 (0.010 mmol s−1). Based on these observations, we concluded that the amino moiety acts as a base and not as a hydrogen-bonding moiety. Furthermore, considering there is no difference between UiO-66, UiO-66-NO2 (an electron withdrawing group) and UiO-66-(OH)2 (a steric electron donating group), there is little to no electronic effect and only a slight steric effect from the linker on the rate limiting step of the hydrolysis reaction (Table 1).| MOF | Initial rate (mM s−1) | Half-life (min) | TOFall (s−1) | TOFsurfacea (s−1) |
|---|---|---|---|---|
| a 400 nm particles of UiO-66 are synthesized. Due to the aperture size of UiO-66 and the relatively larger kinetic diameter of methyl-paraoxon, only the surface sites (ca. 0.75% of the catalyst loading) are catalytically active. | ||||
| UiO-66 | 0.010 | 35 | 0.0077 | 1.0 |
| UiO-66-(OH)2 | 0.0063 | 60 | 0.0047 | 0.62 |
| UiO-66-NO2 | 0.0070 | 45 | 0.0052 | 0.70 |
| UiO-66-NH2 | 0.20 | 1 | 0.15 | 20 |
In order to further investigate the role of the amino moiety, we turned our attention to UiO-67 and its derivatives. In UiO-67 (Fig. 5), the functional groups point away from the node. Under these conditions, the amino moiety can only act as a Brønsted-base around the outer sphere of the active site.
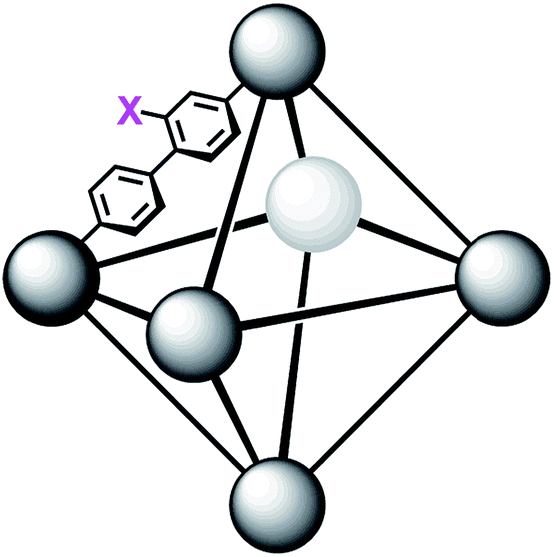 | ||
| Fig. 5 Connectivity of the octahedral pore in UiO-67 (right). X = H for UiO-67, X = NH2 for UiO-67-NH2, and X = N(CH3)2 for UiO-67-NMe2. | ||
In comparison with UiO-66, UiO-67 (Fig. 6a) is a faster catalyst. We suspect that the longer distance between nodes prevents steric crowding around neighbouring nodes allowing for a more active material.74 Nevertheless, as with UiO-66, UiO-67-NH2 and UiO-67-NMe2 are faster than UiO-67 (Fig. 6, Table 2). Due to the fast hydrolysis of UiO-67, the rate enhancement (1.2 times) is not 20 times as observed between UiO-66 and UiO-66-NH2. Due to the fact that UiO-67-NH2 and UiO-67-NMe2 show nearly identical kinetic traces (Fig. 6), we suspect that a different part of the mechanism is now limiting. To illustrate this, the catalyst loading was decreased by a factor of two (Fig. 6b). The hydrolysis rate of UiO-67 decreased by a factor of 3.2 making UiO-67-NH2 3.2 times faster than UiO-67. Furthermore, the hydrolysis rate of UiO-67-NMe2 decreased more than that of UiO-67-NH2 (Fig. 6b); these results support our hypothesis that UiO-67-NH2 and UiO-67-NMe2 are limited by another step in the catalytic cycle when compared to normal catalyst loadings.
