
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAssembly line termination in cylindrocyclophane biosynthesis: discovery of an editing type II thioesterase domain in a type I polyketide synthase†
H.
Nakamura
a,
J. X.
Wang
b and
E. P.
Balskus
*a
aDepartment of Chemistry and Chemical Biology, Harvard University, Cambridge, Massachusetts 02138, USA. E-mail: balskus@chemistry.harvard.edu
bSmall Molecule Mass Spectrometry Facility, FAS Division of Science, Cambridge, Massachusetts 02138, USA
First published on 14th April 2015
Abstract
The termination step is an important source of structural diversity in polyketide biosynthesis. Most type I polyketide synthase (PKS) assembly lines are terminated by a thioesterase (TE) domain located at the C-terminus of the final module, while other PKS assembly lines lack a terminal TE domain and are instead terminated by a separate enzyme in trans. In cylindrocyclophane biosynthesis, the type I modular PKS assembly line is terminated by a freestanding type III PKS (CylI). Unexpectedly, the final module of the type I PKS (CylH) also possesses a C-terminal TE domain. Unlike typical type I PKSs, the CylH TE domain does not influence assembly line termination by CylI in vitro. Instead, this domain phylogenetically resembles a type II TE and possesses activity consistent with an editing function. This finding may shed light on the evolution of unusual PKS termination logic. In addition, the presence of related type II TE domains in many cryptic type I PKS and nonribosomal peptide synthetase (NRPS) assembly lines has implications for pathway annotation, product prediction, and engineering.
Introduction
Polyketides are a large group of secondary metabolites that display a wide range of important biological activities.1 Many polyketides are synthesized by type I polyketide synthases (PKSs), which generate structural diversity and complexity by employing combinations of domains and modules organized into enzymatic assembly lines.2 The core domains of a type I PKS module are the ketosynthase (KS), acyltransferase (AT), and acyl-carrier protein (ACP) domains. The AT domain selects an initial substrate or an extender unit, typically malonyl-coenzyme A (CoA) or methylmalonyl-CoA, and loads it onto the phosphopantetheinyl (ppant) arm of the ACP domain. The KS domain then catalyses a decarboxylative Claisen condensation between the ACP-tethered extender unit and a polyketide intermediate covalently bound to an active site cysteine. The initial β-ketothioester product can be further modified by tailoring domains present in the module (typically ketoreductase (KR), dehydratase (DH), and enoyl reductase (ER) domains). Type I PKSs can either be modular or iterative: each module of a type I modular PKS catalyses one round of elongation and tailoring, while the single module of a type I iterative PKS catalyses multiple rounds of elongation and tailoring.At the end of an assembly line, the nascent polyketide product must be released from the PKS by a terminating enzyme. The type of reaction used for termination has a large influence on the architecture of the final natural product and is therefore an important source of structural diversity in polyketide biosynthesis. There are three major types of termination logic used by type I PKS assembly lines: release by C-terminal thioesterase (TE) domains, release by alternate C-terminal domains, and release by freestanding enzymes in trans. The canonical and most well-studied strategy is C-terminal TE domain-mediated termination. The TE domains that catalyse assembly line terminations are referred to as type I TEs and are typically found at the C-terminus of the final module. TE domains release the nascent polyketide product from the PKS assembly lines by catalysing either hydrolysis or macrocyclization using a conserved Ser-His-Asp catalytic triad (Fig. 1a, top).3 The second termination logic involves product release by C-terminal domains other than TEs. For example, reductase (R) domains catalyse reductive release in multiple PKS assembly lines, including those in xenovulene4 and azanigerone5 biosyntheses. Furthermore, a special type of TE domain called TE/CLC (Claisen-like cyclase) catalyses Claisen-type condensations to form a new ring system in the biosynthesis of the fungal polyketides naphthopyrone6 and aflatoxin.7 Both of these approaches involve the use of C-terminal domains in cis.
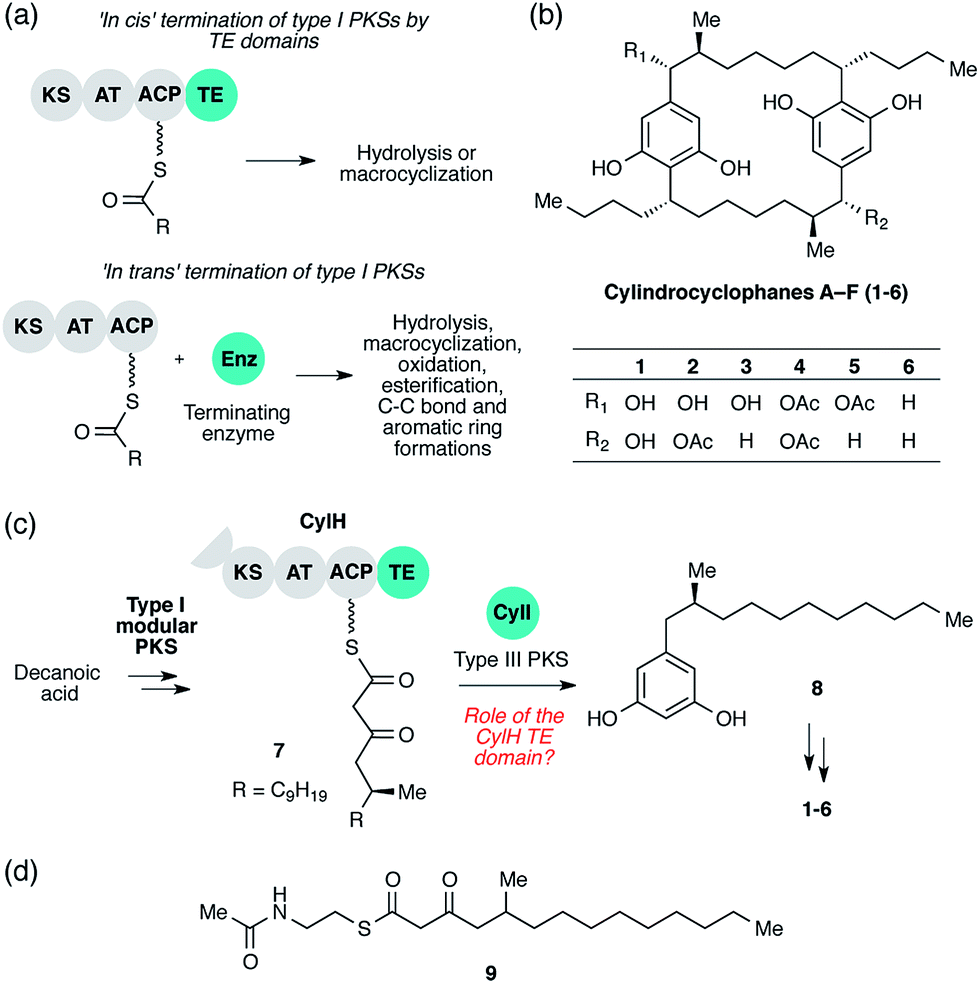 | ||
