
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Graphene–DNAzyme junctions: a platform for direct metal ion detection with ultrahigh sensitivity†
Li
Gao‡
a,
Le-Le
Li‡
b,
Xiaolong
Wang
a,
Peiwen
Wu
b,
Yang
Cao
a,
Bo
Liang
c,
Xin
Li
c,
Yuanwei
Lin
a,
Yi
Lu
*b and
Xuefeng
Guo
*ad
aCenter for Nanochemistry, Beijing National Laboratory for Molecular Sciences, State Key Laboratory for Structural Chemistry of Unstable and Stable Species, College of Chemistry and Molecular Engineering, Peking University, Beijing 100871, P. R. China. E-mail: guoxf@pku.edu.cn
bDepartment of Chemistry, University of Illinois at Urbana-Champaign, Urbana, Illinois 61801, USA. E-mail: yi-lu@illinois.edu
cAdesso Advanced Materials Wuxi Co., Ltd., Huihong Industrial Park, 18 Xishi Road, New District, Wuxi, Jiangsu Province 214000, P. R. China
dDepartment of Materials Science and Engineering, College of Engineering, Peking University, Beijing 100871, P. R. China
First published on 5th February 2015
Abstract
Many metal ions are present in biology and in the human body in trace amounts. Despite numerous efforts, metal sensors with ultrahigh sensitivity (<a few picomolar) are rarely achieved. Here, we describe a platform method that integrates a Cu2+-dependent DNAzyme into graphene–molecule junctions and its application for direct detection of paramagnetic Cu2+ with femtomolar sensitivity and high selectivity. Since DNAzymes specific for other metal ions can be obtained through in vitro selection, the method demonstrated here can be applied to the detection of a broad range of other metal ions.
Introduction
The ability to detect chemical and biological species at ultra-low concentrations is important in many areas, ranging from the diagnosis of life-threatening diseases to the detection of biological agents in warfare or terrorist attacks.1–7 Here, we describe a method to achieve the direct detection of paramagnetic Cu2+ with femtomolar sensitivity and high selectivity by using single-molecule graphene–DNAzyme junctions (Fig. 1). Copper ion is an essential metal ion for many biological functions. Recent studies have shown that bioavailable copper ion in organisms is relatively low. For example, the concentration of free copper ions is said to be ∼10−21 and ∼10−18 M in Escherichia coli and yeast, respectively.8,9 This low level of free copper ions is crucial, as increased copper levels are highly toxic, which can cause gastrointestinal disturbance and liver or kidney damage.10,11 Therefore, direct copper ion detectors with high sensitivity and selectivity are very useful in understanding its roles in biology. Towards this goal, many fluorescence small-organic-molecule-based Cu2+ sensors have been developed based on the changes in their fluorescence intensity upon binding to Cu2+ (ref. 12 and references therein). Most of these sensors, however, require the incorporation of a fluorophore into the metal recognition site, using an organic solvent, and cannot reach the sensitivity required for detection. Only a few such sensors demonstrated nanomolar sensitivity with high selectivity and without using an organic solvent.7,13–22 An efficient way to overcome these problems is to develop nanomaterial-based electrical biosensors that allow ultrasensitive and direct electrical detection of target analytes in a nondestructive manner.23,24 In particular, we are interested in using nanoscale junctions bridged by molecules, such as catalytic DNA or DNAzymes, to build metal sensing platforms offering unique advantages, such as low cost, portability, ultra-high sensitivity, and excellent selectivity.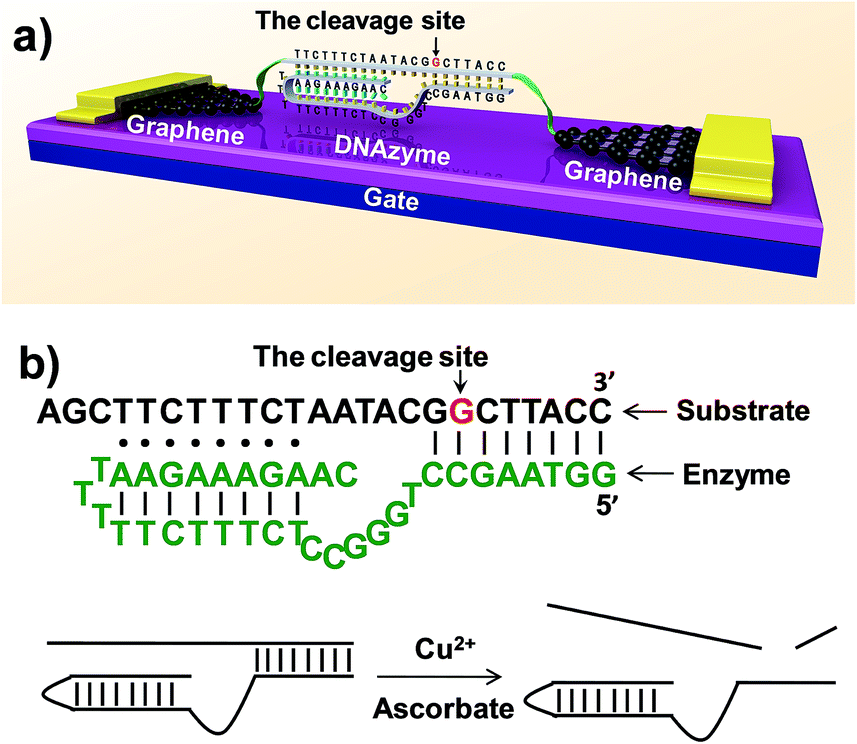 | ||
