
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStructural characterization of holo- and apo-myoglobin in the gas phase by ultraviolet photodissociation mass spectrometry†
Michael B.
Cammarata
and
Jennifer S.
Brodbelt
*
Department of Chemistry, The University of Texas at Austin, 1 University Station A5300, Austin, TX, USA 78712. E-mail: jbrodbelt@cm.utexas.edu
First published on 26th November 2014
Abstract
Ultraviolet photodissociation (UVPD) mass spectrometry is employed to investigate the structure of holo-myoglobin as well as its apo form transferred to the gas phase by native electrospray. UVPD provided insight into the stability of native structural elements of holo-myoglobin. The fragmentation yields from UVPD showed the greatest overall correlation with B-factors generated from the crystal structure of apo-myoglobin, particularly for the more disordered loop regions. Solvent accessibility measurements also showed some correlation with the UVPD fragmentation of holo-myoglobin. Comparison of UVPD of holo- and apo-myoglobin revealed similarities in fragmentation yields, particularly for the lower charge states (8 and 9+). Both holo- and apo-myoglobin exhibited low fragmentation yields for the AGH helical core, whereas regions known to interact with the heme show suppressed fragmentation for holo-myoglobin. The fragment yields from HCD showed the lowest correlation with B-factor values and rather reflected preferential charge-directed backbone cleavages.
Introduction
Protein and macromolecular structure characterization, including the elucidation of protein–protein and protein–ligand interactions, remain significant technological challenges and are critical linchpins in the field of structural biology. In this context, the two most successful techniques for determination of protein structures are X-ray crystallography and nuclear magnetic resonance (NMR) spectrometry.1,2 While these techniques produce unparalleled information about the core structures of proteins, each has specific drawbacks that have propelled the development of other methodologies. For example, crystallization of proteins can be a tedious process that may not always produce a protein crystal even after evaluation of hundreds of conditions. If the proteins are successfully crystallized, the resulting crystal structures reflect static structures which are not necessarily identical to ones in cellular environments and may suffer from artifactual packing.1 NMR spectrometry captures the dynamic structures of proteins but has a practical mass limit of approximately 40 kDa.2Due the nature of these limitations, a number of mass spectrometric methods have been developed for the structural characterization of proteins with high sensitivity and low sample demands. Some of these methods entail using various chemical probe methods on proteins in solution, followed by mass spectrometric detection of products. Hydrogen deuterium exchange (HDX) of the amide hydrogens and covalent labeling of specific amino acids remain at the forefront of mass spectrometric-based strategies for structural biology applications.3 In these methods, the proteins labelled in solution are typically analyzed by a bottom-up type proteomic workflow which entails digestion of the proteins by one or more proteases, separation of the resulting peptides by liquid chromatography and identification by mass spectrometry. The abundances of the modified peptides, which presumably reflect the accessibility of the HDX reagent or chemical probe to specific peptide sites, are quantified relative to the unmodified counterparts to reveal information about the tertiary structure of the protein.4 This process can be simplified by adoption of a top-down workflow which alleviates the proteolytic digestion step but requires a high-resolution/high mass accuracy mass spectrometer to map the incorporation of the covalent labels.5,6 For the top down approach, the deuterium-labelled or probe modified proteins are typically transferred to the gas phase by electrospray ionization (ESI) and fragmented by collisional induced dissociation (CID) or its analog higher energy collisional dissociation (HCD), electron-based activation (ECD or ETD), or ultraviolet photodissociation (UVPD), then the modified sites are determined based on interpretation of the abundances of resulting fragment ions.5,6 Denaturing ESI conditions are typically used to maximize sensitivity. While the top down/labelling approach offers some advantages to the bottom-up methods, the use of any type of labelling method can introduce artifacts during data analysis.
An alternative to labelling methods is to use native spray conditions to ionize proteins for direct analysis in the gas phase. Although the structures of proteins in the gas phase remain a matter of debate and such studies raise concerns about conformational integrity, investigation of intact proteins may offer new insight as well as correlations with known solution structures. It has been shown previously that tertiary intra-protein, protein–ligand and protein–protein interactions can be retained in the gas phase using gentle ESI conditions and buffered solutions.7–23 Several groups have undertaken HDX on the presumed “native” proteins and protein complexes and provided convincing evidence that the gas-phase structures mirror the known crystal or solution structures obtained by X-ray crystallography or NMR.11,12 However, hydrophobic interactions are not well maintained in the gas-phase native structures, and thus electrostatic and van der Waals type interactions play a far more important role. Ion mobility studies have also shown that ions in lower charge states may exhibit similar radii of gyration to that of their solution counterparts.13,14 These studies have also demonstrated that as the charge states increase, the proteins are more elongated, thus showing the impact of charge on denaturation in the gas phase.15–17
There have been a handful of recent studies in which unlabeled native-like proteins have been interrogated by electron-based activation methods, ETD and ECD, to obtain structural insight.18–24 The conclusions from these MS/MS-based studies have been compelling. For example, it was discovered that strong salt bridges were not disrupted by ETD, thus enabling discrimination of different types of salt bridges in the gas phase.19 It was found that specific enhanced backbone cleavage sites could be correlated with regions of the proteins that were known to have high B-factors (a measure of flexibility) from crystallographic studies, suggesting that these regions are also quite disordered (and more flexible) in the gas phase and thus more prone to cleavage.20,21,25,26 Similarly, the abundances of the fragment ions produced by ECD or ETD have been related directly to the stabilities of specific regions of the proteins.25,27–29 There has been one recent report of characterization of native protein complexes by UVPD, ETD and HCD for which UVPD revealed the greatest information about the primary sequences of the proteins in the complexes and also produced non-covalent fragment ions comprised of a portion of the protein bound to the ligand.30 In general, 193 nm UVPD has proven to be an exceptional activation technique for obtaining very high sequence coverage of the protein backbone (for both native and denatured proteins).5,31,32,33 HCD has been applied to top-down analysis of intact proteins (denatured),31,32,34 including one study that showcased the fragmentation of the transmembrane section of integral membrane proteins under denaturing conditions.35
The present study focuses on evaluating the application of 193 nm UVPD to characterize the gas-phase structures of native proteins, as demonstrated for horse heart myoglobin in both its holo and apo (heme-bound) states. Horse heart myoglobin was the first structure to be solved by X-ray crystallography and has become the most popular model protein for development and assessment of new structural methodologies both in apo and holo forms.5,36–41 However, an apo crystal structure has yet to be published. Past studies of myoglobin have been undertaken using an array of mass spectrometric strategies, such as the use of solution phase HDX for examination of structure and kinetic unfolding and ion mobility measurements for investigation of protein cross-section, as well as many computational studies on the stability of the helices in myoglobin.5,6,14,38–40,42 As with past ECD/ETD experiments in which fragment ion abundances were related to the stabilities of particular regions of proteins,9,22,27,29 the same types of correlations are developed in the present study from the examination of UVPD results. The impact of the charge state of the proteins is also examined to determine whether charge-mediated unfolding/elongation is reflected in the resulting fragment ion distributions, and HCD results are evaluated relative to the UVPD results to allow a comparison of the trends observed for a more conventional collision-based activation method (HCD) to photodissociation.