| Fig. 1 Cylindrocyclophane biosynthesis involves unusual type I polyketide synthase (PKS) termination logic. (a) Biosynthetic logic for termination of type I PKS assembly lines. (b) Structures of cylindrocyclophanes isolated from Cylindrospermum licheniforme ATCC 29412. (c) Overview of the hypothesized cylindrocyclophane biosynthetic pathway and proposed termination chemistry. KS = ketosynthase, AT = acyltransferase, ACP = acyl carrier protein, TE = thioesterase. (d) Structure of the N-acetylcysteamine thioester (SNAC) mimic of β-keto ACP thioester 7. | ||
The third termination logic involves PKSs that lack a C-terminal domain for product release, instead employing a separate, freestanding enzyme in trans (Fig. 1a, bottom). For example, a freestanding TE promotes hydrolysis in the biosynthesis of polyether ionophores,8 and a discrete acyltransferase catalyses macrolactamization in rifamycin biosynthesis.9 More recently, a freestanding ketosynthase was postulated to condense two β-ketothioester intermediates, each tethered to a separate type I modular PKS, to form a pyrone ring in myxopyronin biosynthesis.10In trans termination chemistry is frequently found in type I iterative PKS systems. A discrete thioesterase releases a polyene intermediate from a type I iterative PKS in enediyne biosynthesis,11 and an acyltransferase releases an intermediate from an iterative PKS in lovastatin synthesis.12 Other types of enzymes that catalyse product release from PKS assembly lines in trans include a pyridoxal 5′-phosphate (PLP)-dependent enzyme in fumonisin biosynthesis,13 a lactamase in atrochrysone carboxylic acid biosynthesis14 and a Baeyer–Villiger monooxygenase in aurafuron biosynthesis.15 The variety of enzymes that participate in assembly line termination further illustrates the importance of this step to polyketide structural diversity.
The cylindrocyclophanes (1–6) are a family of cyanobacterial polyketides with a rare [7.7]paracyclophane carbon scaffold (Fig. 1b). The biosynthesis of these natural products appears to involve an unusual assembly line termination mechanism.16 In our previous investigation of cylindrocyclophane biosynthesis, we hypothesized that putative biosynthetic precursor 8 would be generated by in trans termination of the type I modular PKS assembly line (CylD–CylH) via freestanding type III PKS CylI (Fig. 1c). In support of this hypothesis, purified CylI converted 9, an N-acetylcysteamine thioester (SNAC) mimic of the CylH-bound β-ketothioester 7 (Fig. 1d), to resorcinol 8. This result implied that CylI accepts acyl-ACP 7 as a substrate in vivo. Type III PKSs typically use acyl-CoA electrophiles and form aromatic products by catalysing iterative decarboxylative Claisen condensations directly with malonyl-CoA extender units.17 Recently several type III PKSs have been shown to accept acyl-ACPs as electrophiles, including ArsB and ArsC from Azobacter vinelandii18 and BpsA from Bacillus subtilis,19 which use ACP-thioesters from fatty acid biosynthesis. Additionally, FtpA from Myxococcus xanthus accepts an acyl-ACP substrate produced by a fatty acyl-AMP ligase FtpD,20 a type III PKS domain fused to a type I fatty acid synthase (FAS) uses an acyl-ACP electrophile in the synthesis differentiation factor DIF-1 by Dictyostelium discoideum,21 and gramicidin synthase from Streptomyces coelicolor accepts both acyl-CoA and acyl-ACP electrophiles in vitro.22 While several type III PKSs are known to utilize acyl-ACP substrates, to our knowledge CylI is the first type III PKS predicted to use an acyl-ACP substrate generated by a type I modular PKS.16
Although there are many examples of PKS termination catalysed by discrete enzymes in trans, all previously characterized pathways with in trans termination logic, to the best of our knowledge, have assembly lines that lack C-terminal TE domains. Cylindrocyclophane biosynthesis, however, is unique in possessing both a freestanding terminating enzyme and a C-terminal TE domain on the final module of its type I modular PKS assembly line (CylH). This is surprising, as termination via a type III PKS should not require a type I TE domain. To understand the unusual termination chemistry involved in this pathway and the role of the TE domain, we have characterized the activity of CylH and CylI in vitro. We confirm that type III PKS CylI accepts a CylH-tethered substrate. We employ mutagenesis to show that the CylH TE domain activity does not influence assembly line termination by CylI or catalyse competing hydrolysis of acyl-ACP 7. Rather, biochemical experiments and bioinformatic analysis of the CylH TE domain reveal similarity to type II TEs, freestanding enzymes that typically play editing roles in polyketide biosynthesis.2,23 Further searches of bacterial genomes reveal that similar editing TE domains are widely incorporated into cryptic PKS and nonribosomal peptide synthetase (NRPS) assembly lines.