| MOF | Initial rate (mM s−1) | Half-life (min) | TOFall (s−1) | TOFsurfacea (s−1) |
|---|---|---|---|---|
| a 400 nm particles of UiO-67 are synthesized; as with UiO-66, only the surface sites are expected to be catalytically active. | ||||
| Full catalyst loading | ||||
| UiO-67 | 0.058 | 4.5 | 0.038 | 5.1 |
| UiO-67-NMe2 | 0.078 | 2 | 0.052 | 7.0 |
| UiO-67-NH2 | 0.067 | 2 | 0.044 | 6.0 |
| Half catalyst loading | ||||
| UiO-67 | 0.018 | 15 | 0.024 | 3.2 |
| UiO-67-NMe2 | 0.049 | 7 | 0.032 | 4.3 |
| UiO-67-NH2 | 0.057 | 3.5 | 0.075 | 10 |
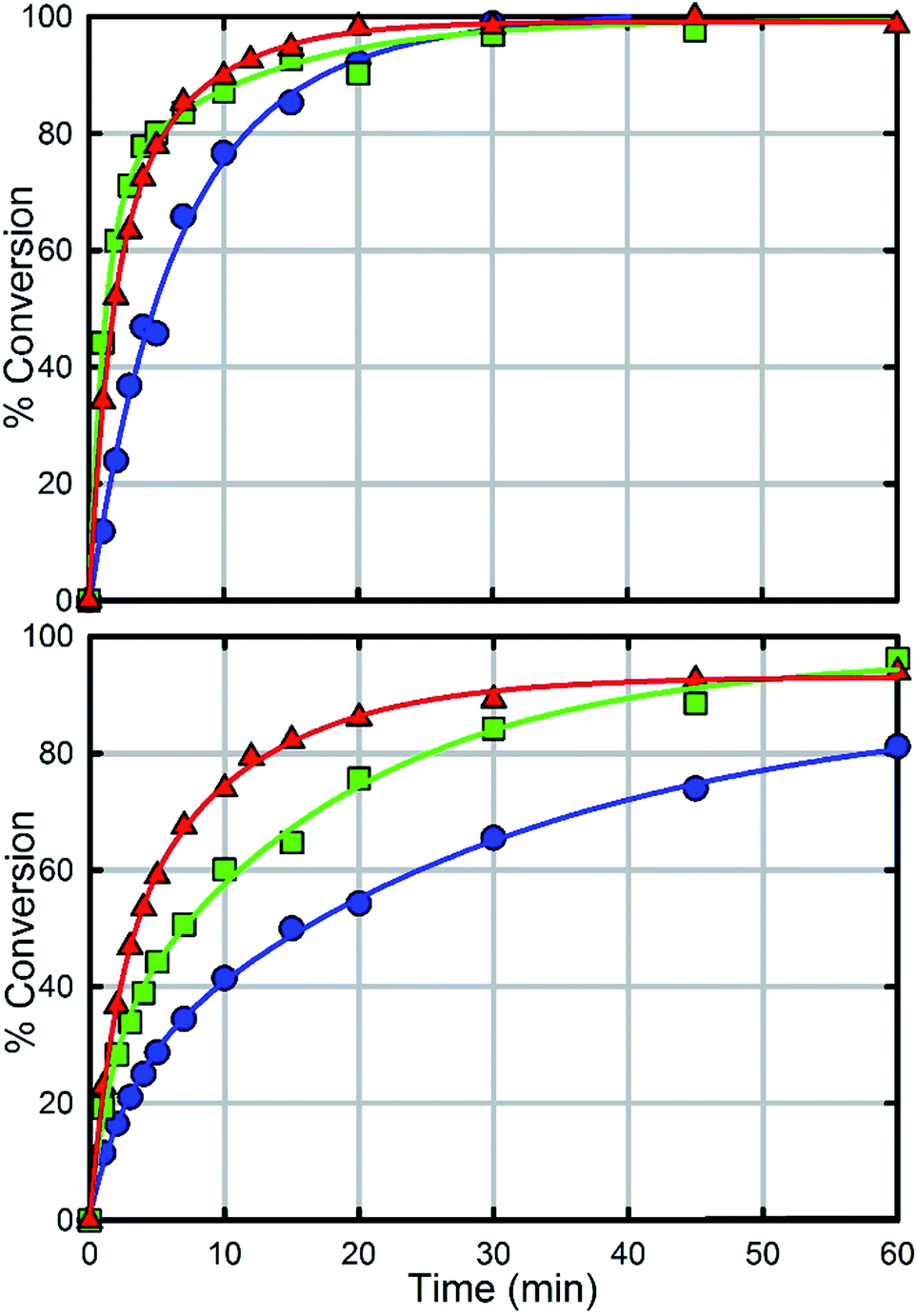 | ||
| Fig. 6 Hydrolysis rate of UiO-67 (blue), UiO-67-NH2 (red) and UiO-67-NMe2 (green) at full catalyst loading (1.5 μmole) with respect to UiO-66 (top) and at half catalyst loading (0.75 μmole) with respect to UiO-66 (bottom). | ||
Thus, given the results above, we propose that the role of amino moiety in these derivatives is to act as a Brønsted-base whereby a proton is transferred to and from the base during the catalytic cycle. This is supported by the UiO-67-NMe2 hydrolysis which is slower than the UiO-67-NH2 hydrolysis likely due to the fact that the dimethylamino moiety is a stronger base and thus it is harder to deprotonate the moiety in order to regenerate the catalyst.
Conclusions
The hydrolysis rate of six UiO-based MOFs was measured in order to probe the importance of various functionalities on the overall hydrolysis rate. We observed that only amino moieties were able to enhance the hydrolysis rate. A rate enhancement of up to 20 times was observed for UiO-66. Due to a careful comparison between UiO-66 and UiO-67 derivatives, we propose that the role of the amino moiety is to act like a Brønsted base, and subsequently a Brønsted acid during the catalytic cycle. With half-life of about 1 minute, UiO-66-NH2, is the fastest methyl-paraoxon hydrolysis MOF-based catalysts reported to date. With these results in hand, we are currently testing the generality of these design rules on other MOF topologies.Acknowledgements