Experimental
Mass spectrometry and data collection
A 5 μM solution of holo-myoglobin (horse heart myoglobin, Sigma, St. Louis, MI) was prepared from lyophilized powder in 5 mM ammonium acetate at pH 5.5 prior native spray. Apo-myoglobin was prepared by butanone extraction as previously described from lyophilized holo-myoglobin.43 After heme extraction the protein was diluted to the same conditions as the holo-form to facilitate ionization of the ligand-free myoglobin in its native state. Denatured apo-myoglobin was generated by infusing 5 μM myoglobin in a solution of 59.9% acetonitrile, 40% water, and 0.1% formic acid at a rate of 1.5 μl min−1. All MS/MS analysis was undertaken using a Thermo Scientific Orbitrap Elite mass spectrometer (Bremen, Germany) outfitted with a 193 nm Coherent Excistar excimer laser for UVPD in the HCD cell as described previously31 and a Prosolia electrospray ionization source (Indianapolis, IN). A voltage of 1.7 kV was applied for ESI, and the heated capillary temperature was maintained at 150 °C. The range was monitored from m/z 220 to 4000 for both survey ESI-MS and all MS/MS scans. Each of the top three most abundant (8+, 9+, and 10+) charge states of the holo and apo protein, as well as the 20+ charge state of the denatured protein, were selected and activated by UVPD. Ions were isolated using a window of 10 m/z. 100 scans were averaged. All results were replicated three to four times. All MS/MS were collected at a resolution of 480 K resolution at m/z 400. UVPD was performed using a single 1.5–2.0 mJ laser pulse in the HCD cell at a nitrogen pressure of approximately 10 mTorr. HCD was performed between 30 to 35% NCE with aforementioned settings.Data analysis
The MS/MS spectra were deconvoluted using the Xtract algorithm with a S/N ratio of 2 and fragment ion identification tolerance of 10 ppm. The deconvoluted spectra were processed using a custom version of ProsightPC 3.0 which searched each spectrum using the nine ion types generated by UVPD (a, a + 1, b, c, x, x + 1, y, Y, z). The identified ion list was tabulated in an Excel format, and the normalized fragmentation yield per amino acid backbone cleavage site was calculated as follows: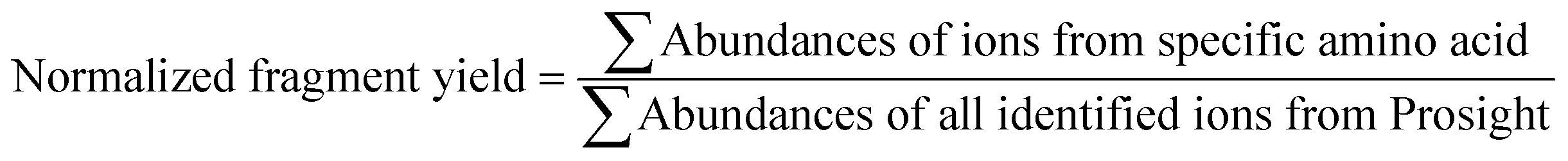
In this approach, all product ions arising from backbone cleavages that occur N-terminal to a specific amino acid (yielding an, bn, and cn ions) are summed with all the complementary C-terminal product ions arising from the same backbone cleavage site (yielding complementary xR−n, yR−n, and zR−n ions) where R is the total number of amino acids in the protein. For example, the abundances of the a70, b70 and c70 ions are summed with the abundances of the x83, y83, and z83 ions, each representing cleavage between residues 70 and 71 for myoglobin (which contains 153 amino acids). In this way, cleavages are categorized via a specific backbone position that is assigned to an amino acid local to the cleavage site. The MS/MS spectra for holo-myoglobin were additionally searched with inclusion of a +615.1695 Da shift corresponding to heme ligand adduction. The identified heme-containing fragment ions were summed with the corresponding apo-fragments from the same spectrum. The resulting fragment yields were also averaged into well-defined demarcated regions and then divided by the length (in terms of number of constituent amino acids) of the regions. These regions are as follows: N-terminus (residues 1–3), A-helix (residues 4–19), B-helix (residues 20–35), C-helix (residues 36–42), CD loop (residues 43–50), D-helix (residues 51–57), E-helix (residues 58–77), EF loop (residues 78–85), F-helix (residues 86 to 94), FG loop (residues 95 to 99), G-helix (residues 100–118), GH loop (residues 119–123), H-helix (residues 124–150), and C-terminus (residues 151–153).38,41,44 Effectively this gives a “fragmentation density” per structural region, a categorization that is beneficial for facilitating discussion about each of the 14 regions at the expense of individual residue-specific changes. The B-factors associated with each amino acid were extracted from the PDB file 1DWR and then averaged for the regions listed above to allow comparisons of B-factors to the fragmentation yields. GetArea was also used to obtain backbone and side-chain solvent accessible surface areas.45