| Fig. 1 (a) Schematic representation of graphene–DNAzyme junctions. (b) The structure of the Cu2+-sensitive DNAzyme and corresponding catalytic activity. The DNA substrate has been functionalized by amines on both ends for molecular connection (see the ESI†). The cleavage site is indicated by an arrow. | ||
DNAzymes are DNA-based biocatalysts that have the ability to perform many chemical and biological reactions.25–27 Most of these reactions require specific metal ions as cofactors. As a result, a number of highly effective fluorescence, colorimetric, and electrochemical sensors based on DNAzymes have been developed for detecting different metal ions,2 such as Pb2+,28–30 UO22+,22 Hg2+,31,32 Cu2+,21 and others.33 Compared to proteins or RNA molecules, DNAzymes are an excellent choice for metal ion detection because of their relatively low cost and high stability towards hydrolysis. In addition, the DNAzymes can still be active even after many cycles of denaturation/renaturation. These properties are ideally suited for electrochemical device engineering and manufacturing. Despite these advantages, DNAzyme-based sensors for ultrasensitive detection of metal ions (less than a few nanomolar) have rarely been achieved. In this study, we aim to demonstrate a new platform for ultrasensitive detection of metal ions by integrating a Cu2+-dependent DNA-cleaving DNAzyme into graphene–molecule junctions (Fig. 1). On the basis of the original DNAzyme sequences,34–36 we designed a Cu2+ electrical sensor consisting of a DNA substrate strand with amines on both ends for the connection to the graphene–molecule junctions, and an enzyme strand that can hybridize to the substrate strand through two base-pairing regions (Fig. 1). The 5′-portion of the enzyme binds the substrate via Watson–Crick base pairs and the 3′-region through the formation of a DNA triplex. Initially, the complex is conductive through π–π stacking.37 In the presence of Cu2+, the substrate is cut at the cleavage site (the deoxyguanosine shown in red and indicated by an arrow in Fig. 1). Because the melting temperatures of the two cleaved fragments are lower than room temperature, the fragments are released (Fig. S1†), leading to the breakage of the junctions, and thus a decrease in device conductance. In addition to employing highly selective DNAzymes, a unique feature of our design is the use of graphene–molecule junctions that consist of one or a small collection of molecules as conductive elements.38,39 This combination can lead to ultrasensitive functional electronic devices and new classes of chemo/biosensors with single-molecule sensitivity.40–49
Results and discussion