O.K.F. and J.T.H. gratefully acknowledges DTRA for financial support (grant HDTRA-1-10-0023). The authors would like to thank Nicolaas Vermeulen for his assistance in the purification of methy-paraoxon.Notes and references
- S. C. L. Kamerlin, P. K. Sharma, R. B. Prasad and A. Warshel, Q. Rev. Biophys., 2013, 46, 1 CrossRef CAS PubMed.
- M. S. Razzaque, Clin. Sci., 2011, 120, 91 CrossRef CAS PubMed.
- A. Anzueto, R. A. deLemos, J. Seidenfeld, G. Moore, H. Hamil, D. Johnson and S. G. Jenkinson, Fundam. Appl. Toxicol., 1990, 14, 676 CrossRef CAS.
- F. R. Sidell and J. Borak, Ann. Emerg. Med., 1992, 21, 865 CrossRef CAS.
- Y.-C. Yang, Acc. Chem. Res., 1999, 32, 109 CrossRef CAS.
- M. Enserink, Science, 2013, 341, 1050 CrossRef CAS PubMed.
- R. Pita and J. Domingo, Toxics, 2014, 2, 391 CrossRef PubMed.
- H. P. Benschop and L. P. A. De Jong, Acc. Chem. Res., 1988, 21, 368 CrossRef CAS.
- For phosphate-based nerve agents, specificiy may be ambiguous. Specificiy may refer to the bond that is being broken (e.g., Me, F, or iso-propyl group in Sarin). Alternatively, specificity may refer to the specific phosphate targeted (i.e., some enzymes are only efficient at hydrolyzing one specific nerve agent and not all nerve agents equilly). Lastly, given the stereogenic phosphorous centre, specificiy may refer to the specific enantiomer (e.g., Sarin), or pairs of enantiomers (e.g., Soman).
- A. A. Neverov, C. T. Liu, S. E. Bunn, D. Edwards, C. J. White, S. A. Melnychuk and R. S. Brown, J. Am. Chem. Soc., 2008, 130, 6639 CrossRef CAS PubMed.