Results
The dominant species observed in the native ESI mass spectrum of both holo- and apo-myoglobin have low charge states (8+ to 10+), unlike the much higher charge states favored by myoglobin under standard denaturing conditions (see Fig. S1†). The ions in low charge states are characteristic of native-like structures, as documented in previous mass spectrometric studies.7–10,14,46 For both holo and apo forms the most abundant charge state is 9+. For the holo (heme-containing) protein there is little to no apo form observed in the native ESI mass spectrum. However, for the apo form low abundance ions in higher charge states are observed (Fig. S1b†) which represent unfolded species. Additionally, some heme-containing species of moderate abundance are observed in the 8+ and 9+ charge states due to incomplete removal of the heme group during preparation of apo-myoglobin. Because the holo and apo forms of myoglobin differ in mass by more than 600 Da, these species are easily resolved and well isolated from each other during subsequent MS/MS analysis. The 8+ and 9+ charge states were determined to have more native-like cross-sections when sprayed between pH 7.0 and 4.0 using native ESI conditions in a recent study by Vahidi et al.14 Each of the low “native” charge states of holo- and apo-myoglobin (i.e. 8+, 9+, 10+) was isolated and activated separately using UVPD. Comparison of the UVPD mass spectra of holo- and apo-myoglobin (9+) is shown in Fig. 1. Expansion of one higher m/z region shows the mass shift associated with retention of the heme group for some of the a and x ions produced by holo-myoglobin, demonstrating the ability to track these non-covalent interactions. Such heme-containing ions were not found upon HCD. The resulting sequence coverages obtained by UVPD for the 8+, 9+, and 10+ charge states were 53%, 87%, and 88% for holo-myoglobin and 64%, 86% and 84% for apo-myoglobin, respectively. The retention of the heme did not notably influence the total sequence coverage obtained. For both holo- and apo-myoglobin there was a shift (an increase) in total sequence coverage from 8+ to 9+, which may be the result of a conformational difference between the charge states in the gas phase or related to the availability of one more proton for charge-mediated fragmentation pathways. The sequence coverage obtained by UVPD of the 20+ species of the denatured protein (heme free) was 77%. Interestingly, the corresponding ETD sequence coverages were low for myoglobin in the native-like charge states (e.g. a sequence coverage of only 18% was attained for 10+). The fragmentation yields were more substantial upon ETD of the denatured 20+ state, giving a sequence coverage of 70% which reflects the well-known charge state dependence of electron activation methods. It has been reported that many salt bridges are not disrupted upon electron activation of native protein structures in low charge states, thus depressing the total sequence coverage of ETD.18 The low overall sequence coverage in the present study confounded deeper correlation of ETD fragmentation with specific structural elements of native states of myoglobin, and thus the ETD results are not discussed further.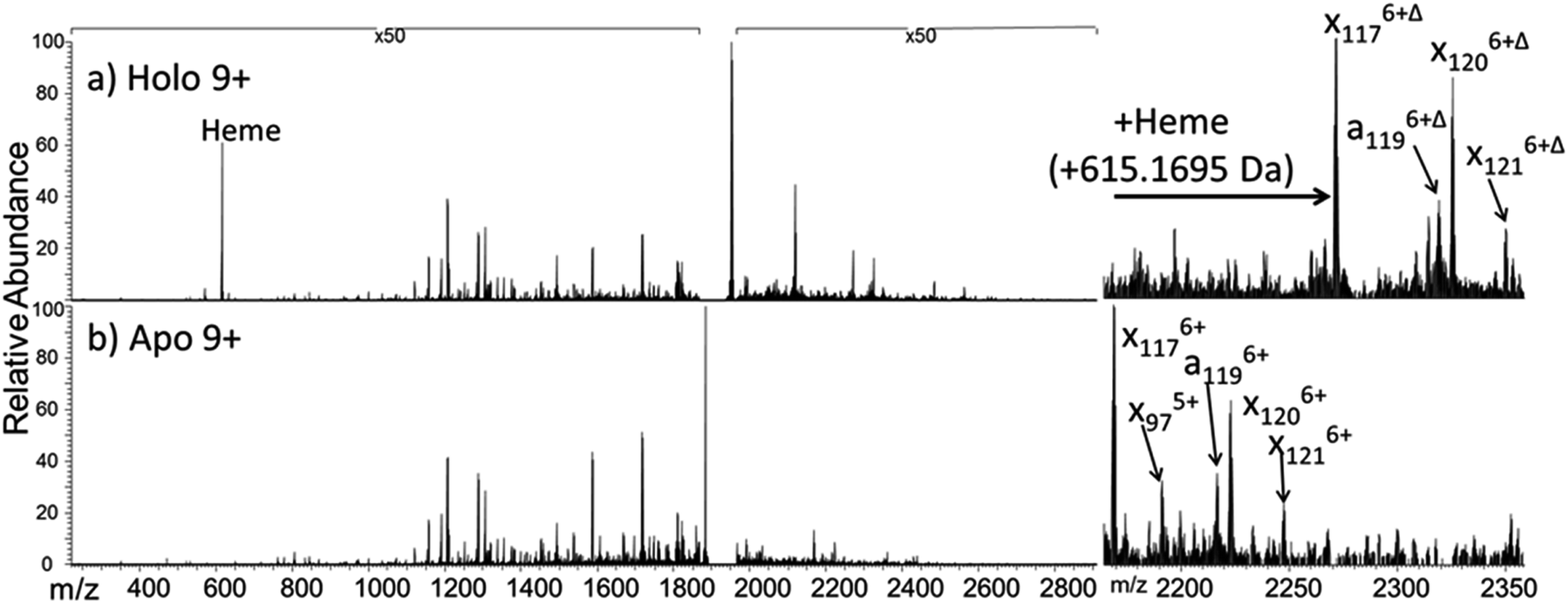 | ||