The graphene–DNAzyme devices were built using a dash-line lithographic (DLL) method described in detail elsewhere.39 The key feature of this DLL technique is the ability to produce nanogapped graphene point contact arrays that can be functionalized by carboxylic acid on each side. These point contacts react with conductive molecules derivatized with amines to form molecular devices in high yields. In addition to this feature, another important advantage of this technique is that the contacts made by covalent amide bond formation are robust and thus can tolerate various kinds of chemical treatments. In conjunction with the electrical properties of graphene electrodes, the ease of device fabrication and the device stability place the graphene–molecule junctions as a promising testbed for molecular electronics.50Under optimized conditions, the maximum connection yield for the DNAzyme molecules was found to be ∼27%, which corresponds to the cutting yield of ∼36%.51 On the basis of these data, the analysis of the number of junctions that contribute to the charge transport, using the binomial distribution, demonstrates that in most cases only one or two junctions contribute to the charge transport of the devices.52Fig. 2a shows the comparison of the I–V curves of a representative DNAzyme-reconnected device before and after cutting. In brief, the black curve shows the S–D current (ID) plotted against the gate voltage (VG) at constant S–D bias voltage (VD = −50 mV) before cutting. The red curve, taken after cutting, shows no conductance down to the noise limit of the measurement (≤100 fA) due to the nanogaps. After molecular connection, we observed the recovery of the original property, albeit at reduced current values (black trace in Fig. 2b). These observations are consistent with our previous cases.39 Interestingly, upon addition of 0.5 nM Cu2+, in the presence of 50 μM ascorbate in HEPES buffer (25 mM, pH 7.0, 750 mM NaCl), the device conductance decreased down to zero (red trace in Fig. 2b) (27 out of 29 devices tested). This is attributable to a Cu2+-catalyzed cleavage of the substrate strand, resulting in a gap between the graphene–molecule junction. We found that the presence of ascorbate is necessary to significantly enhance the reaction rate (Fig. S2†), similar to those observed previously.34–36 Such an enhancement has been ascribed to ascorbate reduction of Cu2+ to form Cu+, which subsequently reacted with oxygen to accelerate the oxidative cleavage of DNA.21,34–36
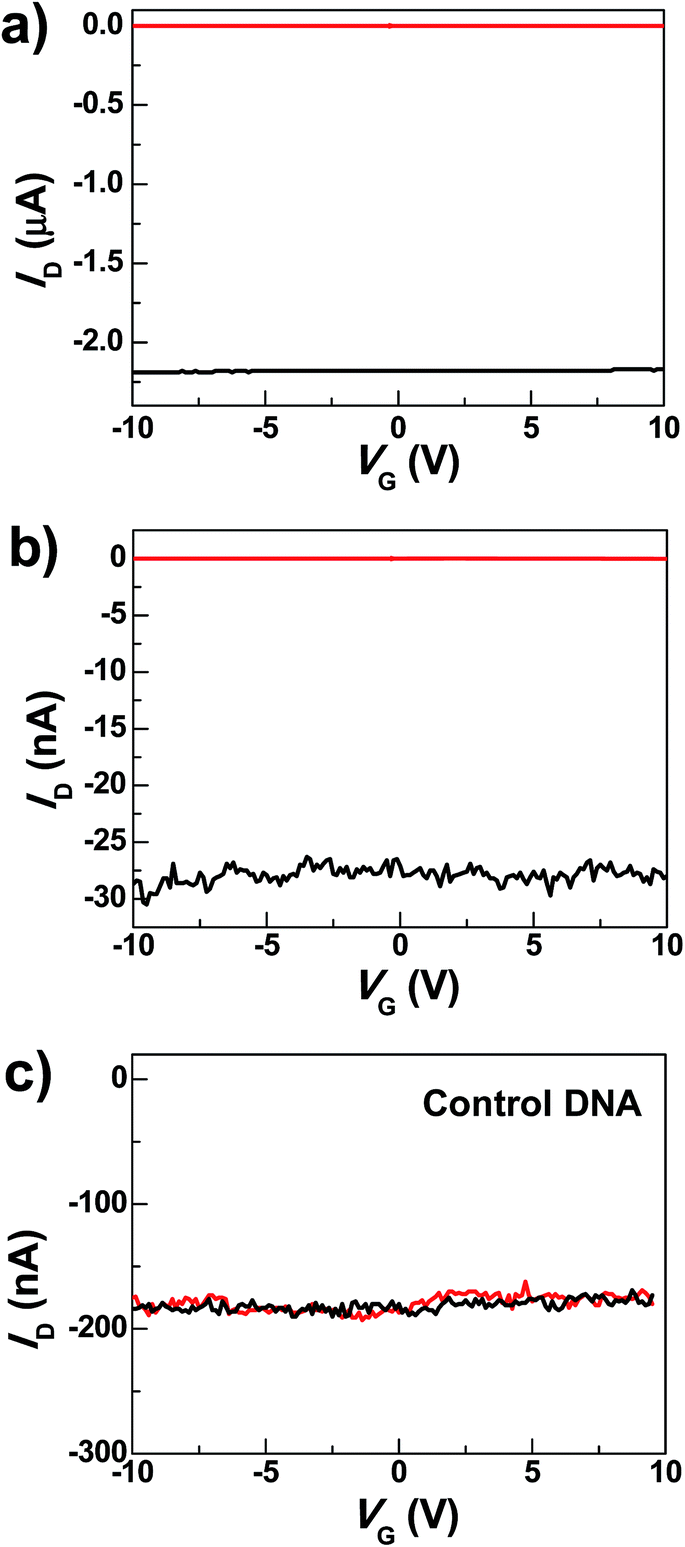 | ||
| Fig. 2 (a) I–V curves of a graphene device before (black) and after (red) cutting. (b) Device characteristics of a representative DNAzyme-reconnected device after DNA connection (black) and further Cu2+ treatments (0.5 nM, 5 min) in the presence of 50 μM ascorbate (red). (c) I–V curves of a graphene device reconnected by control DNA, after DNA connection (black) and after further Cu2+ treatment for 5 min (red) (0.5 nM with 50 μM ascorbate). | ||
To eliminate potential artifacts due to the addition of an electrolyte solution, a control experiment was conducted using 50 μM ascorbate solution without Cu2+ and no obvious conductance changes were observed under the same conditions (Fig. S3†). In addition, to confirm that the observed signal is indeed due to the presence of the Cu2+-specific DNAzyme, we used a control DNA without the Cu2+-dependent catalytic cleavage site to connect the graphene gaps (see the ESI†). In the presence of 0.5 nM Cu2+, no obvious change in conductance was observed (Fig. 2c). These results highlight the unique role of specific DNAzyme sequences in copper detection.