- J. S. W. Tsang, A. A. Neverov and R. S. Brown, Org. Biomol. Chem., 2004, 2, 3457 CAS.
- S. A. Melnychuk, A. A. Neverov and R. S. Brown, Angew. Chem., Int. Ed., 2006, 45, 1767 CrossRef PubMed.
- F. Aguilar-Pérez, P. Gómez-Tagle, E. Collado-Fregoso and A. K. Yatsimirsky, Inorg. Chem., 2006, 45, 9502 CrossRef PubMed.
- T. Liu, A. A. Neverov, J. S. W. Tsang and R. S. Brown, Org. Biomol. Chem., 2005, 3, 1525 CAS.
- E. Stulz, H. B. Bürgi and C. Leumann, Chem.–Eur. J., 2000, 6, 523 CrossRef CAS.
- E. Kimura, Curr. Opin. Chem. Biol., 2000, 4, 207 CrossRef CAS.
- D. R. Edwards, C. T. Liu, G. E. Garrett, A. A. Neverov and R. S. Brown, J. Am. Chem. Soc., 2009, 131, 13738 CrossRef CAS PubMed.
- A. Tamilselvi and G. Mugesh, Chem.–Eur. J., 2010, 16, 8878 CrossRef CAS PubMed.
- K. L. Klinkel, L. A. Kiemele, D. L. Gin and J. R. Hagadorn, J. Mol. Catal. A: Chem., 2007, 267, 173 CrossRef CAS PubMed.
- K. L. Klinkel, L. A. Kiemele, D. L. Gin and J. R. Hagadorn, Chem. Commun., 2006, 2919 RSC.
- S. D. Aubert, Y. Li and F. M. Raushel, Biochemistry, 2004, 43, 5707 CrossRef CAS PubMed.
- T. J. Bandosz, M. Laskoski, J. Mahle, G. Mogilevsky, G. W. Peterson, J. A. Rossin and G. W. Wagner, J. Phys. Chem. C, 2012, 116, 11606 CAS.
- R. K. Totten, Y.-S. Kim, M. H. Weston, O. K. Farha, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2013, 135, 11720 CrossRef CAS PubMed.
- R. K. Totten, P. Ryan, B. Kang, S. J. Lee, L. J. Broadbelt, R. Q. Snurr, J. T. Hupp and S. T. Nguyen, Chem. Commun., 2012, 48, 4178 RSC.
- R. K. Totten, M. H. Weston, J. K. Park, O. K. Farha, J. T. Hupp and S. T. Nguyen, ACS Catal., 2013, 3, 1454 CrossRef CAS.
- B. Kang, J. W. Kurutz, K.-T. Youm, R. K. Totten, J. T. Hupp and S. T. Nguyen, Chem. Sci., 2012, 3, 1938 RSC.
- H.-C. J. Zhou and S. Kitagawa, Chem. Soc. Rev., 2014, 5415 RSC.
- Y. Peng, G. Srinivas, C. E. Wilmer, I. Eryazici, R. Q. Snurr, J. T. Hupp, T. Yildirim and O. K. Farha, Chem. Commun., 2013, 2992 RSC.
- J. E. Mondloch, M. J. Katz, N. Planas, D. Semrouni, L. Gagliardi, J. T. Hupp and O. K. Farha, Chem. Commun., 2014, 50, 8944 RSC.
- D. Feng, Z.-Y. Gu, J.-R. Li, H.-L. Jiang, Z. Wei and H.-C. Zhou, Angew. Chem., Int. Ed., 2012, 51, 10307 CrossRef CAS PubMed.
- Y. Huang, W. Qin, Z. Li and Y. Li, Dalton Trans., 2012, 41, 9283 RSC.
- M. Kandiah, M. H. Nilsen, S. Usseglio, S. Jakobsen, U. Olsbye, M. Tilset, C. Larabi, E. A. Quadrelli, F. Bonino and K. P. Lillerud, Chem. Mater., 2010, 22, 6632 CrossRef CAS.