| Fig. 1 193 nm UVPD mass spectra of the 9+ charge state of the (a) holo and (b) apo form of myoglobin. Panels to the right are expanded regions (m/z 2175 to 2350) with several ions labelled to illustrate the mass shift (+615.1695 Da) affiliated with retention of the heme group. Δ indicates the inclusion of heme in the fragment ions. The 50× amplification applies to both spectra. | ||
Each MS/MS spectrum was processed to calculate the normalized fragmentation yield per amino acid (i.e. based on backbone cleavages between every pair of residues) as described in the experimental section. These values were plotted with their associated standard deviations in Fig. 2 which displays the normalized fragment yield for each native charge state (8+, 9+, 10+) for both forms of myoglobin. The results for the denatured charge state (20+) are included in the apo-myoglobin panel. The general shapes of the fragment distributions in Fig. 2 are similar for the holo- and apo-forms, with clusters of backbone sites showing elevated fragmentation yields relative to others that exhibit little or no yields. The graphs in Fig. 2 also include demarcated regions which are defined based on fourteen known structural regions of myoglobin. If the protein cleaved uniformly at each backbone position, the expected fragmentation yield would be 0.66% (e.g., 100% fragment ion current divided by 152 backbone cleavage sites). Backbone cleavages between residues that were detected to give greater than 0.66% fragmentation yield are considered to be residues exhibiting preferential (enhanced) cleavage. These specific amino acids are highlighted in the primary sequence map (bottom of Fig. 3) and are also superimposed on the myoglobin crystal structure (PDB code 1DWR), as illustrated for each native holo-myoglobin charge state in Fig. 3.
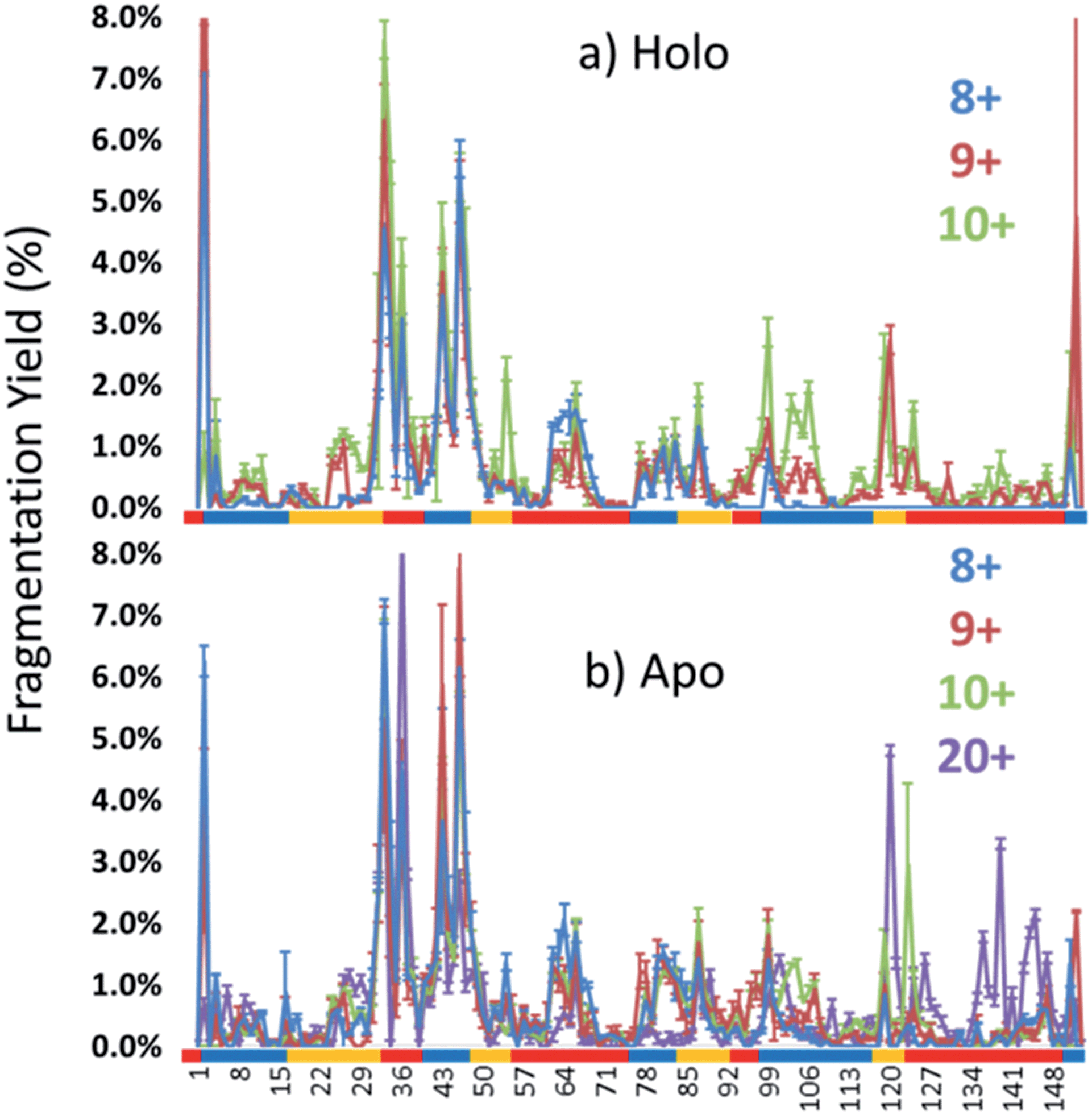 | ||
| Fig. 2 Normalized fragmentation yields upon UVPD of the 8+ to 10+ charge states for both (a) holo and (b) apo forms based on averaging 3–4 replicates. The fragmentation yields for the 20+ denatured state of apo-myoglobin are shown in (b) as well. Structural regions are demarcated below the graphs with colored bars showing the loops and helices. | ||
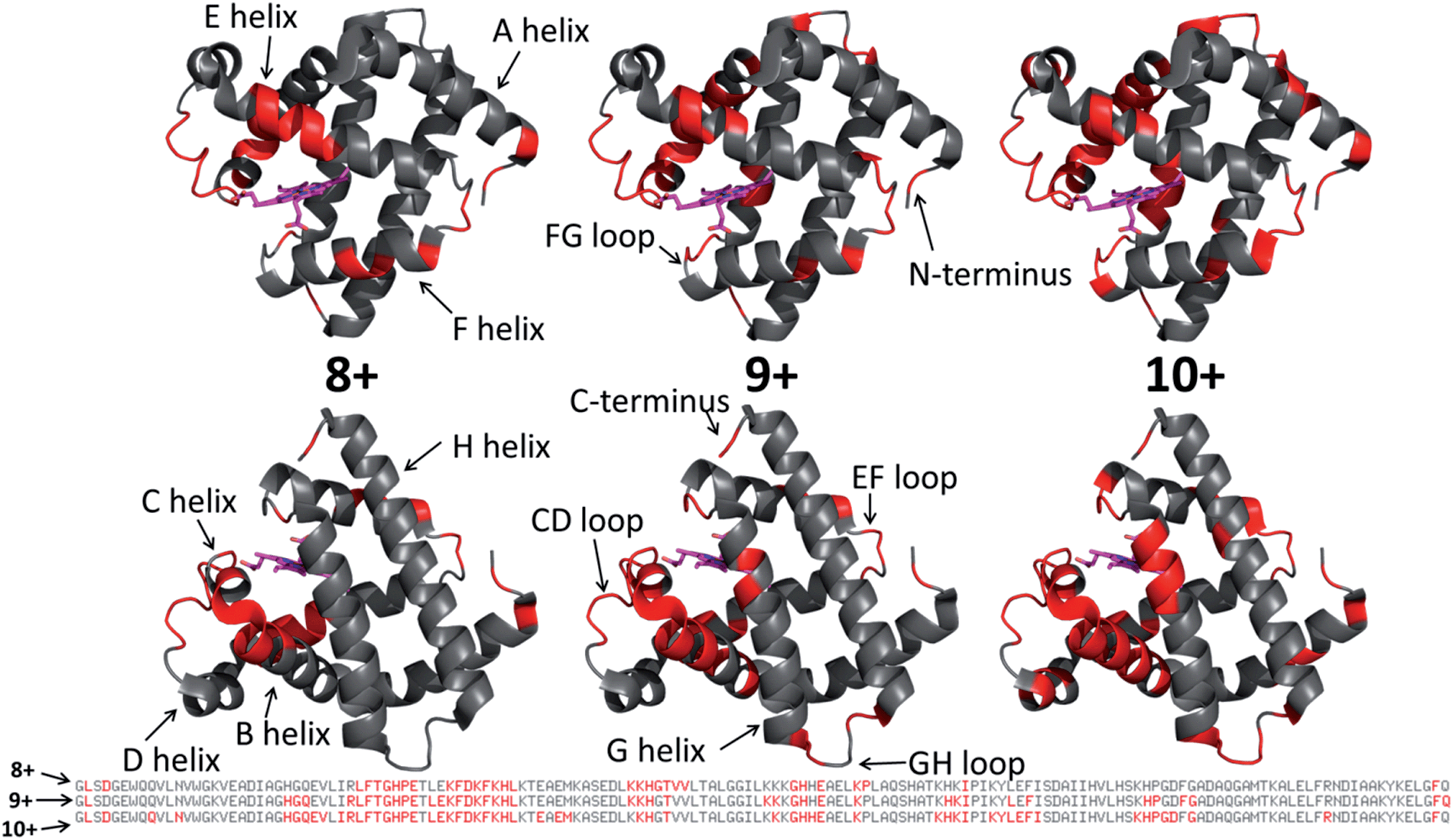 | ||
| Fig. 3 Those residues that were found to have enhanced backbone cleavage frequencies upon UVPD activation are highlighted in red on the holo-myoglobin crystal structure 1DWR, as also indicated in the primary sequence below for the native charge states (8+, 9+, 10+) evaluated. The heme group is shown as a hot pink color. The 1DWR crystal structure has been rotated 180° about the x-axis between the top and bottom structures. | ||
In an effort to streamline the data interpretation in a more structurally meaningful manner, the backbone site-specific fragment yields were grouped and averaged according to the fourteen known structural regions of myoglobin and are displayed in histogram format in Fig. 4 for holo-myoglobin (with structural regions labelled in Fig. S2†) for both HCD and UVPD. The analogous results for apo-myoglobin and denatured myoglobin are shown in Fig. S3.† Essentially this treatment provides fragment yield densities which are a metric of fragmentation localized to specific structural regions of the protein, normalized for the size of the region. This provides a way to visualize the change in fragmentation yield for each activation method and as a function of charge state. To facilitate comparisons, each structural region is assigned an average B-factor value (as described in the Experimental section), which conveys the degree of flexibility in the crystal structure. The B-factors for each defined region of holo-myoglobin (1DWR) are shown in Fig. 4 and 5.
