
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of arsenic-rich Asn ligand complexes from yellow arsenic†‡
C.
Graßl
,
M.
Bodensteiner
,
M.
Zabel
and
M.
Scheer
*
Institut für Anorganische Chemie, Universität Regensburg, Universitätsstraße 31, 93053 Regensburg, Germany. E-mail: manfred.scheer@ur.de
First published on 28th November 2014
Abstract
The reaction of [{η5-Cp′′′Co}2{μ,η4:4-toluene}] with yellow arsenic yields the arsenic-rich Asn ligand complexes [{Cp′′′Co(μ,η2:2-As2)}2] (1), [(Cp′′′Co)4(μ4,η4:4:2:2:1:1-As10)] (2) and [(Cp′′′Co)3(μ3,η4:4:2:1-As12)] (3), which were comprehensively characterized. The molecular structure of 1 show a triple-decker complex with two As2 units forming the middle-deck; compound 2 contains an all-arsenic As10 analogue of dihydrofulvalene in the molecular structure. The As12 ligand in 3 represents the largest Asn ligand complex reported so far.
Introduction
The synthesis of substituent-free Asn ligand complexes was established in the early 1980s. The first arsenic sources to be used for this purpose were cyclo-arsines such as (MeAs)5, and (PhAs)6,1 which formed the first triple-decker sandwich complex A with a distorted As3–As2 middle deck, revealing long As–As contacts of 2.726(3) and 2.752(3) Å.2 Subsequently, polyarsenides were introduced into this chemistry, for which the reaction of As73− with [Cr(CO)3(1,3,5-Me3C6H3)] was reported to yield a norbornadiene-like As7 core in B.3 A similar structural motif was obtained by Goicoechea et al. in [Tl(η2-As7)]2− by the reaction of As73− with TlCl.4 The cobalt arsenic cluster [Co6As12(PEt2Ph)6] (C), containing a cyclo-As6 unit, and [As@Ni12@As20]3− were also synthesized using As73− as the starting material.5,6 Moreover, the neutral nortricyclane derivative, [As7(SiMe3)3], was used as the arsenic source by Fenske et al. in the reaction with [(CpRCoCl)2] (CpR = C5Me5, C5Me4tBu). Here, the cationic clusters [(CpRCo)3(μ3,η4-As6)]2+ (D), [(CpRCo)3(μ3,η4-As6)]+ and [(CpRCo)2(μ2,η4-As4)]2+ were obtained.7 In contrast, the use of yellow arsenic (As4), the unstable allotrope of arsenic, was introduced by cothermolysis reactions with cyclopentadienyl-containing carbonyl complexes, which led to a variety of cyclo-As3, cyclo-As5 and cyclo-As6 complexes.8 For example, the reaction of As4 with [Cp*Co(CO)2] (Cp* = C5Me5) produced two binuclear complexes, [Cp*Co(μ,η2:2-As)2]2 and [Cp*2Co2(μ2,η2:2-As6)], and one trinuclear complex, [Cp*Co(μ,η2:2-As2)]3.9 By reacting As4 with [Cp′′Nb(CO)4] (Cp′′ = 1,3-tBu2C5H3), Scherer et al. obtained [Cp′′Nb2As8] (E), which contains the largest structurally characterized Asn ligand known to date.10 In the reaction of [Cp′′Rh(CO)2] with E4 (E = P, As) the P10 derivative of F was structurally characterized, whereas the As complex F was only characterized by mass spectrometry.11 Larger structurally characterized arsenic scaffolds are present in the polyarsenide anions (the Zintl ions: As42− and As64− and the cage compounds As73−, As113−, As144−) derived from grey arsenic, with the largest reported polyarsenide anion hitherto being As224−.12 These ionic compounds confer thermodynamic stability through factors such as lattice enthalpy, which does not apply to analogous systems involving neutral Asn ligands.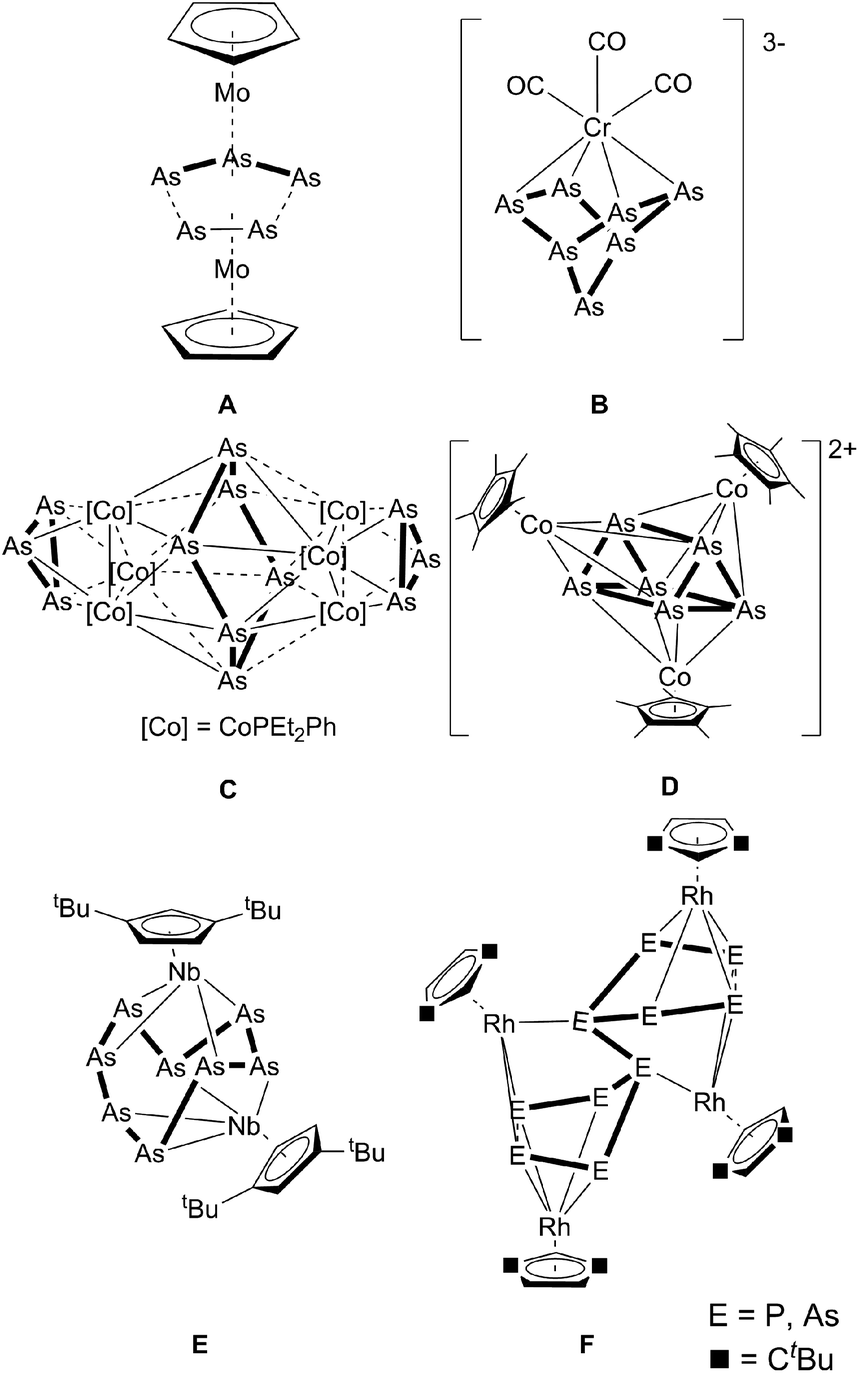
Thus, the question arises whether larger Asn units can be generated. Recently, we reported on the activation of white phosphorus by [{Cp′′′Co}2{μ,η4:4-toluene}] (Cp′′′ = 1,3,5-tBu3C5H2),13 which led to the formation of extended polyphosphorus scaffolds. Since [{Cp′′′Co}2{μ,η4:4-toluene}] dissociates in solution into the unsaturated 14 VE complex [Cp′′′Co], this reactive moiety allows one to work under mild reaction conditions at low temperatures, yielding complexes containing P16 and P24 ligands, respectively, by consuming P4 moieties for aggregation.14
This success with phosphorus raised the question of approaching As-rich ligand complexes by a similar methodology using yellow arsenic. However, the poor solubility of yellow arsenic in common solvents combined with its extreme light sensitivity with respect to the formation of grey arsenic, are in strong contrast to the properties of white phosphorus. This complicates the use of As4 as an arsenic source at ambient temperature and below. Moreover, in the few reports where As4 has been used in reactions with transition metal compounds at room temperature and below, conversions to relatively small As1 or As2 units15 or to a butterfly As42− moiety16 were described. These observations raise important questions and challenges about whether solutions of As4 be prepared in sufficiently high concentrations to form extended polyarsenic units that are larger than those currently known. Our findings on this topic are reported herein.