- J. B. DeCoste, M. H. Weston, P. E. Fuller, T. M. Tovar, G. W. Peterson, M. D. LeVan and O. K. Farha, Angew. Chem., Int. Ed., 2014, 53, 14092 CrossRef CAS PubMed.
- S. Ma and H.-C. Zhou, Chem. Commun., 2010, 46, 44 RSC.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030 CAS.
- Y.-S. Bae, A. M. Spokoyny, O. K. Farha, R. Q. Snurr, J. T. Hupp and C. A. Mirkin, Chem. Commun., 2010, 46, 3478 RSC.
- Y.-S. Bae, B. G. Hauser, Y. J. Colón, J. T. Hupp, O. K. Farha and R. Q. Snurr, Microporous Mesoporous Mater., 2013, 169, 176 CrossRef CAS PubMed.
- C. Y. Lee, Y.-S. Bae, N. C. Jeong, O. K. Farha, A. A. Sarjeant, C. L. Stern, P. Nickias, R. Q. Snurr, J. T. Hupp and S. T. Nguyen, J. Am. Chem. Soc., 2011, 133, 5228 CrossRef CAS PubMed.
- J.-R. Li, R. J. Kuppler and H.-C. Zhou, Chem. Soc. Rev., 2009, 38, 1477 RSC.
- J.-R. Li, J. Sculley and H.-C. Zhou, Chem. Rev., 2012, 112, 869 CrossRef CAS PubMed.
- Z.-Y. Gu, C.-X. Yang, N. Chang and X.-P. Yan, Acc. Chem. Res., 2012, 45, 734 CrossRef CAS PubMed.
- Z. Hu, B. J. Deibert and J. Li, Chem. Soc. Rev., 2014, 43, 5815 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105 CrossRef CAS PubMed.
- Q. Han, L. Zhang, C. He, J. Niu and C. Duan, Inorg. Chem., 2012, 51, 5118 CrossRef CAS PubMed.
- J. B. DeCoste and G. W. Peterson, Chem. Rev., 2014, 114, 5695 CrossRef CAS PubMed.
- F.-J. Ma, S.-X. Liu, C.-Y. Sun, D.-D. Liang, G.-J. Ren, F. Wei, Y.-G. Chen and Z.-M. Su, J. Am. Chem. Soc., 2011, 133, 4178 CrossRef CAS PubMed.
- H. G. T. Nguyen, M. H. Weston, O. K. Farha, J. T. Hupp and S. T. Nguyen, CrystEngComm, 2012, 14, 4115 RSC.
- C. Y. Lee, O. K. Farha, B. J. Hong, A. A. Sarjeant, S. T. Nguyen and J. T. Hupp, J. Am. Chem. Soc., 2011, 133, 15858 CrossRef CAS PubMed.
- J.-L. Wang, C. Wang and W. Lin, ACS Catal., 2012, 2, 2630 CrossRef CAS.
- M. C. So, S. Jin, H.-J. Son, G. P. Wiederrecht, O. K. Farha and J. T. Hupp, J. Am. Chem. Soc., 2013, 135, 15698 CrossRef CAS PubMed.
- C. A. Kent, D. Liu, L. Ma, J. M. Papanikolas, T. J. Meyer and W. Lin, J. Am. Chem. Soc., 2011, 133, 12940 CrossRef CAS PubMed.
- M. J. Katz, Z. J. Brown, Y. J. Colón, P. W. Siu, K. A. Scheidt, R. Q. Snurr, J. T. Hupp and O. K. Farha, Chem. Commun., 2013, 49, 9449 RSC.
- M. J. Katz, J. E. Mondloch, R. K. Totten, J. K. Park, S. T. Nguyen, O. K. Farha and J. T. Hupp, Angew. Chem., Int. Ed., 2013, 497 Search PubMed.
- S. Wang, L. Bromberg, H. Schreuder-Gibson and T. A. Hatton, ACS Appl. Mater. Interfaces, 2013, 5, 1269 CAS.