These discoveries have broad implications for how we think about the roles of TE domains in biosynthesis. Previous work has revealed the existence of type II-like TE domains in type I PKS assembly lines, including those that make ajudazole and jerangolid,24–26 but their biochemical functions have been largely unexplored. The discovery of an in cis TE domain with a dedicated editing role raises the possibility that similar domains are found in other assembly lines. This prospect overturns canonical enzymatic assembly line biosynthetic logic, which assumes that in cis TE domains mediate only termination chemistry, and should influence how we annotate newly discovered assembly lines. Our findings also raise the possibility that in cis editing is important for optimal function of certain assembly lines, an observation that could be exploited to better engineer these systems. Overall, the discovery of an editing C-terminal TE domain in a type I PKS assembly line has implications both for the evolution of unusual termination logic and our understanding of the functions of these important domains.
Results and discussion
Assessment of the CylH TE domain's role in assembly line termination
We previously showed that type III PKS CylI converted acyl-SNAC substrate 9 to resorcinol 8via two extension cycles using malonyl-CoA as an extender unit, followed by cyclization, decarboxylation and aromatization.16 The direct use of 9 by CylI suggested that the C-terminal TE domain of the final PKS module of CylH was not necessary for assembly line termination. In this work, we aimed to investigate whether the CylH TE domain is catalytically active and to elucidate the role of the TE domain in the context of the type I PKS assembly line. We cloned, heterologously expressed and purified the final PKS module of CylH (CylHPKS = KS-AT-ACP-TE) and the excised CylH TE domain (CylHTE), as well as their corresponding TE active site serine to alanine mutants (CylHPKS-S1201A and CylHTE-S95A) using E. coli as an expression host (Fig. S1–S3, Tables S1 and S2†). Production of CylHPKS and CylHPKS-S1201A required co-expression of chaperones GroES and GroEL. Successful posttranslational modification of the ACP domain of CylHPKS and the CylHPKS-S1201A mutant by Sfp, a promiscuous phosphopantetheinyl transferase,27 was verified using the BODIPY-CoA loading assay (Fig. S4 and Table S3†).We performed in vitro assays to examine the elongation activity of CylHPKS (Fig. 2a). Holo CylHPKS was prepared in situ by incubation with CoA and Sfp, and the resulting holo enzyme was incubated with acyl-SNAC 10 and malonyl-CoA. The assay mixtures were quenched by the addition of 1 M KOH, followed by heating at 65 °C to ensure complete hydrolysis of acyl-ACP 7 to β-keto acid 11. We did not observe significant amounts of product decarboxylation under these conditions (Fig. S5†). CylHPKS catalysed elongation of substrate 10, leading to the formation of 11 in full assays but not negative controls lacking either enzyme or substrate (Fig. S6 and Table S4†). Addition of type III PKS CylI to the assay mixture resulted in the production of resorcinol 8 (Fig. 2b). We observed a small amount of resorcinol formation in the absence of CylHPKS due to promiscuous activity of CylI toward substrate 10, which undergoes three extension cycles prior to cyclization, decarboxylation and aromatization to give resorcinol 8. The substantial increase in the rate of resorcinol formation in the presence of CylHPKS, however, shows that CylI uses the β-keto ACP thioester 7 as a preferred substrate. Overall, these assays confirmed that CylHPKS catalysed elongation and that CylI could utilize a CylH-tethered substrate.
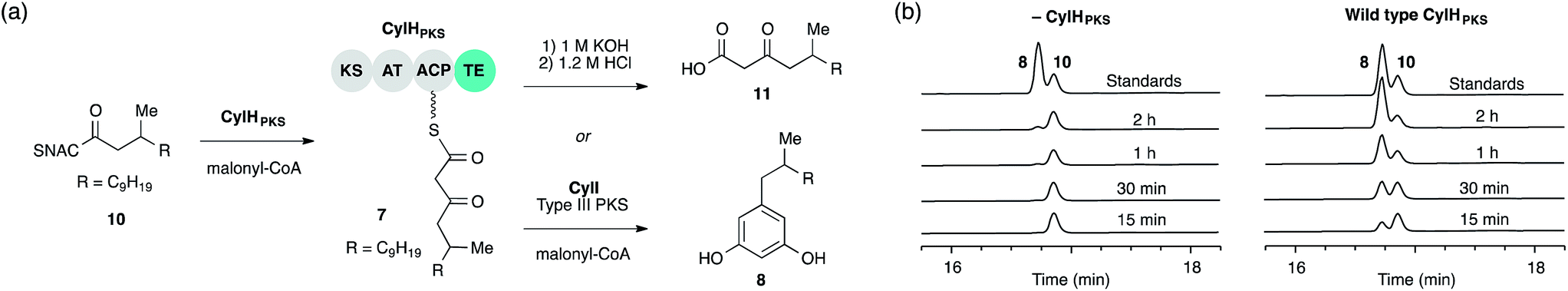 | ||
| Fig. 2 The final module of the cylindrocyclophane PKS assembly line generates a substrate for type III PKS CylI. (a) Assays used to study the elongation activity of CylHPKS and the resorcinol formation by CylI using acyl-ACP substrate 7. (b) HPLC assays of CylI-catalysed resorcinol formation show that CylH increases reaction efficiency (monitored at 210 nm). | ||
To determine whether TE domain activity impacted the rate of resorcinol formation, we performed the elongation assay with CylHPKS-S1201A. The TE domain mutation did not appear to affect the qualitative rate of resorcinol formation, suggesting that the TE domain is not directly involved in either aromatic ring formation or a competing hydrolysis (Fig. 3a and S7†). If the TE domain is catalytically active and has a competing hydrolytic activity for acyl-ACP 7, we should observe accumulation of the hydrolysis product 11. We therefore quantified the amount of resorcinol 8 and acyl-ACP hydrolysis product 11 in assays containing CylI and either CylHPKS or CylHPKS-S1201A by LC-MS (Fig. 3b, S8 and S9†). In the full assays containing CylI, very small quantities of hydrolysis product 11 were detected (∼0.3 µM for both CylHPKS and CylHPKS-S1201A) compared to resorcinol 8, indicating that the CylH TE domain does not have a competing hydrolytic activity in assembly line termination. The trace amount of hydrolysis product 11 observed in these assays can also result from background hydrolysis of acyl-ACP 7, if the transfer of nascent polyketide to CylI is inefficient. This is likely the reason for the higher concentrations of 11 present in assays lacking CylI.