Results and discussion
In contrast to the situation with P4 solutions (vide supra), the concentration of As4 in solution is too low at −30 °C. Therefore, the reaction of [{Cp′′′Co}2{μ,η4:4-toluene}]13 was performed at room temperature, with a saturated arsenic solution in toluene yielding three products (Scheme 1). After column chromatographic workup, 1 was isolated in 54% yield as the main product, followed by the As12 complex 3 (14%) and the As10 complex 2 (8%) (Scheme 1). Complex 1 can be formed selectively when the reaction takes place at 70 °C in good yields (82%) with no evidence for 2 and 3. Similar complexes of 1 have been reported as minor products of the cothermolysis between [CpRCo(CO)2] (CpR = C5Me5 (Cp*), C5Me4Et (Cp′)) and yellow arsenic at 190 °C (for [Cp*CoAs2]2 6% and for [Cp′CoAs2]2 2% yield).9 | ||
| Scheme 1 Synthesis of compounds 1–3. | ||
The 1H NMR spectra of 1–3 show the corresponding signals for the tBu groups and the signals for the aromatic protons. Due to the rotation of the cyclopentadienyl ligands in 2 only broad signals for the tBu groups as well as the aromatic protons are observed in the 1H NMR spectra. In the FDI mass spectra the molecular ion peaks of 1–3 are observed, and in the case of 1 further fragmentation was not detected. For 2 and 3 two fragments, [(Cp′′′Co)2As6]+ and [(Cp′′′Co)2As4]+, and in addition for 3 the fragment [(Cp′′′Co)2As5]+ can be further found in the mass spectra (FDI-MS).
Compound 1 crystalizes from a saturated hexane solution as dark green blocks. The solid state structure (Fig. 1) shows a triple decker complex with two As2 units as middle deck. The bond length As1–As2 of 2.2795(5) Å is shorter as a single bond determined for As4 by electron diffraction in the gas phase (2.44 Å (ref. 17) and 2.435(4) Å (ref. 18)) and by DFT calculations (2.437 Å (ref. 15b)), respectively. The As1–As2 distance can be compared with the distance in the diarsene [{(Me3Si)3CAs}2], which contains an arsenic–arsenic double bond of 2.245(1) Å.19 In contrast, the distance between the two As2 units of 2.8209(4) Å is beyond what can reasonably be considered a bond, however it is closer than the sum of the van-der-Waals-radii (3.7 Å). The distance between the two As2 units in [Cp′CoAs2]2 was found to be 2.844(1) Å, which is comparable to the distance in 1.