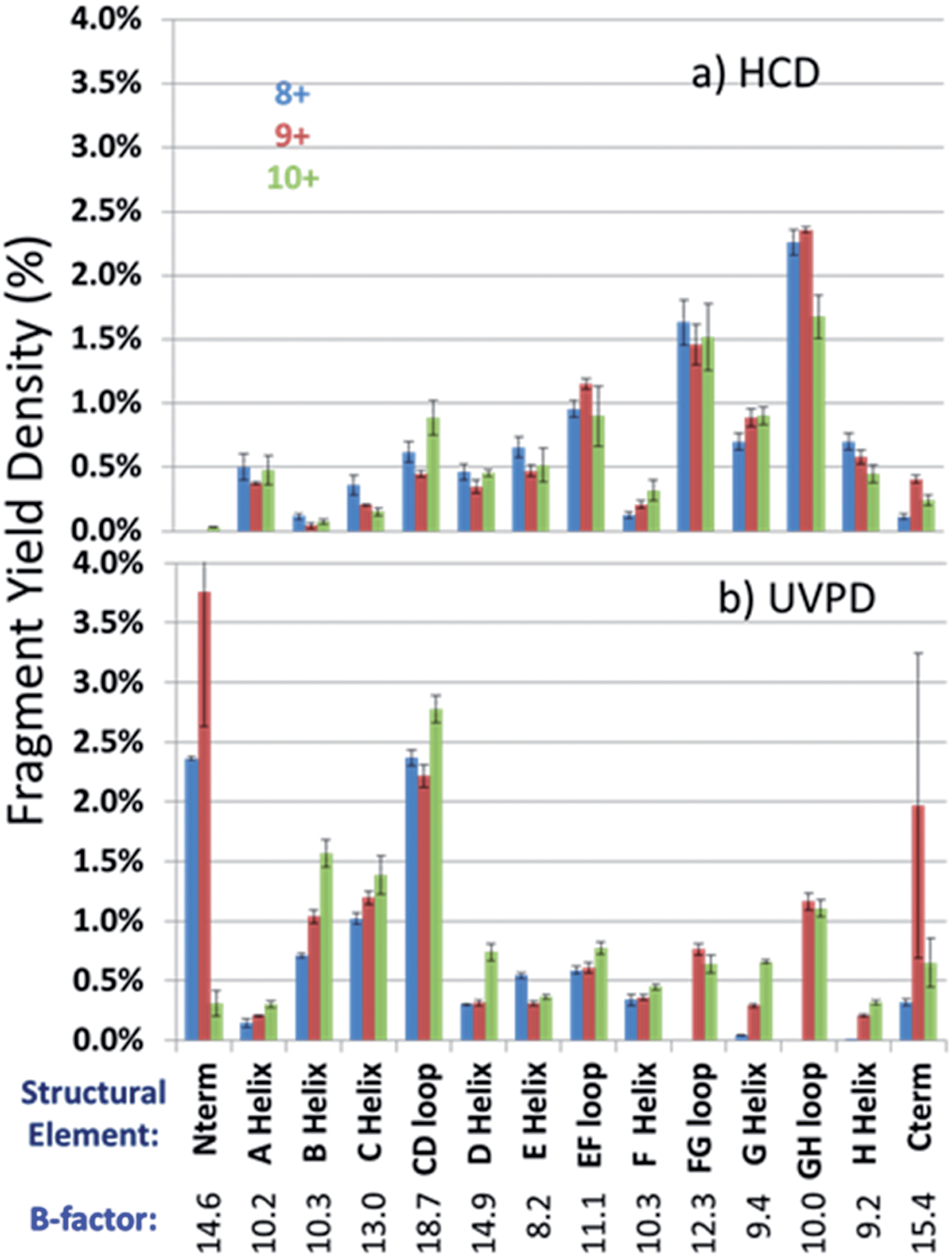 | ||
| Fig. 4 Normalized fragmentation yields averaged over the total number of amino acids in each structural element for charge states 8+ (blue bars), 9+ (red bars), and 10+ (green bars) of holo-myoglobin for (a) HCD and (b) UVPD. Numbers under the structural regions show the calculated average B-factors for holo-myoglobin. | ||
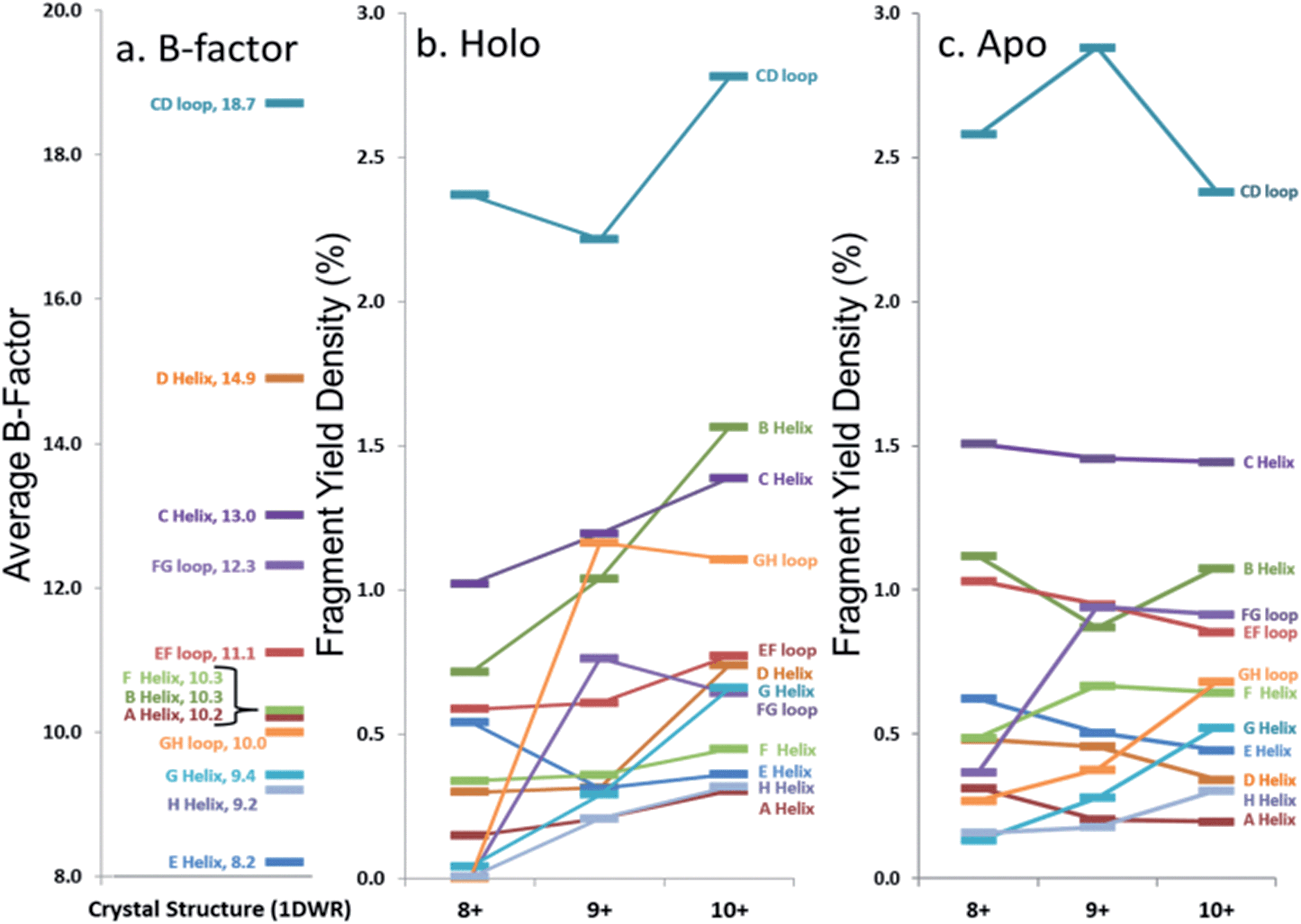 | ||
| Fig. 5 Comparison of average (a) B-factor values and fragment yield densities of respective labelled regions from the (b) holo and (c) apo forms of myoglobin for the three native-like charge states. Regions with greater B-factors or fragmentation yield densities indicate less stable regions whereas those with lower values indicate more stable regions. The C- and N-termini have been removed from the figure. Standard deviations for fragment yield densities are given in Fig. 4 and S2† for holo and apo forms, respectively. | ||
The sequence or order of unfolding of myoglobin based upon molecular dynamics simulations, CD experiments, NMR, and HDX mass spectrometric data provides another perspective on the structural stability of the protein.38–41,47 As briefly summarized here based on published findings,37–40,46 the F-helix is the most unstable structural element, thus the first to unfold. This is followed by unfolding of the C, D, and E helices, prior to the unfolding of the B-helix. The last helices to unfold are the A, G, and H helices or the AGH core. Not surprisingly, this AGH core is found to be consistently the most stable and least flexible, with the exception of the N-terminus of the G-helix, which has been shown by computational modeling to fray.40B-factors from the 1DWR crystal structure were also analyzed and used to correlate the fragmentation yields in the present study to a measured and quantified flexibility value (B-factor). The averaged B-factors of the previously defined demarcated regions are summarized in Fig. 5a. Those regions with high B-factor values are flexible regions of the protein, while those with low B-factor values are more rigid and more stable. The charge state-based fragmentation yield data obtained from UVPD of both the holo and apo forms is shown in Fig. 5b and c, respectively. The UVPD results obtained for the apo-form should not be compared directly to the B-factor values because the latter includes the impact of the heme group. However, comparison of the B-factor values with the UVPD data for holo-myoglobin and comparison of the UVPD data for the holo and apo proteins provides interesting insight, as described in this study. A more detailed comparison of the UVPD results for the apo and holo forms as a function of charge state is shown in Fig. S4,† also discussed in detail later.