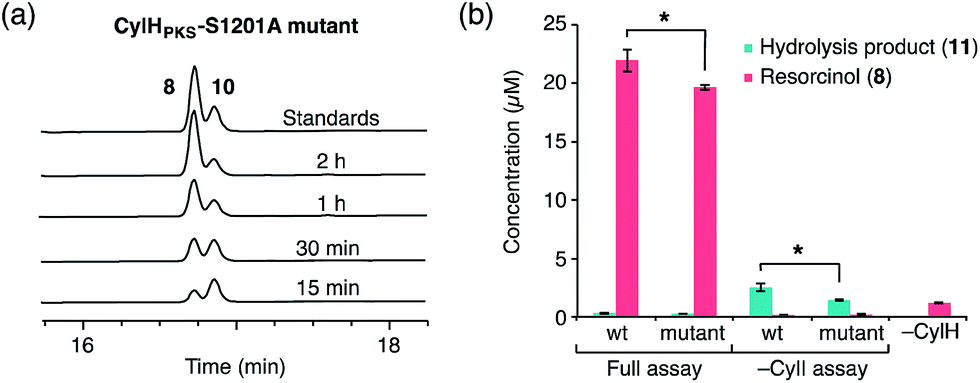 | ||
| Fig. 3 The activity of the CylH TE domain does not impact assembly line termination by CylI. (a) HPLC assay of CylI-catalysed resorcinol formation with CylHPKS-S1201A mutant (monitored at 210 nm). (b) LC-MS quantitation of resorcinol 8 and hydrolysis product 11 in assays containing CylI and either wild type CylHPKS or the CylHPKS-S1201A mutant. Mean values ± standard deviation of assays done in triplicate are shown, and p-values <0.05 are marked with *. | ||
We also tested whether addition of CylHTE impacted the efficiency of CylI-catalysed resorcinol formation using SNAC substrate 9, and observed no qualitative difference in reaction efficiency in the presence of CylHTE or the CylHTE-S95A mutant (Fig. S10†). These results support the hypothesis that the CylH TE domain does not influence termination in the PKS assembly line.
Bioinformatic and phylogenetic analyses of the CylH TE domain
To determine whether the apparent lack of CylH TE domain activity arose from mutations to key active site residues, specifically the conserved Ser–His–Asp catalytic triad, we analysed this domain using bioinformatics. When we aligned the amino acid sequence of CylHTE with those of characterized type I TEs, the catalytic Asp residue appeared to be absent (Fig. S1†). However, when we performed the alignment with sequences of characterized type II TEs, the catalytic triad of CylHTE was conserved. This discrepancy between the two alignments suggested that despite its position at the C-terminus of a type I modular PKS, the CylH TE domain might be more closely related to type II TEs. Additional support for this hypothesis included an HHPred structure homology search, which identified type II TEs RifR28 and RedJ29 as the top two hits. Using the crystal structure of RifR as a template, we constructed a CylHTE homology model, which possesses a well-aligned catalytic triad (Fig. 4a).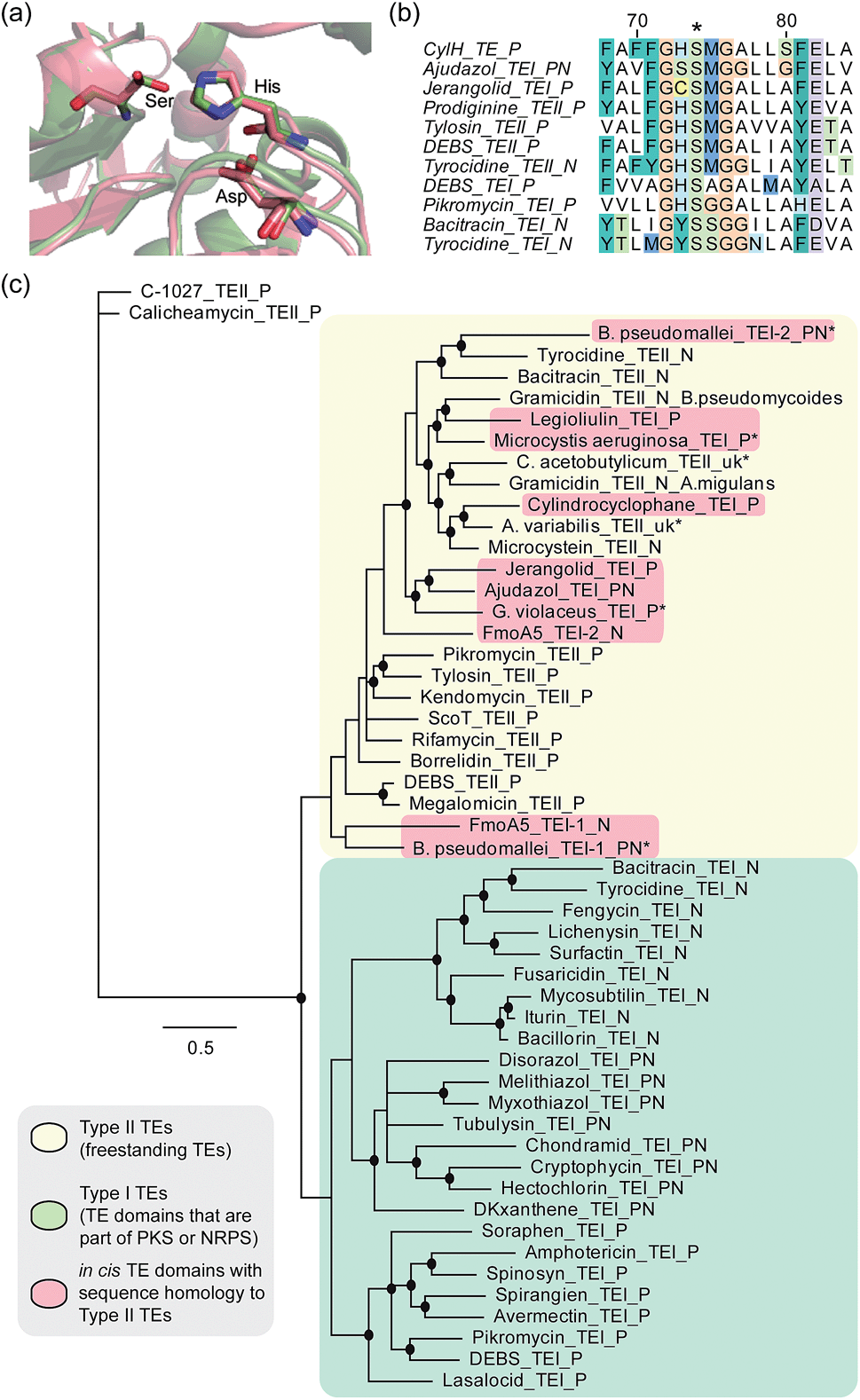 | ||
| Fig. 4 Bioinformatic analysis of the CylH TE domain reveals similarity to type II thioesterases. (a) Homology model of the CylH TE domain (green) overlaid with type II TE RifR (pink) shows conservation of the catalytic triad (Asp–His–Ser). (b) Multiple sequence alignment of the hydrolase signature sequence of characterized TEs shows that the CylH TE domain has the GHSMG motif common in type II TEs. (c) Phylogenetic tree of characterized TE domains using Bayesian estimation reveals clustering of the CylH TE domain with type II TEs (all bootstrap values were greater than 50. Ones with values above 75 are marked with •). TEI = type I TE, TEII = type II TE, P = PKS, N = NRPS, PN = hybrid PKS-NRPS, uk = unknown. TEs that have not been previously characterized are marked with *. The accession numbers of the proteins used in the analysis are listed in Table S7†). | ||
As in the case of CylHTE, C-terminal TE domains in the ajudazole (AjuTE) and jerangolid (JerTE) biosyntheses were previously reported to resemble type II TEs.24 In fact, CylHTE appears to have high sequence homology to AjuTE (53% similarity, 33% identity) and JerTE (55% similarity, 39% identity). Interestingly, the hydrolase signature sequence (GXSXG) of CylHTE matches that of type II TEs perfectly (GHSMG), while the His residue in the signature sequence of AjuTE and JerTE are replaced by Ser and Cys respectively.24 This His residue has been shown to stabilize the active site by forming a hydrogen bond with the backbone carbonyl of the catalytic His residue in the crystal structure of RifR.28 The hydrolase signature sequence of CylHTE provides another evidence for its closer homology to type II TEs.
Finally, we constructed a phylogenetic tree following the method used by Buntin and co-workers24 and found that CylHTE clusters with freestanding type II TEs, not C-terminal type I TEs (Fig. 4c). Overall, these analyses show that the CylH TE domain has the catalytic residues needed for activity, but is more closely related to type II TEs rather than the type I TEs more typically associated with modular PKSs.