 | ||
| Fig. 1 Molecular structure of 1. H atoms are omitted for clarity. Anisotropic displacement parameters are depicted at 50% probability level. Selected bond lengths [Å] and angles [°]: As1–As2 2.2795(5), As1⋯As2′ 2.8209(4), Co–As1 2.4355(5), Co–As2 2.4211(5), Co–Ccentr. 2.092(3); As2–As1–As2′ 89.747(13). | ||
Compound 2 crystallizes from a saturated dichlormethane solution and constitutes the all-pnictogen analogue As10 of dihydrofulvalene, which acts formally as a 16-electron donor ligand. The main feature of the structure of 2 (Fig. 2) is an As10 ligand consisting of two As5 units bonded by an As–As bond. In each of the As5 rings four arsenic atoms coordinate to a [Cp′′′Co] fragment whereas a second [Cp′′′Co] fragment is coordinated by two arsenic atoms of one As5 ring and one arsenic atom of the second As5 ring. Accordingly, there are two types of [Cp′′′Co] fragments, one coordinates via π bonds to four As atoms (av. As–Co 2.456(2) Å) and the other [Cp′′′Co] fragment is coordinated formally by its lone pair to the atom Co1 (As5–Co1 2.273(2) Å), and side-on to an As–As bond (As1–Co1, As2–Co1 2.350(3) Å). Viewing the bond distance alternations, the shorter lone pair donation leads obviously to a longer π-type bond inclusive of the longer As–As bond of the linking As atoms (vide infra). However, formal coordination of the arsenic lone pair to cobalt occurs in the range of 2.326(1)–2.350(2) Å in compounds such as [Co2{μ-(C2(CO2Me)2)}{μ-(AsMe2)2S}(CO)4] and [Co2(R′CCR′′){μ-(AsPh2)2S}(CO)4] (R′, R′′ = CO2Me, Ph).20 The As–As bond lengths of 2 are in the range characteristic of single bonds (for details see ESI‡), and only two distances between As1–As2 (As1′–As2′) are longer 2.705(2) Å (Fig. 2). However, these elongated distances are shorter than the As–As distances found in [(Cp*Fe)2(Cp*Co)As6], where two As3 triangles are connected by As–As bonds with distances of 2.800(2) to 2.871(1) Å.21 In the triple-decker sandwich complex A there are As–As distances in the range of 2.726(3) and 2.752(3) Å which are regarded as being bonds with the bond order of 0.5.2 Therefore, one can speculate of a weak interaction between the atoms As1 and As2.
 | ||
| Fig. 2 Molecular structure of 2·4CH2Cl2. The H atoms and the solvent molecules are omitted for clarity. Anisotropic displacement parameters are depicted at 50% probability level. | ||
Single crystals of 3 were obtained from a saturated hexane solution as black needles. The structure of 3 (Fig. 3) can be derived from that of 2 in which one As5(CoCp′′′)2 unit is replaced by a norbornane-like As7CoCp′′′ fragment. All bond lengths are in the range of As–As single bonds, with the exception of the linking distance of the two Asn moieties at the atoms As1–As2 (2.6684(5) Å). This distance is comparable with the corresponding bond length in 2 (2.705(2) Å). The elongation of the As–As bond through the coordination of two [Cp′′′Co] fragments is comparable with the phosphorus analogue of 3, where a similar behavior is observed.16 Also in 3 the formal lone-pair coordination of Co to As (Co2–As6 2.2628(6) Å) is slightly shorter than the other Co–As distances (average 2.4314(6) Å). The As12 ligand, which is the largest yet obtained, can be described as a 12-electron donor.
 | ||
| Fig. 3 Molecular structure of 3. H atoms are omitted for clarity. Anisotropic displacement parameters are depicted at 50% probability level. | ||
Conclusions
In summary, it has been shown that the use of the Co complex [{Cp′′′Co}2{μ,η4:4-toluene}] can initiate mild activation of yellow arsenic. Using this method, arsenic-rich Asn ligand complexes could be synthesized. Complexes 2 and 3 contain As10 and As12 ligands, which are the largest substituent-free polyarsenic ligands yet observed in transition metal complexes, and have been unambiguously characterized by X-ray crystallography for the first time.Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft.Notes and references
- A.-J. Dimaio and A. L. Rheingold, Chem. Rev., 1990, 90, 169–190 CrossRef CAS.
- A. L. Rheingold, M. J. Foley and P. J. Sullivan, J. Am. Chem. Soc., 1982, 104, 4727–4729 CrossRef CAS.