To test the sensitivity of these graphene–DNAzyme junctions, we investigated the responses of newly-prepared DNAzyme-rejoined devices to Cu2+ in different concentrations (0.5 nM, 0.5 pM, 0.05 pM, 5 fM, and 0.5 fM) (Fig. 3). Even though the ID values vary depending on the device fabrications, the reaction time is not affected, because it is characteristic of the Cu2+-induced cleavage of the DNAzyme. As shown in Fig. 3e, the reaction time is longer when the Cu2+ concentration is lower. For example, we observed the device breakage in less than 5 min for 0.5 nM Cu2+ compared to ∼60 min for 0.5 fM Cu2+. The detailed dynamics of the cleavage process were measured by monitoring the current change ratios as a function of time at different concentrations (Fig. 3f). We found that the diffusion and binding of metal ions are concentration-dependent while the rate of the breakage is similar, completed within ∼240 s. The time-dependent sensing behaviors can be explained as follows. For a chemical reaction nA + mB → pP + qQ, the reaction rate obeys the following kinetic formula: r = KTCACB, where K is the rate constant and C is the concentration of each reactant. Since our device consists of only one or a few DNA molecules spanning the nanogaps, the DNA concentration can be considered constant for this reaction. Furthermore, the rate of the binding reaction between DNA and metal ions is directly proportional to the concentration of the metal ions although the actual diffusion rate doesn't change. If the concentration of Cu2+ is lower, r is smaller, therefore resulting in the longer reaction time. Conversely, the fact that the breakage process after binding didn't show concentration dependence demonstrates the single-event sensitivity, which is of crucial importance to future single-molecule biodetection. The current increase we observed before DNA cleavage is attributable to the rigidification of DNA conformation during the initial metal binding, improving π–π stacking between base pairs and thus increasing the DNA conductivity.47,49 After the Cu2+ binding and conformational change, the Cu2+-promoted DNAzyme cleavage resulted in gaps between the two electrodes and thus the gradual decrease of the current down to zero. Remarkably, the Cu2+ in all concentrations investigated resulted in the complete breakdown of the drain current, even at 0.5 fM, although the conductance values varied from device to device. This detection limit is significantly lower than those of previously reported Cu2+ sensors, such as the lateral flow nucleic acid biosensors (10 nM),53 phosphorescence sensor (35 nM),14 optical chemosensors (10 nM),54 ratiometric fluorescence sensors (3 μM),20 and DNAzyme catalytic beacon sensors, which represent one of the most sensitive turn-on Cu2+ sensors (0.6 nM).21,55 The realization of atomic level precision in the cutting procedure and precise control of the molecular conformation on the substrate within the graphene gaps and the contact configuration are challenges for future studies to overcome.