In all previous studies that have characterized in cis type II-like TE domains in enzymatic assembly lines, the TE domain is thought to play a role in product release. The C-terminal TE domain in ajudazol has been found to catalyse isochromanone ring formation,24 and the homologous TE domains in jerangolid30 and legioliulin25 biosyntheses have been implicated in promoting lactonization and isochromanone ring formation, respectively. The excised AjuTE and JerTE domains also possess hydrolytic activity toward short chain acyl p-nitrophenyl esters, substrates commonly used to assess the editing function of type II TEs.24 More recently, the NRPS assembly line that constructs 4-methyloxazoline-containing natural products was found to contain an unusual module (FmoA5) with two tandem internal TE domains (C-T-TE-TE-A-T).26 The second TE domain was reported to have homology to type II TEs, but the activity of these TE domains has not yet been determined.26 These unusual in cis type II TE domains clustered with other freestanding type II TEs along with the CylH TE domain in our phylogenetic tree.
Investigation of the editing function of the CylH TE domain
Unlike AjuTE and JerTE, the activity of the CylH TE domain does not support a role in assembly line termination. Consequently, we investigated the possibility that this domain has an editing role in cylindrocyclophane biosynthesis. In our LC-MS quantification assay, we observed a slight but statistically significant difference in the hydrolytic activities of CylHPKS and CylHPKS-S1201A mutant in the absence of CylI (2.5 µM with CylHPKSvs. 1.4 µM with CylHPKS-S1201A, Fig. 3b). This activity appears to be consistent with one of the functions of type II editing TEs to reactivate impaired assembly lines by hydrolysing acyl-ACP intermediates with prolonged half-lives.23c–f In addition, CylI appears to produce a slightly greater amount of resorcinol 8 when utilizing substrate generated by the wild type CylHPKS (21.9 µM with CylHPKSvs. 19.5 µM with CylHPKS-S1201A). This increase in activity may be a result of the TE domain editing activity rescuing stalled CylHPKS. However, as the observed differences were slight we performed additional experiments to study the function of the CylH TE domain.The editing roles of characterized type II TEs have been largely inferred from their ability to hydrolyze short chain acyl-SNAC substrates that mimic the acyl-ACP thioesters generated on stalled assembly lines. Such acyl-ACP thioesters could result from either premature decarboxylation of the extender unit23c,f or mispriming of the apo enzyme by promiscuous phosphopantetheinyl transferases using an acyl-CoA instead of free CoA.23b,e,f We therefore examined the activity of CylHTE and CylHTE-S95A toward acetyl-SNAC using a previously reported assay and compared its hydrolysis kinetics to that of TycF, a type II TE with known activity toward this substrate (kcat/Km = 3.6 ± 0.7 M−1 s−1, Km = 47.9 ± 9.7 mM).23e CylHTE displayed hydrolytic activity toward acetyl-SNAC (kcat/Km = 1.0 ± 0.4 M−1 s−1, Km = 4.5 ± 1.7 mM), while CylHTE-S95A was inactive (Fig. S11 and Table S5†). However, CylHTE activity was lower than that of TycF and tylosin type II TE (kcat/Km = 2.5 M−1 s−1).23c In addition, we tested the hydrolytic activity of CylHTE on other short-chain acyl-SNACs (propionyl, butyryl, pentanoyl and hexanoyl). CylHTE displayed the highest activity toward acetyl-SNAC, followed by propionyl-SNAC, and was less active toward longer acyl-SNACs (Fig. S12 and Table S6†). The preference of CylHTE for shorter acyl-SNACs is consistent with this domain having evolved to remove acyl-ACP that could result from extender unit decarboxylation or mispriming. Together these experiments confirm that CylHTE is catalytically active and support a role in assembly line editing.