For the present study, our goal was to assess whether the UVPD fragmentation yields (Fig. 2 and 4) reflected the local stabilities of the tertiary and secondary structural features of myoglobin (i.e. structures in Fig. 3) as suggested previously in a number of publications utilizing electron activation and 266 nm UVPD for other proteins.23,27,29,48 In essence, these previous studies indicated that those regions of a protein that displayed high fragmentation frequencies and thus resulted in high fragment yields were presumed to be conformationally flexible and/or have few retained intramolecular interactions. Those which had low fragmentation yields were conformationally stable elements, such as those with strong intramolecular interactions in the gas phase. A number of substantial variations in relative fragmentation yields are observed in Fig. 5 and S4† as a function of charge state and apo/holo state of myoglobin, and visualized for the 9+ species in Fig. S5.†
Discussion
UVPD of holo-myoglobin
Most of the preferential fragmentation upon UVPD of the three native charge states of both holo- and apo-myoglobin occurred in the loop regions (Fig. 4b and S3b†), which typically have larger B-factors compared to helices. There appears to be good agreement between both the B-factors and the sequence of unfolding in solution with the 193 nm UVPD results, particularly with the lowest charge state (8+) interrogated (Fig. 5). For the 8+ charge state of holo-myoglobin, the AGH core helices exhibited the lowest fragmentation yields and in fact mirrored the order of stability predicted from the B-factors. The GH loop also has one of the lowest UVPD fragmentation yields, again consistent with the low B-factor value. At the other extreme, the CD loop and C helix had the highest UVPD fragmentation yields, outcomes also reflected in the B-factors which are two of the highest among all the structural elements. There are other structural elements for which the UVPD fragmentation yields and B values did not agree so well. These include the FG loop and D helix which had low UVPD fragmentation yields but relatively high B values, and the E helix exhibited a moderate fragmentation yield but had the lowest B value of all structural elements.When comparing the UVPD fragment yields obtained for the 8+ charge state to the 9+ charge state, fragmentation of the FG and GH loop regions was greatly enhanced, along with significant increases in the fragmentation of the connecting helices G and H (in all cases going from virtually no detectable fragmentation for the 8+ charge state to moderate/significant fragmentation for the 9+ charge state) (Fig. 4b and 5b). The fragmentation yields of the B and C neighboring helices also increased with charge state. The CD loop and the E helix are two regions that showed moderate decreases in fragmentation going from the 8+ to 9+ charge state, and several other structural regions, including the A, D and F helices and the EF loop, exhibited no significant change as a function of charge state. Collective grouping of these regions (those showing decreasing fragmentation or no change with charge state) reveals that all are important for heme binding.49 It has also been noted that absorption cross-sections of proteins change with charge density and conformation.50 This prior finding might explain in part some of the changes observed in the UVPD fragmentation yields as a function of charge state in the present study, namely a possible charge-density induced change in absorption cross-section in addition to a degree of protein unfolding as the charge state increases. Upon transitioning to the 10+ charge state, the most stable regions predicted by UVPD (e.g. lowest fragmentation yields) are the A, H, E and F helices, all of which increased slightly from the fragmentation yields determined for the 9+ charge state. The G helix displays a more significant increase in fragmentation yield for the 10+ charge state, an outcome attributed to N-terminal fraying as a consequence of unfolding of the FG loop that was also noted from the 8+ to 9+ charge states. This type of fraying has been noted in prior solution studies.40,47 Several of the structural regions show an increase in fragmentation for the 10+ charge state, suggestive of unfolding related to the D, G, C, and B helices along with EF and CD loops. This nearly global increase in fragmentation with charge state tracks well with the concept of electrostatic-induced elongation. Even with this alpha helical fraying and expansion in the regions from B to the D helix, the maintenance of the E and F helices as well as the AGH core of the protein allow retention of interactions with the heme group.
Solvent accessibility is another physical parameter that has shown positive correlation with B-factors of proteins51,52 as well as with ETD fragmentation trends for native proteins,18 and thus was also evaluated in the context of the UVPD data in the present study. A plot of the solvent accessible surface area (SASA) for both the backbone and side chains generated from the 1DWR crystal structure is shown in Fig. S6† (along with the B-factor values and UVPD fragmentation yields for ready comparison). There is some general correlation backbone SASA and the UVPD fragmentation yield for those regions having SASA above 50%. Those regions that have both low SASA values for the side-chain and backbone regions also have the lowest UVPD fragmentation yields, particularly for the 8+ charge state. The A-helix region shows the worst correlation between SASA and UVPD fragmentation yields, with rather prominent SASA values for the side-chains of the A-helix region of the protein but yet very low UVPD yields. However, the SASA values of the backbone in this A-helix region are very low.
The observation that the loop regions tend to fragment more readily than the helices by UVPD may also be related in part to the larger molar absorptivities of loops compared to helices at 193 nm. Tsai et al. reported that the λmax of a typical alpha-helix occurred between 189–204 nm in solution with a molar absorptivity of 4.1 cm2 mol × 10−3, whereas the molar absorptivities of coiled or loop regions were around 6.9 cm2 mol × 10−3 with a similar λmax (187–190 nm).51 Interestingly, beta-sheets were reported to have an even greater molar absorptivity;50 however, myoglobin contains no beta-sheets.53 Although the exact mechanism of the UV activation and dissociation process for large molecules like proteins remains unknown in the gas phase, it is conceivable that local absorptivities of certain structural elements of proteins that are retained in the gas phase could have an impact on energy deposition and re-distribution prior to fragmentation. More general trends about fragmentation efficiencies of helical and non-helical regions of native proteins may emerge as additional proteins are analyzed by UVPD.
UVPD fragmentation behavior of holo- versus apo-myoglobin
Apo-myoglobin adopts a structure similar to holo-myoglobin except with greater disorder and partial unfolding of the F helix, adjacent EF and FG loops, N-terminus of the G helix and H helix.54,55 UVPD fragmentation trends were constructed for apo-myoglobin (produced via chemically-induced removal of heme in solution, followed by native ESI of the heme-free protein). The normalized fragmentation yields for the backbone-specific cleavages are shown in Fig. 2b, and the averaged fragmentation yields grouped by the 14 structural elements are illustrated in Fig. S3b† and tracked versus charge state in Fig. 5c. The trends for the holo- versus apo-forms of myoglobin are summarized in Fig. S4† for the three native charge states, thus allowing a more detailed assessment of the impact of the heme ligand. Previous circular dichroism (CD) analysis of holo and apo myoglobin has shown that there is a reduction in alpha helical content for the apo form relative to the holo form,56–58 so this feature could account for some variations in the observed UVPD fragmentation yields. For example, virtually all regions of the heme-free protein have greater fragmentation yields than the holo-form for the 8+ charge state, an outcome that is consistent both with the CD and spectroscopic solution data and the differences in molar absorptivity attributed to the increasing loop and decreasing alpha helical content. Relative to holo-myoglobin, the most dramatic increases in fragmentation density for apo-myoglobin occur for the B and C helices as well as the EF loop regions, followed by less significant increases in fragmentation yields in the CD, FG and GH loops. The larger changes in fragmentation density in these regions may be indicative of unfolding or partial unfolding events in regions which were stabilized by the heme group binding for the holo form of the protein. Aside from these increases in fragmentation density or unfolding for the holo form, the AGH core and FH/GH loops still exhibit the lowest fragmentation yields for apo-myoglobin.One might anticipate a general across-the-board increase in UVPD fragmentation yields for apo-myoglobin relative to holo-myoglobin because the collisional cross-section of myoglobin increases, albeit slightly, upon transition from the holo to myo form.14 Although there are modest increases in UVPD yields for many of the structural elements (AGH helical core, D, E, F helices) going from the holo to myo form, a few regions (EF loop, B and C helices, CD, EF and FG loops) show more significant increases. The increases in the C, D, E and F helix fragmentation yields for the apo form may reflect the lack of stabilizing non-covalent interactions with the heme group that also allows the intervening loop regions to release and helices to unfold. For the 9+ and 10+ charge states, two regions showed a decrease in UVPD fragmentation for the apo form: the B helix and GH loop, as well as the CD loop and D helix for the 10+ charge state. The substantial amount of change in fragmentation yields for the 10+ charge state of holo- versus apo-myoglobin may reflect significant differences in secondary and possibly tertiary structures. The agreement in the UVPD trends between the holo and apo forms of myoglobin with a particular strong similarity in fragmentation behavior of the AGH core helices suggesting that this helical bundle is stabilized in both of these forms.