- V. E. Lewis, W. J. Donarski, J. R. Wild and F. M. Raushel, Biochemistry, 1988, 27, 1591 CrossRef CAS.
- K.-Y. Wong and J. Gao, Biochemistry, 2007, 46, 13352 CrossRef CAS PubMed.
- J. G. Zalatan and D. Herschlag, J. Am. Chem. Soc., 2006, 128, 1293 CrossRef CAS PubMed.
- P. Imhof, S. Fischer, R. Krämer and J. C. Smith, J. Mol. Struct.: THEOCHEM, 2005, 713, 1 CrossRef CAS PubMed.
- S.-L. Chen, W.-H. Fang and F. Himo, J. Phys. Chem. B, 2007, 111, 1253 CrossRef CAS PubMed.
- A. J. Kirby and W. P. Jencks, J. Am. Chem. Soc., 1965, 87, 3209 CrossRef CAS.
- V. K. Ol'khovik, A. A. Pap, V. A. Vasilevskii, N. A. Galinovskii and S. N. Tereshko, Russ. J. Org. Chem., 2008, 44, 1172 CrossRef.
- J. Casado, M. A. Lopez-Quintela and F. M. Lorenzo-Barral, J. Chem. Educ., 1986, 63, 450 CrossRef CAS.
- G. C. Shearer, S. M. Chavan, J. Ethiraj, J. G. Vitillo, S. Svelle, U. Olsbye, C. Lamberti, S. Bordiga and K. P. Lillerud, Chem. Mater., 2014, 26, 4068 CrossRef CAS.
- H. R. Abid, H. Tian, H.-M. Ang, M. O. Tade, C. E. Buckley and S. Wang, Chem. Eng. J., 2012, 187, 415 CrossRef CAS PubMed.
- S. Chavan, J. G. Vitillo, D. Gianolio, O. Zavorotynska, B. Civalleri, S. Jakobsen, M. H. Nilsen, L. Valenzano, C. Lamberti, K. P. Lillerud and S. Bordiga, Phys. Chem. Chem. Phys., 2012, 14, 1614 RSC.
- M. J. Cliffe, W. Wan, X. Zou, P. a. Chater, A. K. Kleppe, M. G. Tucker, H. Wilhelm, N. P. Funnell, F.-X. Coudert and A. L. Goodwin, Nat. Commun., 2014, 5, 4176 CAS.
- S. J. Garibay and S. M. Cohen, Chem. Commun., 2010, 46, 7700 RSC.
- D. I. Kolokolov, A. G. Stepanov, V. Guillerm, C. Serre, B. Frick and H. Jobic, J. Phys. Chem. C, 2012, 116, 12131 CAS.
- S. Øien, D. Wragg, H. Reinsch, S. Svelle, S. Bordiga, C. Lamberti and K. P. Lillerud, Cryst. Growth Des., 2014, 14, 5370 Search PubMed.
- A. Schaate, P. Roy, A. Godt, J. Lippke, F. Waltz, M. Wiebcke and P. Behrens, Chem.–Eur. J., 2011, 17, 6643 CrossRef CAS PubMed.
- L. Valenzano, B. Civalleri, S. Chavan, S. Bordiga, M. H. Nilsen, S. Jakobsen, K. P. Lillerud and C. Lamberti, Chem. Mater., 2011, 23, 1700 CrossRef CAS.
- J. H. Cavka, S. Jakobsen, U. Olsbye, N. Guillou, C. Lamberti, S. Bordiga and K. P. Lillerud, J. Am. Chem. Soc., 2008, 130, 13850 CrossRef PubMed.
- Filtration of UiO-66-NH2 after 1.8 min of catalysis (Fig. S5 in the ESI†) demonstrates that the hydrolysis is heterogeneous. Furthermore, no leached zirconium is observed via ICP-AES analysis of the remaining solution.
- Scanning-Electron Micrographs (SEMs) of UiO-66 and UiO-67 demonstrate that the particles sizes of these MOFs are similar.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03613a |
| This journal is © The Royal Society of Chemistry 2015 |