To investigate the editing function of the CylH TE domain in the context of the cylindrocyclophane PKS assembly line, we tested its ability to rescue a stalled version of CylHPKS. We generated 14C-acetyl CylHPKS and CylHPKS-S1201A in situ by loading [acetyl-1-14C]-CoA onto the respective apo enzymes using Sfp. The amount of radiolabel on each of the enzymes was monitored by autoradiography (Fig. 5a). Comparison of the radiolabelling of CylHPKS and CylHPKS-S1201A over time revealed a loss of radiolabel from CylHPKS and accumulation of radiolabel on CylHPKS-S1201A (Fig. 5b). This observation is consistent with the CylH TE domain hydrolyzing the 14C-acetyl-ACP intermediate, an activity analogous to that of discrete type II TEs. Overall, the result of this assay supports the hypothesis that the CylH TE domain plays an editing role in cylindrocyclophane biosynthesis.
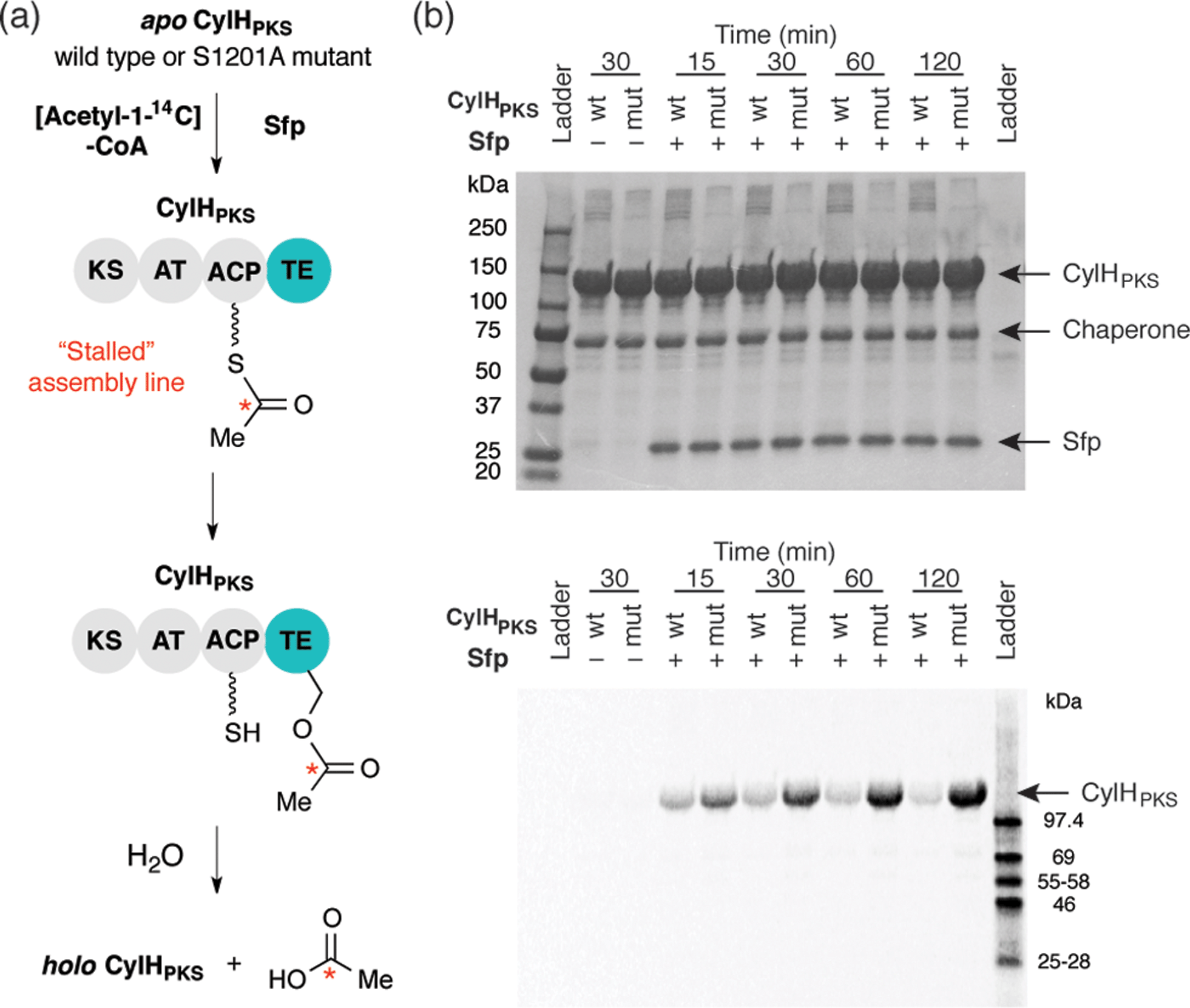 | ||
| Fig. 5 The CylH TE domain hydrolyzes an acetyl-ACP intermediate on a stalled version of CyH. (a) Design of a gel autoradiography assay to assess the ability of CylHPKS to hydrolyze an [acetyl-1-14C]-ACP intermediate. (b) Autoradiogram of these reaction time course for hydrolysis of 14C-acetyl-ACP-loaded wild type CylHPKS and the CylHPKS-S1201A mutant (top image = Coomassie stained gel, bottom = autoradiogram). | ||
To the best of our knowledge, this work represents the first demonstration of editing activity for an in cis TE domain that lacks a role in assembly line termination. There are several potential explanations for this observation. The activity of CylH TE domain could have evolved to maintain a higher proportion of holo enzyme during cylindrocyclophane PKS assembly line function. As formation of monomeric resorcinol 8 by the cylindrocyclophane PKS assembly line appears to be tightly coupled to the subsequent macrocyclizing, dimerization event,16 we hypothesize that the editing function of the CylH TE domain may be important for controlling the rate of monomer generation. In addition, this assembly line arrangement is intriguing from an evolutionary perspective. The CylH TE domain may have evolved through fusion of a freestanding type II TE to the C-terminus of the type I modular PKS. Alternately, this domain could represent a snapshot in the evolution of a non-canonical in trans termination mechanism, with the presence of the C-terminal TE domain a relic from an ancestral terminating PKS module.