- B. W. Eichorn, R. C. Haushalter and J. C. Huffmann, Angew. Chem., Int. Ed., 1989, 28, 1032–1033 CrossRef.
- C. M. Knapp, J. S. Large, N. H. Rees and J. M. Goicoechea, Dalton Trans., 2011, 40, 735–745 RSC.
- R. Ahlrichs, D. Fenske, K. Fromm, H. Krautscheid, U. Krautscheid and O. Treutler, Chem.–Eur. J., 1996, 2, 238–244 CrossRef CAS.
- M. J. Moses, J. C. Fettinger and B. W. Eichhorn, Science, 2003, 300, 778–780 CrossRef CAS PubMed.
- C. von Hähnisch, D. Fenske, F. Weigend and R. Ahlrichs, Chem.–Eur. J., 1997, 3, 1494–1498 CrossRef.
- O. J. Scherer, Acc. Chem. Res., 1999, 32, 751–762 CrossRef CAS.
- O. J. Scherer, K. Pfeiffer, G. Heckmann and G. Wolmershäuser, J. Organomet. Chem., 1992, 425, 141–149 CrossRef CAS.
- O. J. Scherer, R. Winter, G. Heckmann and G. Wolmershäuser, Angew. Chem., Int. Ed., 1991, 7, 850–852 CrossRef.
- O. J. Scherer, B. Höbel and G. Wolmershäuser, Angew. Chem., Int. Ed., 1992, 31, 1027–1028 CrossRef.
- (a) S. Scharfe, F. Kraus, S. Stegmaier, A. Schier and T. F. Fässler, Angew. Chem., Int. Ed., 2011, 50, 3630–3670 CrossRef CAS PubMed; (b) R. C. Haushalter, B. W. Eichhorn, A. L. Rheingold and S. Geib, J. Chem. Soc., Chem. Commun., 1988, 1027–1028 RSC.
- J. J. Schneider, D. Wolf, C. Janiak, O. Heinemann, J. Rust and C. Krüger, Chem.–Eur. J., 1998, 4, 1982–1991 CrossRef CAS.
- F. Dielmann, M. Sierka, A. V. Virovets and M. Scheer, Angew. Chem., Int. Ed., 2010, 49, 6860–6864 CrossRef CAS PubMed.
- (a) J. J. Curley, N. A. Piro and C. C. Cummins, Inorg. Chem., 2009, 48, 9599–9601 CrossRef CAS PubMed; (b) H. A. Spinney, N. A. Piro and C. C. Cummins, J. Am. Chem. Soc., 2009, 131, 16233–16243 CrossRef CAS PubMed.
- (a) C. Schwarzmaier, A. Y. Timoshkin, G. Balázs and M. Scheer, Angew. Chem., Int. Ed., 2014, 53, 9077–9081 CrossRef CAS PubMed; (b) S. Heinl and M. Scheer, Chem. Sci., 2014, 5, 3221–3225 RSC.
- L. R. Maxwell, S. B. Hendrick and V. M. Mosley, Chem. Phys., 1935, 3, 699–709 CAS.
- Y. Morino, T. Ukaji and T. Ito, Bull. Chem. Soc. Jpn., 1966, 39, 64–71 CrossRef CAS.
- A. H. Cowley, N. C. Normann and M. Pakulski, J. Chem. Soc., Dalton Trans., 1985, 1, 383–386 RSC.
- J. D. King, M. J. Mays, M. MacPartlin, G. A. Solan and C. L. Stone, J. Organomet. Chem., 2003, 681, 102–114 CrossRef CAS.
- G. Friedrich, O. J. Scherer and G. Wolmershäuser, Z. Anorg. Allg. Chem., 1996, 622, 1478–1486 CrossRef CAS.
Footnotes |
| † Dedicated to Professor Martin Jansen on the occasion of his 70's birthday. |
| ‡ Electronic supplementary information (ESI) available: Experimental and X-ray data. CCDC 1002519–1002521. For ESI and crystallographic data in CIF or other electronic format. see DOI: 10.1039/c4sc03543g |
| This journal is © The Royal Society of Chemistry 2015 |