HCD of holo- and apo-myoglobin
A final comparison was undertaken to evaluate the fragmentation trends of myoglobin obtained by HCD, another activation method that has shown promise for characterization of intact proteins in top-down studies. To date, HCD has not been reported to cause fragmentation that is specific for native protein structures, but in general it has not yet been extensively explored.8,29 The HCD fragmentation results are shown for both holo and apo forms in Fig. 4a and S3a,† respectively, and exhibit similar trends. Each displays modest variation in the fragmentation yields among the three lowest charge states and a far more drastic change for the elongated protein in the 20+ charge state. For the latter 20+ charge state, fragmentation was mainly localized near the termini of the protein (A- and H-helices, GH loop), an outcome that has been noted previously for HCD of a variety of proteins in high charge states (i.e. the type of denatured proteins commonly analyzed in top-down proteomics studies). HCD of the native-like charge states showed more extensive fragmentation of the EF and FG loop regions in holo-myoglobin compared to UVPD. In many cases, the regions that give the lowest fragmentation yields by HCD were not those with the lowest B-factors (most stable) nor did the trend mirror the established sequence of protein unfolding. For example the AGH helices, which are known to be the most stable core elements of myoglobin, exhibited among the highest fragmentation yields upon HCD. The GH loop is considered the most stable loop element in light of its low B-factor value (10.0), yet its HCD fragmentation yield was very high. The B, C, D and F helices displayed the lowest fragmentation yields upon HCD which opposes the trend in the order of unfolding. Interestingly, several of the regions that exhibited the most extensive fragmentation upon UVPD (B, C helices and CD loop) had among the lowest HCD yields, and similarly the AGH helical core and GH loop were found to exhibit substantial fragmentation upon HCD but not upon UVPD. The lack of consistent correlation between the HCD fragmentation yields and the order of solution unfolding and B-factors, suggests that the impact of the mobile protons which facilitate collisional activated backbone cleavages and residue-specific preferential cleavages (i.e. those affiliated with the presence of Asp, Glu, Pro, Gly, Ile, Leu and Val residues) substantially modulates HCD fragmentation in the gas phase, even for those proteins assumed to be in native-like conformations (low charge states).Conclusions
For the low charge states of myoglobin (presumed native-like structures), the trend in the UVPD fragmentation yields (from largest yield to lowest yield) showed good agreement with the average B-factor trend (from highest B-factor to lowest), especially for the 8+ and 9+ states of holo-myoglobin. The UVPD fragmentation trends appear to reflect little dependence on side-chain interactions and rather on secondary and tertiary interactions with amide hydrogens. For example, in a folded helix the backbone amide-hydrogens are involved in strong hydrogen bonding interactions, whereas in a loop region these amide-hydrogens are not heavily involved in stabilizing interactions. The corresponding UVPD fragment yields suggested that cleavages were favored at backbone positions for which the amides were not involved in hydrogen bonding interactions. As the charge state increased, the UVPD fragmentation yields increased for the N-terminus region of the G-helix, the F-helix and the FG loop. Additionally, UVPD led to preferential backbone cleavages in regions of the protein with higher relative B-factors, such as loop regions and frayed helical ends. The increase in fragment yields for specific structural regions upon UVPD of the 8+ to 10+ states of myoglobin are consistent with unfolding of the G and F helices and FG and GH loops. Differences in fragmentation may also arise in part from the difference in molar absorptivities between the various secondary structures in which loops have greater absorptivities than helices. Comparison of UVPD fragmentation yields for the holo and apo forms revealed similarities in the AGH helical core of the protein, whereas the fragmentation yields were suppressed in holo-myoglobin for those regions known to participate in heme binding. HCD displayed enhanced backbone fragmentation in regions that were more likely reflective of the known preferential charge-directed residue-specific backbone cleavages affiliated with collisional activation rather than correlation with the least stable regions based on B-factors. These results suggest that UV photodissociation of native-like proteins may be a promising avenue for elucidating the most flexible and the more rigid regions of proteins in the gas phase. Additional case studies are warranted to explore this strategy in more detail, especially to decipher the impact of charge state and the influence of bound ligands on the ability to derive correlations between UV photodissociation yields and structural issues.Acknowledgements
Funding from the NSF (CHE-1402753) and the Welch Foundation (F-1155) is acknowledged.Notes and references
- K. R. Acharya and M. D. Lloyd, Trends Pharmacol. Sci., 2005, 26, 10–14 CrossRef CAS PubMed.
- H. Yu, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 332–334 CrossRef CAS.
- L. Konermann, S. Vahidi and M. A. Sowole, Anal. Chem., 2014, 86, 213–232 CrossRef CAS PubMed.
- J. P. O'Brien, L. K. Mayberry, P. A. Murphy, K. S. Browning and J. S. Brodbelt, J. Proteome Res., 2013, 12, 5867–5877 CrossRef PubMed.
- M. Cammarata, K.-Y. Lin, J. Pruet, H.-W. Liu and J. Brodbelt, Anal. Chem., 2014, 86, 2534–2542 CrossRef CAS PubMed.
- J. Pan, J. Han, C. H. Borchers and L. Konermann, J. Am. Chem. Soc., 2009, 131, 12801–12808 CrossRef CAS PubMed.
- A. J. R. Heck, Nat. Methods, 2008, 5, 927–933 CrossRef CAS PubMed.
- M. E. Belov, E. Damoc, E. Denisov, P. D. Compton, S. Horning, A. A. Makarov and N. L. Kelleher, Anal. Chem., 2013, 85, 11163–11173 CrossRef CAS PubMed.
- K. Breuker and F. W. McLafferty, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18145–18152 CrossRef CAS PubMed.
- M. Sharon and C. V. Robinson, Annu. Rev. Biochem., 2007, 76, 167–193 CrossRef CAS PubMed.
- T. D. Wood, R. A. Chorush, F. M. Wampler, D. P. Little, P. B. O'Connor and F. W. McLafferty, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 2451–2454 CrossRef CAS.
- M. A. Freitas, C. L. Hendrickson, M. R. Emmett and A. G. Marshall, Int. J. Mass Spectrom., 1999, 185–187, 565–575 CrossRef.
- K. B. Shelimov, D. E. Clemmer, R. R. Hudgins and M. F. Jarrold, J. Am. Chem. Soc., 1997, 119, 2240–2248 CrossRef CAS.
- S. Vahidi, B. B. Stocks and L. Konermann, Anal. Chem., 2013, 85, 10471–10478 CrossRef CAS PubMed.
- C. A. Scarff, K. Thalassinos, G. R. Hilton and J. H. Scrivens, Rapid Commun. Mass Spectrom., 2008, 22, 3297–3304 CrossRef CAS PubMed.