Identification of in cis type II TE domains in other PKS and NRPS assembly lines
We performed additional bioinformatic analyses to determine the prevalence of in cis type II TE domains in type I PKS or NRPS assembly lines. A BLASTP search of sequenced bacterial genomes using the CylH TE domain as a query identified 84 unique type I PKS and NRPS assembly lines containing in cis type II TE domains, including AjuTE and JerTE (Table S8†). Many of the hits have unusual domains or domain organizations, such as a hybrid PKS–NRPS from Burkholderia mallei that contains TE domains at both its N- and C-termini. The ubiquity of in cis type II TE domains, as well as the diverse activities of those that have been biochemically characterized, suggests that these domains may possess new, unexplored functions and could be important new tools for combinatorial biosynthesis. These TE domains may also display editing activity at particular steps in biosynthesis that could be important for maintaining the efficiency of these assembly line enzymes. While the function of the TE/CLC domain in maintaining assembly line fidelity of fungal type I iterative PKS from aflatoxin biosynthesis is well-characterised,31 the editing roles of TE domains in type I modular PKS assembly lines have not been widely explored. A better understanding of the functions of these domains could be applied to improve the efficiency of engineered assembly lines through the strategic introduction of editing activity. Finally, the wide distribution of C-terminal type II TE domains has implications for future characterization and annotation of cryptic multimodular assembly line pathways in bacterial genomes, as one can no longer assume that these domains follow canonical termination logic.Conclusions
In summary, we have discovered and characterized the role of a C-terminal in cis type II TE domain in cylindrocyclophane biosynthesis. We have shown that the activity of this domain does not influence assembly line termination by type III PKS CylI. However, the CylH TE domain is catalytically active and can hydrolyze acetyl-SNAC and acetyl-ACP, as well as other short-chain acyl-SNACs, which is consistent with a role in preserving assembly line function via editing. The activity of freestanding type II TEs are often important for maintaining the efficiency of enzymatic assembly lines. Likewise, the CylH TE domain may also be necessary for maintaining the productivity of the type I modular PKS in cylindrocyclophane biosynthesis. Bioinformatic analyses suggest that such atypical in cis type II TE domains are widespread in other multimodular assembly lines. Further efforts to understand the functions of these domains will improve both our understanding of modular enzymatic assembly line function and our ability to engineer and utilize the chemical capabilities of these systems.Acknowledgements
We acknowledge Li Zha for help with the autoradiography experiments, Zebulon Levine for the synthesis of acetyl-SNAC, and Jonathan Marks for help with the bioinformatics. We received financial support for Harvard University and the Searle Scholars Program. H.N. acknowledges fellowship support from the NSF GRFP (DGE1144152).Notes and references
- (a) J. Staunton and K. J. Weissman, Nat. Prod. Rep., 2001, 18, 380 RSC; (b) B. J. Rawlings, Nat. Prod. Rep., 2001, 18, 190 RSC; (c) B. J. Rawlings, Nat. Prod. Rep., 2001, 18, 231 RSC.
- M. A. Fischbach and C. T. Walsh, Chem. Rev., 2006, 106, 3468 CrossRef CAS PubMed.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255 RSC.
- A. M. Bailey, R. J. Cox, K. Harley, C. M. Lazarus, T. J. Simpson and E. Skellam, Chem. Commun., 2007, 4053 RSC.
- A. O. Zabala, W. Xu, Y.-H. Chooi and Y. Tang, Chem. Biol., 2012, 19, 1049 CrossRef CAS PubMed.
- I. Fujii, A. Watanabe, U. Sankawa and Y. Ebizuka, Chem. Biol., 2001, 8, 189 CrossRef CAS.
- T. P. Korman, J. M. Crawford, J. W. Labonte, A. G. Newman, J. Wong, C. A. Townsend and S.-C. Tsai, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 6246 CrossRef CAS PubMed.
- (a) T. Liu, D. You, C. Valenzano, Y. Sun, J. Li, Q. Yu, X. Zhou, D. E. Cane and Z. Deng, Chem. Biol., 2006, 13, 945 CrossRef CAS PubMed; (b) B. M. Harvey, H. Hong, M. A. Jones, Z. A. Hughes-Thomas, R. M. Goss, M. L. Heathcote, V. M. Bolanos-Garcia, W. Kroutil, J. Staunton, P. F. Leadlay and J. B. Spencer, ChemBioChem, 2006, 7, 1435 CrossRef CAS PubMed; (c) T. Liu, X. Lin, X. Zhou, Z. Deng and D. E. Cane, Chem. Biol., 2008, 15, 449 CrossRef CAS PubMed.
- T.-W. Yu, Y. Shen, Y. Doi-Katayama, L. Tang, C. Park, B. S. Moore, C. R. Hutchinson and H. G. Floss, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 9051 CrossRef CAS.
- H. Sucipto, S. C. Wenzel and R. Müller, ChemBioChem, 2013, 14, 1581 CrossRef CAS PubMed.