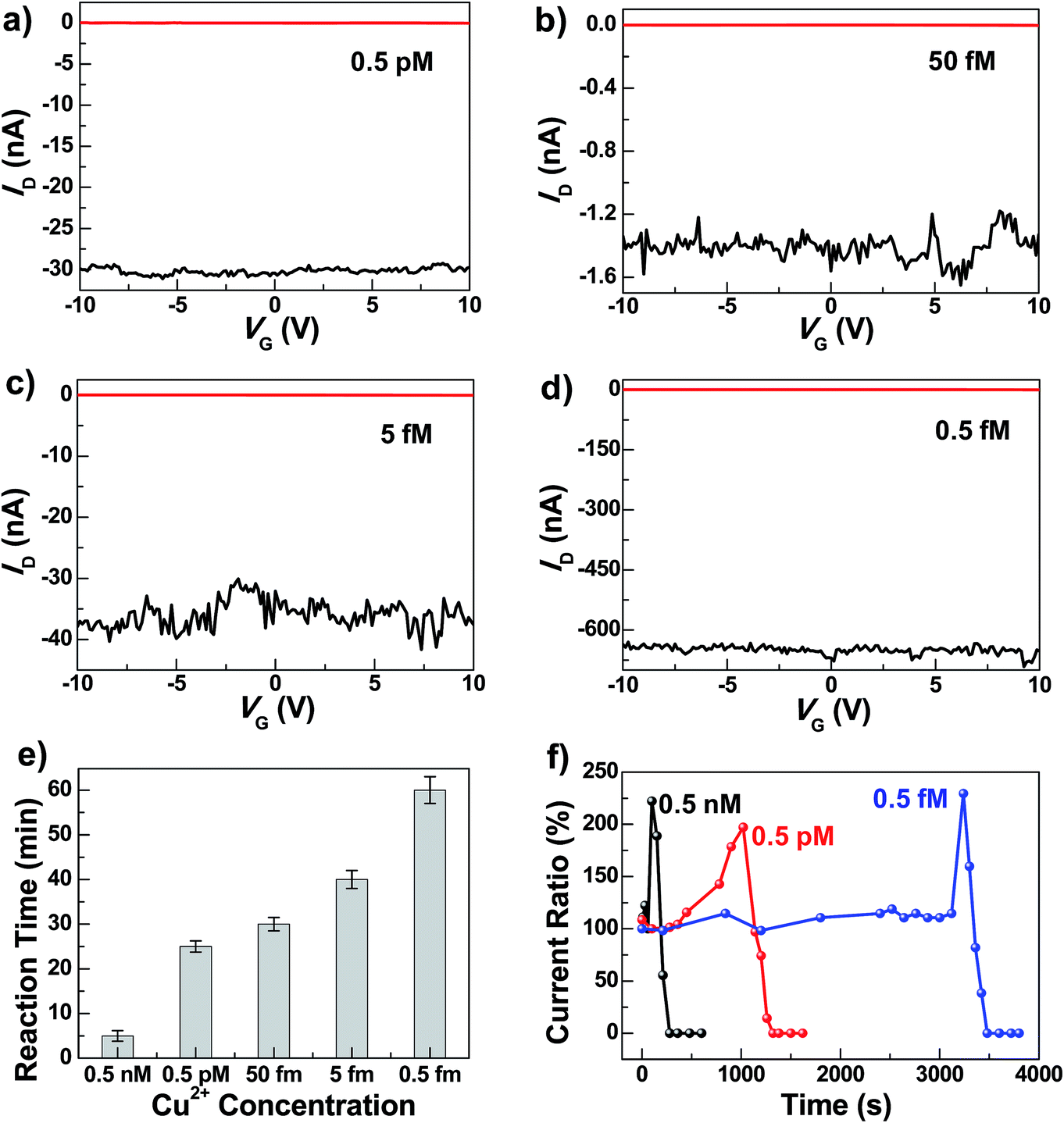 | ||
| Fig. 3 (a–d) Device characteristics of different graphene–DNAzyme junctions after DNA connection (black) and further Cu2+ treatments of different concentrations in the presence of 50 μM ascorbate (red) (a: 0.5 pM; b: 50 fM; c: 5 fM; d: 0.5 fM). (e) Statistical reaction times for the complete Cu2+ catalytic reactions at different concentrations. (f) Concentration-dependent dynamics of the Cu2+ catalytic reactions. All the measurements were performed at VD = −50 mV. | ||
Beside high sensitivity, high selectivity is also crucial for sensing. To evaluate the selectivity of DNAzyme-bridged graphene devices, we measured the conductance changes of freshly prepared working devices after adding Pb2+ (0.5 nM), Zn2+ (0.5 nM), Mg2+ (0.5 nM), Ca2+ (0.5 nM), Fe2+ (0.5 nM), Fe3+ (0.5 nM), Ni2+ (0.5 nM), K+ (5 mM), Na+ (135 mM) and Al3+ (60 nM) for 5 min under the same conditions. As illustrated in Fig. 4 and S4,† the responses of the devices to these metal ions were essentially unchanged in the presence or absence of these metal ions. In contrast, further treatment of the above systems with 0.5 nM Cu2+ in the presence of 50 μM ascorbate for 5 min resulted in the loss of device conductance. Therefore, these results demonstrate excellent selectivity of these DNAzyme-functionalized devices towards Cu2+.