- C. Uetrecht, R. J. Rose, E. van Duijn, K. Lorenzen and A. J. R. Heck, Chem. Soc. Rev., 2010, 39, 1633–1655 RSC.
- M. Vonderach, M. Winghart, L. MacAleesem, F. Chirot, R. Antoine, P. Dugourd, P. Weis, O. Hampe and M. M. Kappes, Phys. Chem. Chem. Phys., 2014, 16, 3007–3013 RSC.
- F. Lermyte, A. Konijnenberg, J. P. Williams, J. M. Brown, D. Valkenborg and F. Sobott, J. Am. Soc. Mass Spectrom., 2014, 25, 343–350 CrossRef CAS PubMed.
- Z. Zhang, S. J. Browne and R. W. Vachet, J. Am. Soc. Mass Spectrom., 2014, 25, 604–613 CrossRef CAS PubMed.
- H. Zhang, W. Cui, J. Wen, R. E. Blankenship and M. L. Gross, Anal. Chem., 2011, 83, 5598–5606 CrossRef CAS PubMed.
- H. Zhang, W. Cui and M. L. Gross, Int. J. Mass Spectrom., 2013, 354–355, 288–291 CrossRef CAS PubMed.
- K. Breuker, S. Brüschweiler and M. Tollinger, Angew. Chem., Int. Ed., 2011, 50, 873–877 CrossRef CAS PubMed.
- M. Schennach and K. Breuker, Angew. Chem., Int. Ed., 2014, 53, 164–168 CrossRef CAS PubMed.
- H. Li, P. Wongkongkathep, S. L. V. Orden, R. R. O. Loo and J. A. Loo, J. Am. Soc. Mass Spectrom., 2014, 25, 343–350 CrossRef PubMed.
- K. Breuker, H. Oh, D. M. Horn, B. A. Cerda and F. W. McLafferty, J. Am. Chem. Soc., 2002, 124, 6407–6420 CrossRef CAS PubMed.
- H. Li, J. J. Wolff, S. L. Van Orden and J. A. Loo, Anal. Chem., 2014, 86, 317–320 CrossRef CAS PubMed.
- O. S. Skinner, F. W. McLafferty and K. Breuker, J. Am. Soc. Mass Spectrom., 2012, 23, 1011–1014 CrossRef CAS PubMed.
- E. W. Robinson, R. D. Leib and E. R. Williams, J. Am. Soc. Mass Spectrom., 2006, 17, 1470–1480 CrossRef PubMed.
- H. Oh, K. Breuker, S. K. Sze, Y. Ge, B. K. Carpenter and F. W. McLafferty, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 15863–15868 CrossRef CAS PubMed.
- J. P. O'Brien, W. Li, Y. Zhang and J. S. Brodbelt, J. Am. Chem. Soc., 2014, 136, 12920–12928 CrossRef PubMed.
- J. R. Cannon, M. B. Cammarata, S. A. Robotham, V. C. Cotham, J. B. Shaw, R. T. Fellers, B. P. Early, P. M. Thomas, N. L. Kelleher and J. S. Brodbelt, Anal. Chem., 2014, 86, 2185–2192 CrossRef CAS PubMed.
- J. B. Shaw, W. Li, D. D. Holden, Y. Zhang, J. Griep-Raming, R. T. Fellers, B. P. Early, P. M. Thomas, N. L. Kelleher and J. S. Brodbelt, J. Am. Chem. Soc., 2013, 135, 12646–12651 CrossRef CAS PubMed.
- J. R. Cannon, C. Kluwe, A. Ellington and J. S. Brodbelt, Proteomics, 2014, 14, 1165–1173 CrossRef CAS PubMed.
- A. D. Catherman, K. R. Durbin, D. R. Ahlf, B. P. Early, R. T. Fellers, J. C. Tran, P. M. Thomas and N. L. Kelleher, Mol. Cell. Proteomics, 2013, 12, 3465–3473 CAS.
- O. S. Skinner, A. D. Catherman, B. P. Early, P. M. Thomas, P. D. Compton and N. L. Kelleher, Anal. Chem., 2014, 86, 4627–4634 CrossRef CAS PubMed.
- D. C. Phillips, Nature, 1958, 181, 662–666 CrossRef.
- S. Schwarzinger, R. Mohana-Borges, G. J. A. Kroon, H. J. Dyson and P. E. Wright, Protein Sci., 2008, 17, 313–321 CrossRef CAS PubMed.
- D. Eliezer, J. Yao, H. J. Dyson and P. E. Wright, Nat. Struct. Mol. Biol., 1998, 5, 148–155 CAS.
- D. A. Simmons, S. D. Dunn and L. Konermann, Biochemistry, 2003, 42, 5896–5905 CrossRef CAS PubMed.
- P. Dasmeh and K. P. Kepp, PLoS One, 2013, 8, e80308 Search PubMed.
- F. M. Hughson, P. E. Wright and R. L. Baldwin, Science, 1990, 249, 1544–1548 CAS.
- R. S. Johnson and K. A. Walsh, Protein Sci., 1994, 3, 2411–2418 CrossRef CAS PubMed.
- F. W. Teale, Biochim. Biophys. Acta, 1959, 35, 543 CrossRef CAS.
- M. T. Reymond, H. J. Dyson, P. E. Wright and G. Merutka, Protein Sci., 1997, 6, 706–716 CrossRef CAS PubMed.
- R. Fraczkiewicz and W. Braun, J. Comput. Chem., 1998, 19, 319–333 CrossRef CAS.
- X. Lin, W. Zhao and X. Wang, J. Mass Spectrom., 2010, 45, 618–626 CAS.
- J. Tirado-Rives and W. L. Jorgensen, Biochemistry, 1993, 32, 4175–4184 CrossRef CAS.
- T. Ly and R. R. Julian, J. Am. Chem. Soc., 2010, 132, 8602–8609 CrossRef CAS PubMed.
- T. L. Whitaker, M. B. Berry, E. L. Ho, M. S. Hargrove, G. N. Phillips, N. H. Komiyama, K. Nagai and J. S. Olson, Biochemistry, 1995, 34, 8221–8226 CrossRef CAS.
- C. Brunet, R. Antoine, P. Dugourd, F. Canon, A. Giuliani and L. Nahon, J. Chem. Phys., 2013, 138, 064301 CrossRef PubMed.
- Y.-T. Chien, J.-K. Hwang and S.-W. Huang, in Protein Structure, ed. E. Faraggi, InTech, 2012 Search PubMed.
- H. Zhang, T. Zhang, K. Chen, S. Shen, J. Ruan and L. Kurgan, Proteins: Struct., Funct., Bioinf., 2009, 76, 617–636 CrossRef CAS PubMed.
- C. S. Tsai, Biomacromolecules: Introduction to structure, function and informatics, John Wiley & Sons, 2007 Search PubMed.
- D. Eliezer and P. E. Wright, J. Mol. Biol., 1996, 263, 531–538 CrossRef CAS PubMed.
- C. Nishimura, H. J. Dyson and P. E. Wright, J. Mol. Biol., 2006, 355, 139–156 CrossRef CAS PubMed.
- D. H. A. Correa and C. H. I. Ramos, Afr. J. Biochem. Res., 2009, 3, 164–173 CAS.
- Y. Kawamura-Konishi, H. Kihara and H. Suzuki, Eur. J. Biochem., 1988, 170, 589–595 CrossRef CAS PubMed.
- F. Wang and X. Tang, Biochemistry, 1996, 35, 4069–4078 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ESI mass spectra of holo- and apo-myoglobin, schematic representation of holo-myoglobin, histograms of HCD and UVPD fragmentation yields of apo-myoglobin, B-factors and solvent accessibilities. See DOI: 10.1039/c4sc03200d |
| This journal is © The Royal Society of Chemistry 2015 |