- (a) R. Kong, L. P. Goh, C. W. Liew, Q. S. Ho, E. Murugan, B. Li, K. Tang and Z.-X. Liang, J. Am. Chem. Soc., 2008, 130, 8142 CrossRef CAS PubMed; (b) J. Zhang, S. G. Van Lanen, J. Ju, W. Liu, P. C. Dorrestein, W. Li, N. L. Kelleher and B. Shen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1460 CrossRef CAS PubMed; (c) M. Kotaka, R. Kong, I. Qureshi, Q. S. Ho, H. Sun, C. W. Liew, L. P. Goh, P. Cheung, Y. Mu, J. Lescar and Z.-X. Liang, J. Biol. Chem., 2009, 284, 15739 CrossRef CAS PubMed; (d) K. Belecki, J. M. Crawford and C. A. Townsend, J. Am. Chem. Soc., 2009, 131, 12564 CrossRef CAS PubMed.
- X. Xie, M. J. Meehan, W. Xu, P. C. Dorrestein and Y. Tang, J. Am. Chem. Soc., 2009, 131, 8388 CrossRef CAS PubMed.
- R. Gerber, L. Lou and L. Du, J. Am. Chem. Soc., 2009, 131, 3148 CrossRef CAS PubMed.
- T. Awakawa, K. Yokota, N. Funa, F. Doi, N. Mori, H. Watanabe and S. Horinouchi, Chem. Biol., 2009, 16, 613 CrossRef CAS PubMed.
- B. Frank, S. C. Wenzel, H. B. Bode, M. Scharfe, H. Blöcker and R. Müller, J. Mol. Biol., 2007, 374, 24 CrossRef CAS PubMed.
- H. Nakamura, H. A. Hamer, G. Sirasani and E. P. Balskus, J. Am. Chem. Soc., 2012, 134, 18518 CrossRef CAS PubMed.
- M. B. Austin and J. P. Noel, Nat. Prod. Rep., 2003, 20, 79 RSC.
- A. Miyanaga, N. Funa, T. Awakawa and S. Horinouchi, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 871 CrossRef CAS PubMed.
- C. Nakano, H. Ozawa, G. Akanuma, N. Funa and S. Horinouchi, J. Bacteriol., 2009, 191, 4916 CrossRef CAS PubMed.
- T. Hayashi, Y. Kitamura, N. Funa, Y. Ohnishi and S. Horinouchi, ChemBioChem, 2011, 12, 2166 CrossRef CAS PubMed.
- M. B. Austin, T. Saito, M. E. Bowman, S. Haydock, A. Kato, B. S. Moore, R. R. Kay and J. P. Noel, Nat. Chem. Biol., 2006, 2, 494 CrossRef CAS PubMed.
- (a) L. Song, F. Barona-Gomez, C. Corre, L. Xiang, D. W. Udwary, M. B. Austin, J. P. Noel, B. S. Moore and G. L. Challis, J. Am. Chem. Soc., 2006, 128, 14754 CrossRef CAS PubMed; (b) S. Grüschow, T. J. Buchholz, W. Seufert, J. S. Dordick and D. H. Sherman, ChemBioChem, 2007, 8, 863 CrossRef PubMed; (c) J. A. Chemler, T. J. Buchholz, T. W. Geders, D. L. Akey, C. M. Rath, G. E. Chlipala, J. L. Smith and D. H. Sherman, J. Am. Chem. Soc., 2012, 134, 7359 CrossRef CAS PubMed.
- (a) A. R. Butler, N. Bate and E. Cundliffe, Chem. Biol., 1999, 6, 287 CrossRef CAS; (b) D. Schwarzer, H. D. Mootz, U. Linne and M. A. Marahiel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14083 CrossRef CAS PubMed; (c) M. L. Heathcote, J. Staunton and P. F. Leadlay, Chem. Biol., 2001, 8, 207 CrossRef CAS; (d) B. S. Kim, T. A. Cropp, B. J. Beck, D. H. Sherman and K. A. Reynolds, J. Biol. Chem., 2002, 277, 48028 CrossRef CAS PubMed; (e) E. Yeh, R. M. Kohli, S. D. Bruner and C. T. Walsh, ChemBioChem, 2004, 5, 1290 CrossRef CAS PubMed; (f) M. Kotowska and K. Pawlik, Appl. Microbiol. Biotechnol., 2014, 98, 7735 CrossRef CAS PubMed.
- K. Buntin, K. J. Weissman and R. Müller, ChemBioChem, 2010, 11, 1137 CrossRef CAS PubMed.
- T. Ahrendt, M. Miltenberger, I. Haneburger, F. Kirchner, M. Kronenwerth, A. O. Brachmann, H. Hilbi and H. B. Bode, ChemBioChem, 2013, 14, 1415 CrossRef CAS PubMed.
- A. Muliandi, Y. Katsuyama, K. Sone, M. Izumikawa, T. Moriya, J. Hashimoto, I. Kozone, M. Takagi, K. Shin-ya and Y. Ohnishi, Chem. Biol., 2014, 21, 923 CrossRef CAS PubMed.
- L. E. N. Quadri, P. H. Weinreb, M. Lei, M. M. Nakano, P. Zuber and C. T. Walsh, Biochemistry, 1998, 37, 1585 CrossRef CAS PubMed.
- H. B. Claxton, D. L. Akey, M. K. Silver, S. J. Admiraal and J. L. Smith, J. Biol. Chem., 2009, 284, 5021 CrossRef CAS PubMed.
- J. R. Whicher, G. Florova, P. K. Sydor, R. Singh, M. Alhamadsheh, G. L. Challis, K. A. Reynolds and J. L. Smith, J. Biol. Chem., 2011, 286, 22558 CrossRef CAS PubMed.
- B. Julien, Z.-Q. Tian, R. Reid and C. D. Reeves, Chem. Biol., 2006, 13, 1277 CrossRef CAS PubMed.
- A. L. Vagstad, S. B. Bumpus, K. Belecki, N. L. Kelleher and C. A. Townsend, J. Am. Chem. Soc., 2012, 134, 6865 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S12; Tables S1–S8, full experimental details and procedures, 1H and 13C NMR spectral data and HRMS data of compounds 10 and 11 and the internal standards. See DOI: 10.1039/c4sc03132f |
| This journal is © The Royal Society of Chemistry 2015 |