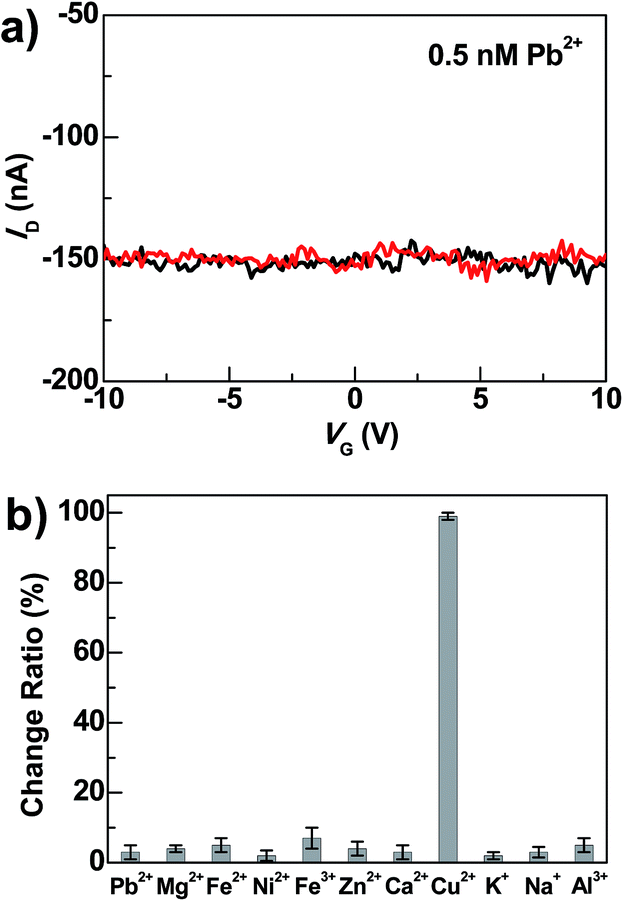 | ||
| Fig. 4 (a) I–V curves of DNAzyme-bridged graphene devices after DNAzyme connection (black) and after further treatments with Pb2+ (∼0.5 nM) under the same conditions in the presence of 50 μM ascorbate (red). (b) Statistical comparisons of conductance changes under the same conditions in the presence of 50 μM ascorbate (0.5 nM of Pb2+, Mg2+, Fe2+, Ni2+, Fe3+, Zn2+, Ca2+ and Cu2+; 5 mM of K+; 135 mM of Na+; 60 nM of Al3+). All the measurements were performed at VD = −50 mV. | ||
Conclusions
In summary, we have demonstrated a platform of using single-molecule graphene–DNAzyme junctions to achieve direct electrical detection of paramagnetic Cu2+ with ultra-high femtomolar sensitivity and high selectivity. While most metal ion sensors reported previously relied on optical properties that require the labeling of DNAzymes with either fluoresencent or colorimetric groups, and these optical properties can often be interfered by background fluorescence or colors, the current system integrated the DNAzymes directly into electrical circuits without labeling them, and with electrical signals that are much less vulnerable to interference. Even though several examples of DNAzyme-based metal ion sensors have been published, none of the reported DNAzyme sensors have achieved ultra-high sensitivity as reported in this work. We accomplished this task for using a novel signal transduction mechanism using a graphene–DNAzyme junction. Just like organic molecule-based metal ion sensors, despite numerous papers published in the field for many years, there is still a major advance possible for better performance if a new signal transduction mechanism can be introduced. Since DNAzymes selective for a variety of metal ions can be obtained through in vitro selection, the sensing system demonstrated here can be applied to the detection of many other metal ions. Finally, since the graphene devices are compatible to current complementary metal oxide semiconductor (CMOS) technologies, our system has the potential for the development of direct, low-cost, high-throughput and real-time detection arrays for chemical and biological reactions.Acknowledgements
We acknowledge primary financial support from MOST (2012CB921404) and NSFC (21225311, 51121091, 91333102, and 21373014) (to X.G.) and US National Institutes of Health (ES16865 to Y.L.).Notes and references
- F. Patolsky, G. Zhang and C. M. Lieber, Nat. Protoc., 2006, 1, 1711 CrossRef CAS PubMed.
- J. Liu, Z. Cao and Y. Lu, Chem. Rev., 2009, 109, 1948 CrossRef CAS PubMed.
- D. R. Kauffman and A. Star, Chem. Soc. Rev., 2008, 37, 1197 RSC.
- M. H. Lim and S. J. Lippard, Acc. Chem. Res., 2007, 40, 41 CrossRef CAS PubMed.
- L. Finney, Y. Chishti, T. Khare, C. Giometti, A. Levina, P. A. Lay and S. Vogt, ACS Chem. Biol., 2010, 5, 577 CrossRef CAS PubMed.
- C. Jia and X. Guo, Chem. Soc. Rev., 2013, 42, 5642 RSC.
- X. Xie, W. Xu and X. Liu, Acc. Chem. Res., 2012, 45, 1511 CrossRef CAS PubMed.
- A. Changela, K. Chen, Y. Xue, J. Holschen, C. E. Outten, T. V. O'Halloran and A. Mondragon, Science, 2003, 301, 1383 CrossRef CAS PubMed.
- Z. G. Xiao, F. Loughlin, G. N. George, G. J. Howlett and A. G. Wedd, J. Am. Chem. Soc., 2004, 126, 3081 CrossRef CAS PubMed.
- C. L. Dupont, G. Grass and C. Rensing, Metallomics, 2011, 3, 1109 RSC.
- P. G. Georgopoulos, A. Roy, M. J. Yonone-Lioy, R. E. Opiekun and P. J. Lioy, J. Toxicol. Environ. Health, Part B, 2001, 4, 341 CAS.
- Y. Tang, Y. Qu, Z. Song, X. He, J. Xie, J. Hua and G. Chen, Org. Biomol. Chem., 2012, 10, 555 CAS.
- J. Liu, J. Karpus, S. V. Wegner, P. R. Chen and C. He, J. Am. Chem. Soc., 2013, 135, 3144 CrossRef CAS PubMed.
- V. Dujols, F. Ford and A. W. Czarnik, J. Am. Chem. Soc., 1997, 119, 7386 CrossRef CAS.
- Y. You, Y. Han, Y. Lee, S. Y. Park, W. Nam and S. J. Lippard, J. Am. Chem. Soc., 2011, 133, 11488 CrossRef CAS PubMed.
- L. Zeng, E. W. Miller, A. Pralle, E. Y. Isacoff and C. J. A. Chang, J. Am. Chem. Soc., 2006, 128, 10 CrossRef CAS PubMed.
- Q. Wu and E. V. Anslyn, J. Am. Chem. Soc., 2004, 126, 14682 CrossRef CAS PubMed.
- L. Marbella, B. Serli-Mitasev and P. Basu, Angew. Chem., Int. Ed., 2009, 48, 3996 CrossRef CAS PubMed.
- Z. C. Wen, R. Yang, H. He and Y. B. Jiang, Chem. Commun., 2006, 106 RSC.
- Y. Chen, C. Zhu, J. Cen, J. Li, W. He, Y. Jiao and Z. Guo, Chem. Commun., 2013, 7632 RSC.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2007, 129, 9838 CrossRef CAS PubMed.
- J. Liu, A. K. Brown, X. Meng, D. M. Cropek, J. D. Istok, D. B. Watson and Y. Lu, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2056 CrossRef CAS PubMed.
- F. Patolskym and C. M. Lieber, Mater. Today, 2005, 8, 20 CrossRef.
- X. Guo, Adv. Mater., 2013, 25, 3397 CrossRef CAS PubMed.
- R. R. Breaker, Nat. Biotechnol., 1997, 15, 427 CrossRef CAS PubMed.
- Y. Lu, Chem.–Eur. J., 2002, 8, 4588 CrossRef CAS.
- S. K. Silverman, Nucleic Acids Res., 2005, 33, 6151 CrossRef CAS PubMed.
- J. Li and Y. Lu, J. Am. Chem. Soc., 2000, 122, 10466 CrossRef CAS.
- J. Liu and Y. Lu, J. Am. Chem. Soc., 2003, 125, 6642 CrossRef CAS PubMed.
- Y. Xiao, A. A. Rowe and K. W. Plaxco, J. Am. Chem. Soc., 2007, 129, 262 CrossRef CAS PubMed.
- J. Liu and Y. Lu, Angew. Chem., Int. Ed., 2007, 46, 7587 CrossRef CAS PubMed.
- S. V. Wegner, A. Okesli, P. Chen and C. He, J. Am. Chem. Soc., 2007, 129, 3474 CrossRef CAS PubMed.
- S. A. McManus and Y. Li, J. Am. Chem. Soc., 2013, 135, 7181 CrossRef CAS PubMed.
- N. Carmi, L. A. Shultz and R. R. Breaker, Chem. Biol., 1996, 3, 1039 CrossRef CAS.
- N. Carmi and R. R. Breaker, Bioorg. Med. Chem., 2001, 9, 2589 CrossRef CAS.
- N. Carmi, H. R. Balkhi and R. R. Breaker, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 2233 CrossRef CAS.
- J. G. Genereux and J. K. Barton, Chem. Rev., 2010, 110, 1642 CrossRef CAS PubMed.
- X. Guo, J. P. Small, J. E. Klare, Y. Wang, M. S. Purewal, I. W. Tam, B. H. Hong, R. Caldwell, L. Huang, S. O'Brien, J. Yan, R. Breslow, S. J. Wind, J. Hone, P. Kim and C. Nuckolls, Science, 2006, 311, 356 CrossRef CAS PubMed.
- Y. Cao, S. Dong, S. Liu, L. He, L. Gan, X. Yu, M. L. Steigerwald, X. Wu, Z. Liu and X. Guo, Angew. Chem., Int. Ed., 2012, 51, 12228 CrossRef CAS PubMed.
- S. Sorgenfrei, C. Y. Chiu, R. Gonzalez, Y. J. Yu, P. Kim, C. Nuckolls and K. L. Shepard, Nat. Nanotechnol., 2011, 6, 126 CrossRef CAS PubMed.
- T. Takada, M. Fujitsuka and T. Majima, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11179 CrossRef CAS PubMed.
- Y. Choi, I. S. Moody, P. C. Sims, S. R. Hunt, B. L. Corso, G. A. Weiss and P. G. Collins, Science, 2012, 335, 319 CrossRef CAS PubMed.
- H. Wang, N. B. Muren, D. Ordinario, A. A. Gorodetsky, J. K. Barton and C. Nuckolls, Chem. Sci., 2012, 3, 62 RSC.
- B. Xu, P. Zhang, X. Li and N. Tao, Nano Lett., 2004, 4, 1105 CrossRef CAS.
- P. C. Sims, I. S. Moody, Y. Choi, C. Dong, M. Iftikhar, B. L. Corso, O. T. Gul, P. G. Collins and G. A. Weiss, J. Am. Chem. Soc., 2013, 135, 7861 CrossRef CAS PubMed.
- C. Jia, J. Wang, C. Yao, Y. Cao, Y. Zhong, Z. R. Liu, Z. F. Liu and X. Guo, Angew. Chem., Int. Ed., 2013, 52, 8666 CrossRef CAS PubMed.
- S. Liu, X. Zhang, W. Luo, Z. Wang, X. Guo, M. L. Steigerwald and X. Fang, Angew. Chem., Int. Ed., 2011, 50, 2496 CrossRef CAS PubMed.
- S. Sorgenfrei, C. Y. Chiu, M. Johnston, C. Nuckolls and K. L. Shepard, Nano Lett., 2011, 11, 3739 CrossRef CAS PubMed.
- S. P. Liu, S. H. Weisbrod, Z. Tang, A. Marx, E. Scheer and A. Erbe, Angew. Chem., Int. Ed., 2010, 49, 3313 CrossRef CAS PubMed.
- L. Sun, Y. A. Diaz-Fernandez, T. A. Gschneidtner, F. Westerlund, S. Lara-Avilab and K. Moth-Poulsen, Chem. Soc. Rev., 2014, 43, 7378 RSC.
- We define the cutting yield as the fraction of graphene transistors on a chip that are electrically disconnected after oxygen plasma etching, and the connection yield as the fraction of the completely-broken devices that get reconnected after molecular connection.
- To address the number of junctions that contribute to the charge transport, we theoretically calculated the probability of the reconnected devices with n-rejoined junctions by using the binomial distribution. If the connection yield is among 20–30%, the ratio of single-junction devices to the overall reconnected devices is ∼82–89%. These suggest that in most cases, only one or two junctions contribute to the charge transport of the devices. The detailed calculation can be found in ref. 39.
- Z. Fang, J. Huang, P. Lie, Z. Xiao, C. Ouyang, Q. Wu, Y. Wu, G. Liu and L. Zeng, Chem. Commun., 2010, 46, 9043 RSC.
- Y. Xiang, Z. Li, X. Chen and A. Tong, Talanta, 2008, 74, 1148 CrossRef CAS PubMed.
- H. Li, X. Huang, D. Kong, H. Shen and Y. Liu, Biosens. Bioelectron., 2013, 42, 225 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03612c |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |