
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Dehydrogenation, disproportionation and transfer hydrogenation reactions of formic acid catalyzed by molybdenum hydride compounds†
Michelle C.
Neary
and
Gerard
Parkin
*
Department of Chemistry, Columbia University, New York, New York 10027, USA. E-mail: parkin@columbia.edu
First published on 14th January 2015
Abstract
The cyclopentadienyl molybdenum hydride compounds, CpRMo(PMe3)3−x(CO)xH (CpR = Cp, Cp*; x = 0, 1, 2 or 3), are catalysts for the dehydrogenation of formic acid, with the most active catalysts having the composition CpRMo(PMe3)2(CO)H. The mechanism of the catalytic cycle is proposed to involve (i) protonation of the molybdenum hydride complex, (ii) elimination of H2 and coordination of formate, and (iii) decarboxylation of the formate ligand to regenerate the hydride species. NMR spectroscopy indicates that the nature of the resting state depends on the composition of the catalyst. For example, (i) the resting states for the CpMo(CO)3H and CpMo(PMe3)(CO)2H systems are the hydride complexes themselves, (ii) the resting state for the CpMo(PMe3)3H system is the protonated species [CpMo(PMe3)3H2]+, and (iii) the resting state for the CpMo(PMe3)2(CO)H system is the formate complex, CpMo(PMe3)2(CO)(κ1-O2CH), in the presence of a high concentration of formic acid, but CpMo(PMe3)2(CO)H when the concentration of acid is low. While CO2 and H2 are the principal products of the catalytic reaction induced by CpRMo(PMe3)3−x(CO)xH, methanol and methyl formate are also observed. The generation of methanol is a consequence of disproportionation of formic acid, while methyl formate is a product of subsequent esterification. The disproportionation of formic acid is a manifestation of a transfer hydrogenation reaction, which may also be applied to the reduction of aldehydes and ketones. Thus, CpMo(CO)3H also catalyzes the reduction of a variety of ketones and aldehydes to alcohols by formic acid, via a mechanism that involves ionic hydrogenation.
Introduction
The growing demand for energy requires the development of alternative energy sources that are more sustainable than fossil fuels and have a reduced impact on the environment. As such, both the sun and the wind are considered to be major candidates for the production of renewable energy.1 However, since energy production from these sources would vary with location and time, it is essential to couple the production of energy with efficient methods for storage and transportation.2 In this regard, an attractive energy storage medium is provided by hydrogen,3 which can, for example, be consumed in a fuel cell with only water as a byproduct. Unfortunately, a problem with using hydrogen in this manner is that present storage and transportation techniques are inadequate.4 For example, not only does storing liquid hydrogen present a safety risk, considerable energy is also required to liquefy the hydrogen and maintain it in this form.2cIn view of these issues, efforts have focused on the use of physisorption and chemical methods to provide hydrogen on demand.5 Of these approaches, the use of formic acid as a chemical medium for storing H2 has garnered much attention.2,6,7 Specifically, although formic acid contains only 4.4% hydrogen by mass, it is an attractive storage medium because (i) it is a liquid at room temperature and is therefore easy to handle and transport, (ii) it is commercially available on a large scale8,9 and (iii) the byproduct of H2 release is carbon dioxide which, in principle, can be trapped10 and either recycled11,12 or used as a C1 source for other chemicals.12 With respect to achieving the release of H2 from formic acid, much research has been directed towards the discovery of both heterogeneous13 and homogeneous14–22 catalyst systems. However, since most of these catalyst systems feature precious metals (e.g. Pd, Pt, Au), an important current thrust is the discovery of catalysts that utilize earth abundant nonprecious metals.19–22 Therefore, we report here a series of molybdenum compounds that are not only effective for the catalytic release of H2 from formic acid, but are also capable of (i) forming methanol via disproportionation of formic acid and (ii) using formic acid as a reagent for the transfer hydrogenation of aldehydes and ketones.
Results and discussion
We have previously described the first example of a molybdenum compound, namely Cp*Mo(PMe3)2(CO)H,20,23 to serve as a catalyst for the dehydrogenation (decarboxylation) of formic acid (Scheme 1).24 In order to understand the factors that influence dehydrogenation, we sought to extend the investigation to the series of compounds, CpMo(PMe3)3−x(CO)xH and Cp*Mo(PMe3)3−x(CO)xH (x = 0, 1, 2 or 3), in which the substitution of (i) PMe3 ligands by CO and (ii) Cp* ligands by Cp would be expected to exert significant electronic and structural effects, as illustrated by the fact that the oxidation potentials of the CpMo(PMe3)3−x(CO)xH/[CpMo(PMe3)3−x(CO)xH]+ couples span a range of ca. 2 V.25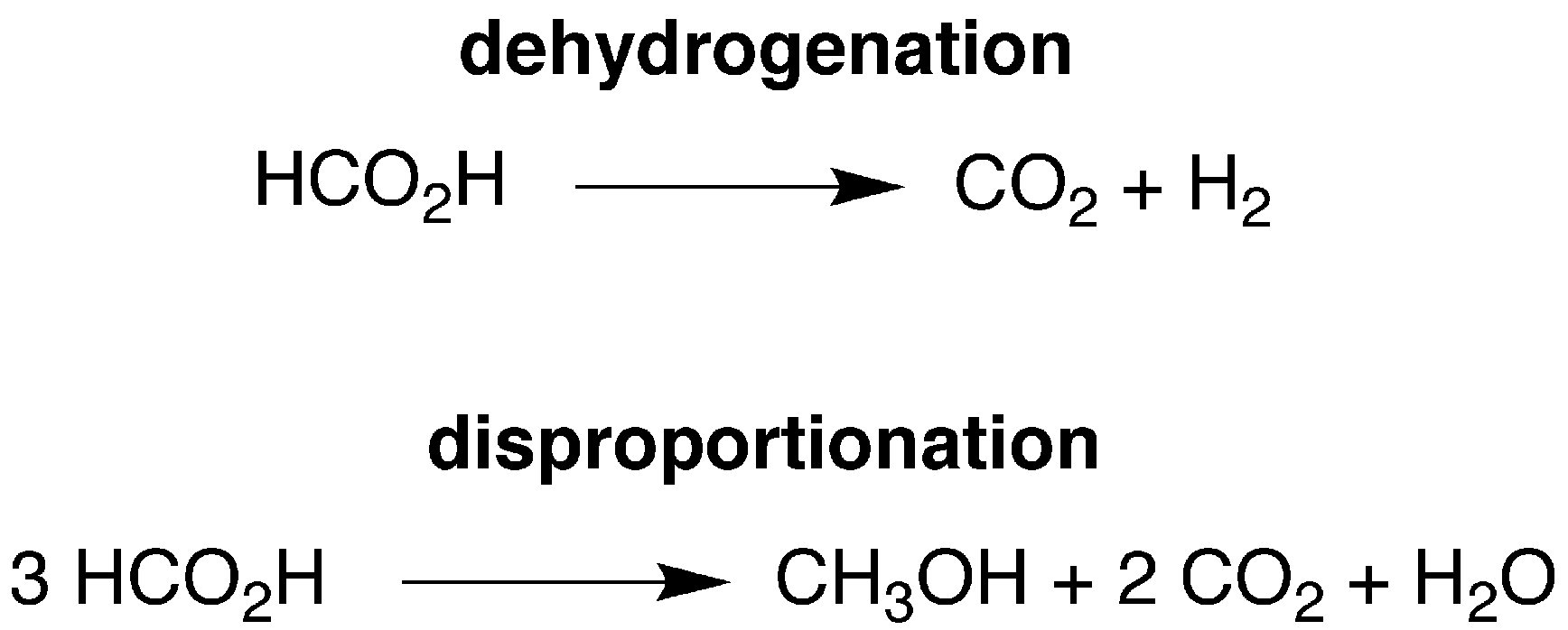 | ||
| Scheme 1 Dehydrogenation and disproportionation of HCO2H. | ||
Indeed, comparison of CpMo(PMe3)3H,26 CpMo(PMe3)2(CO)H,27 CpMo(PMe3)(CO)2H27,28 and CpMo(CO)3H29 indicates that substitution of a CO ligand by PMe3 has a profound effect on the rate of decarboxylation and release of H2 (Table 1).30 For example, while the reaction catalyzed by the tricarbonyl derivative, CpMo(CO)3H, requires temperatures of 100 °C in order to proceed efficiently in benzene, the bis(phosphine) derivative, CpMo(PMe3)2(CO)H, is an effective catalyst at room temperature.31 Most interestingly, the trend is not monotonic, such that the tris(phosphine) derivative CpMo(PMe3)3H exhibits no significant catalytic activity at room temperature. Thus, of the series of compounds CpMo(PMe3)3−x(CO)xH, the bis(phosphine) derivative, CpMo(PMe3)2(CO)H, has the greatest activity.
| Cp | Cp* | |
|---|---|---|
| a [CpRMo(PMe3)3−x(CO)xH] = 0.016 M, [HCO2H]initial = 0.39 M, C6D6, values at 50% conversion. | ||
| CpRMo(PMe3)2(CO)H | 31 | 54 |
| CpRMo(PMe3)(CO)2H | 0.64 | 1.2 |
| CpRMo(CO)3H | 0.67 | 0.33 |
Although CpMo(PMe3)3H exhibits little activity at room temperature, it does, nevertheless, induce the decarboxylation of formic acid at elevated temperatures. However, it is evident that CpMo(PMe3)3H is not necessarily the catalytically active species because it is converted to the monocarbonyl compound CpMo(PMe3)2(CO)H under these conditions (Scheme 2).32
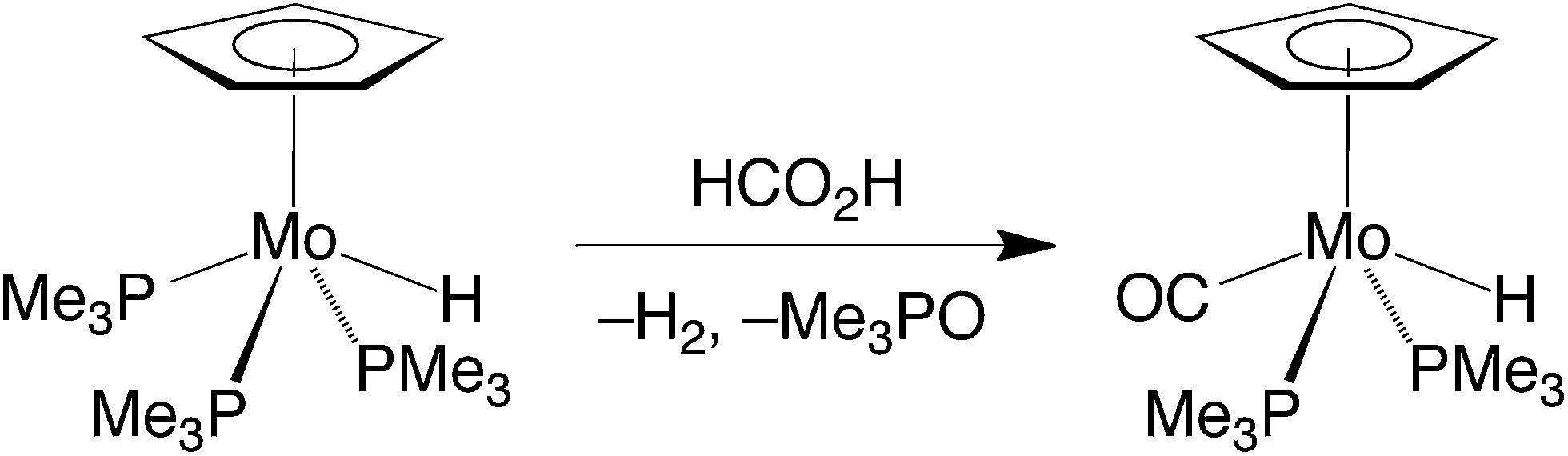 | ||
| Scheme 2 Conversion of CpMo(PMe3)3H to CpMo(PMe3)2(CO)H by formic acid. | ||
The nature of the substituents on the cyclopentadienyl ring also influences the reactivity, with Cp*Mo(PMe3)2(CO)H being a more active catalyst than CpMo(PMe3)2(CO)H (Table 1).31 The difference is not, however, as pronounced as that achieved by variation of the number of PMe3 and CO ligands.
The essential features of the mechanism proposed for the catalytic cycle are illustrated in Scheme 3 and involve two main sequences, namely (i) protonation, elimination of H2 and coordination of formate, and (ii) decarboxylation of the formate ligand to regenerate the hydride species. In order to obtain evidence for such a mechanism, the nature of the molybdenum-containing compounds that form upon treatment of CpMo(PMe3)3−x(CO)xH (x = 0, 1, 2, or 3) with formic acid has been probed by NMR spectroscopy.
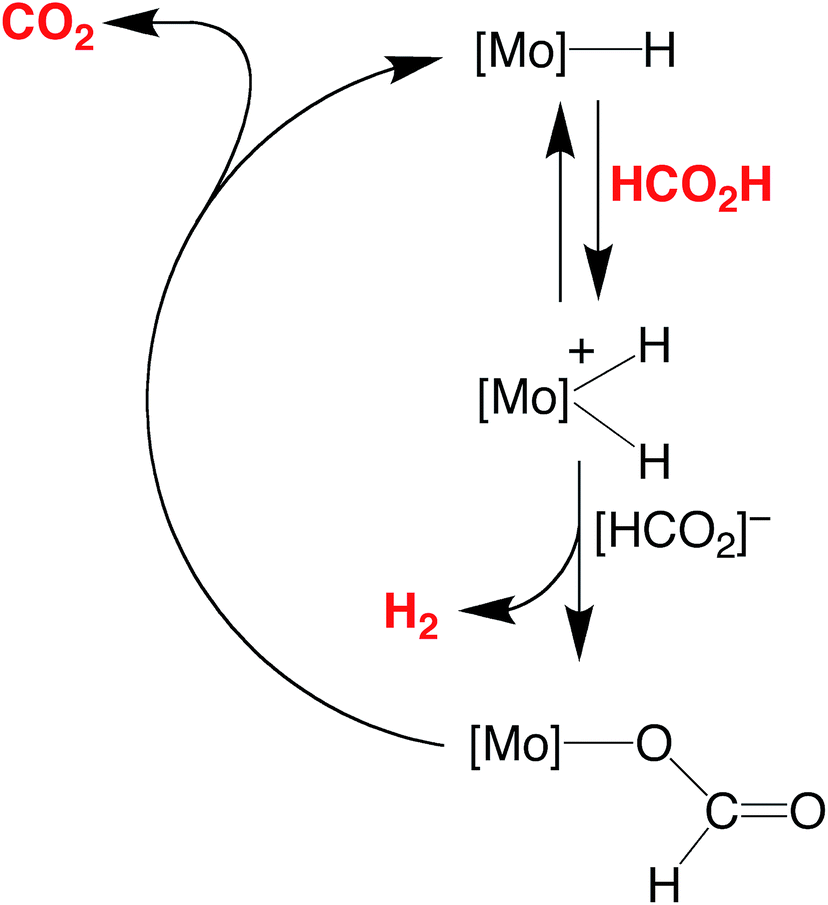 | ||
| Scheme 3 Essential features for the proposed mechanism of dehydrogenation of HCO2H by CpRMo(PMe3)3−x(CO)xH. | ||
Evidence that CpMo(PMe3)3−x(CO)xH may be protonated by formic acid is provided by the observation that treatment of CpMo(PMe3)3H with formic acid results in the formation of [CpMo(PMe3)3H2][HCO2], which is characterized by two hydride signals in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio at −2.57 [m, 2JP–H = 54, 2JP–H = 49 and 2JH–H = 8] and −5.74 [m, 2JP–H = 46, 2JP–H = 8 and 2JH–H = 8] in the 1H NMR spectrum (CD3CN) at 239 K.33 Support for the identification of [CpMo(PMe3)3H2][HCO2] is provided by previous studies in which [CpMo(PMe3)3H2]+ has been generated by the reaction of CpMo(PMe3)3H with HBF4.25,26a Furthermore, we have also structurally characterized the chloride derivative [CpMo(PMe3)3H2][Cl] by X-ray diffraction (Fig. 1), which reinforces its identification as a dihydride rather than a dihydrogen complex.34
:1 ratio at −2.57 [m, 2JP–H = 54, 2JP–H = 49 and 2JH–H = 8] and −5.74 [m, 2JP–H = 46, 2JP–H = 8 and 2JH–H = 8] in the 1H NMR spectrum (CD3CN) at 239 K.33 Support for the identification of [CpMo(PMe3)3H2][HCO2] is provided by previous studies in which [CpMo(PMe3)3H2]+ has been generated by the reaction of CpMo(PMe3)3H with HBF4.25,26a Furthermore, we have also structurally characterized the chloride derivative [CpMo(PMe3)3H2][Cl] by X-ray diffraction (Fig. 1), which reinforces its identification as a dihydride rather than a dihydrogen complex.34
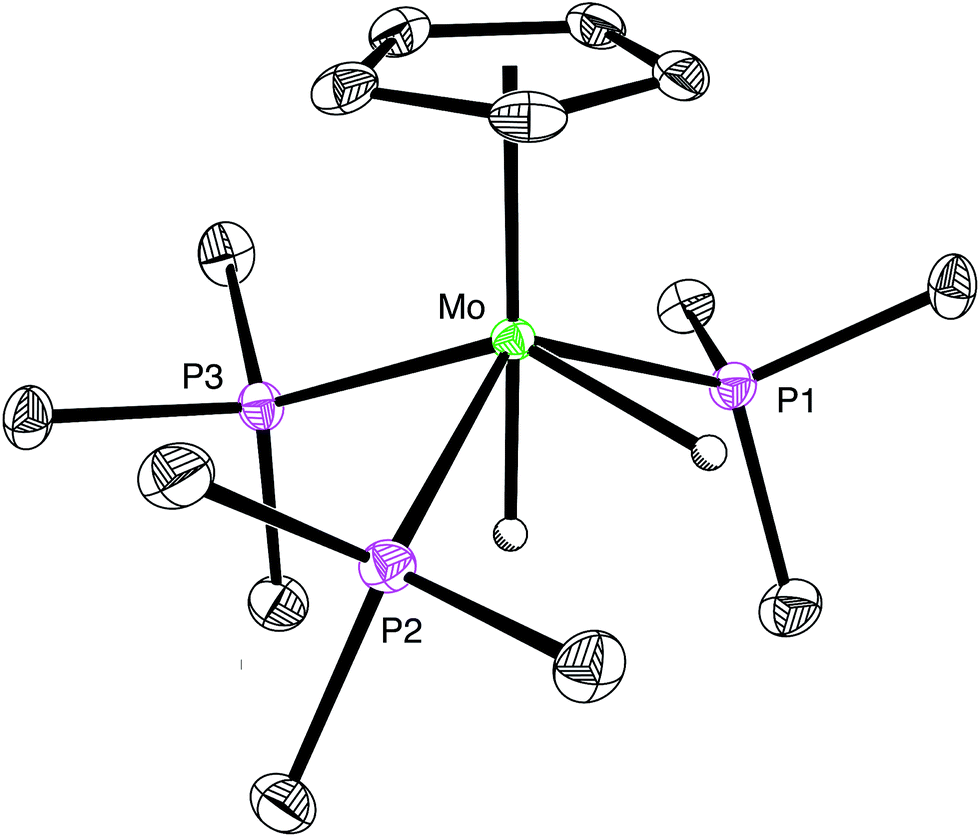 | ||
| Fig. 1 Molecular structure of [CpMo(PMe3)3H2][Cl] (only the cation is shown). | ||
However, despite the fact that CpMo(PMe3)3H is readily protonated, it is not, as noted above, an effective catalyst for decarboxylation of formic acid at room temperature. Presumably, the catalytic inactivity of CpMo(PMe3)3H is a consequence of dissociation of H2 from [CpMo(PMe3)3H2][HCO2] being non-facile at room temperature. Supporting this suggestion, the tetrafluoroborate derivative, [CpMo(PMe3)3H2][BF4], is stable with respect to dissociation of H2 at 80 °C.25
Evidence for the formation of a protonated species has also been obtained for the reaction of CpMo(PMe3)2(CO)H with formic acid. Specifically, treatment of CpMo(PMe3)2(CO)H in d8-toluene with an excess of formic acid at low temperature (276 K) allows for observation of a protonated species, [CpMo(PMe3)2(CO)H2]+, which is characterized by a triplet signal assignable to the hydride ligands at −3.33 ppm [t, 2JP–H = 43 Hz] in the 1H NMR spectrum.35,36 In contrast to [CpMo(PMe3)3H2]+, however, [CpMo(PMe3)2(CO)H2]+ does not persist at room temperature, dissociating H2 and coordinating formate to give CpMo(PMe3)2(CO)(κ1-O2CH). The latter compound is also unstable and only exists for a prolonged period in the presence of excess formic acid because it slowly dissociates CO2 and regenerates CpMo(PMe3)2(CO)H.37 It is, therefore, evident that both protonation and release of H2 is facile in this system, such that decarboxylation is the turnover-limiting step of the catalytic cycle in benzene during the initial stages. However, as the formic acid is consumed, the pseudo first order rate constant for protonation of CpMo(PMe3)2(CO)H decreases relative to that for decarboxylation of CpMo(PMe3)2(CO)(κ1-O2CH), such that CpMo(PMe3)2(CO)H becomes the resting state during the latter stage of the catalytic transformation.
Unlike CpMo(PMe3)3H and CpMo(PMe3)2(CO)H, the dicarbonyl and tricarbonyl compounds, CpMo(PMe3)(CO)2H and CpMo(CO)3H, do not react with formic acid in benzene at room temperature. As such, elevated temperatures are required for efficient catalysis. However, although it is unreactive towards formic acid at room temperature, previous studies have shown that CpMo(PMe3)(CO)2H reacts with a stronger acid, namely HBF4, to release H2, with no observation of the dihydride cation, [CpMo(PMe3)(CO)2H2]+.38,39 Likewise, CpMo(CO)3H rapidly eliminates H2 upon treatment with TfOH to afford CpMo(CO)3OTf, with no observation of [CpMo(CO)3H2]+.39a,40 On the basis of these observations, it is evident that the reduced basicity of the metal center resulting from the increased replacement of the PMe3 ligands by CO41,42 causes protonation to be turnover-limiting.
The above observations clearly indicate that, for the series of CpMo(PMe3)3−x(CO)xH compounds, the turnover-limiting step depends on the distribution of PMe3 and CO ligands. Specifically, (i) the catalytic activities of CpMo(CO)3H and CpMo(PMe3)(CO)2H are limited by their susceptibility to protonation by formic acid, (ii) the activity of CpMo(PMe3)3H is limited by the inability of [CpMo(PMe3)3H2]+ to dissociate H2, and (iii) the activity of CpMo(PMe3)2(CO)H is limited by decarboxylation of the formate intermediate, CpMo(PMe3)2(CO)(κ1-O2CH), in the presence of a high concentration of formic acid, but by protonation of CpMo(PMe3)2(CO)H when the concentration of acid is low.
The different forms of the catalyst resting states provide some insight into the non-monotonic variation in activity upon substituting PMe3 for carbonyl ligands. Thus, while CpMo(CO)3H has a low activity because the metal center is not susceptible to protonation, CpMo(PMe3)3H has a low activity because the protonated derivative [CpMo(PMe3)3H2]+ is relatively stable with respect to elimination of H2. As such, the hybrid carbonyl/phosphine derivatives, CpRMo(PMe3)2(CO)H, have the greatest reactivity.
Interestingly, while CO2 and H2 are the principal products of the catalytic conversion of formic acid by CpRMo(PMe3)3−x(CO)xH (R = H, Me), methanol and methyl formate are also observed. The formation of methanol is a consequence of disproportionation of formic acid (Scheme 1), while the methyl formate is a result of subsequent esterification. The observation of this additional pathway is of note in view of the fact that the generation of methanol is a key component of the potential methanol economy,43 and yet there are only two reports for the formation of methanol by the homogeneous catalytic disproportionation of formic acid.15g,17c,44 Furthermore, both of these reports describe catalysts that contain precious metals, namely iridium17c and ruthenium.15g As such, the ability to catalyze this transformation by compounds containing the nonprecious metal, molybdenum, is noteworthy. The selectivity for the catalytic formation of methanol and methyl formate relative to decarboxylation by CpRMo(PMe3)3−x(CO)xH depends on the nature of the cyclopentadienyl substituents and the number of carbonyl ligands, and is greatest for CpMo(CO)3H, which can achieve a selectivity45 (21%) that is intermediate between the values for the iridium (12%)17c and ruthenium (27%)15g,46 systems.
A simplified mechanism for the formation of methanol is illustrated in Scheme 4 and is based on that previously proposed for the catalytic ionic hydrogenation of ketones.34,39,47–49 Thus, it is proposed that CpRMo(PMe3)3−x(CO)xH delivers a hydride ligand to the protonated form of formic acid, [HC(OH)2]+, which is present due to autoionization50 or generated incipiently, thereby forming methylene diol. The formation of methanol from CH2(OH)2 can be achieved in principle by either (i) disproportionation to methanol and formic acid,51 or by (ii) dehydration to formaldehyde followed by catalytic ionic hydrogenation akin to that for the first reduction sequence of formic acid (Scheme 4).
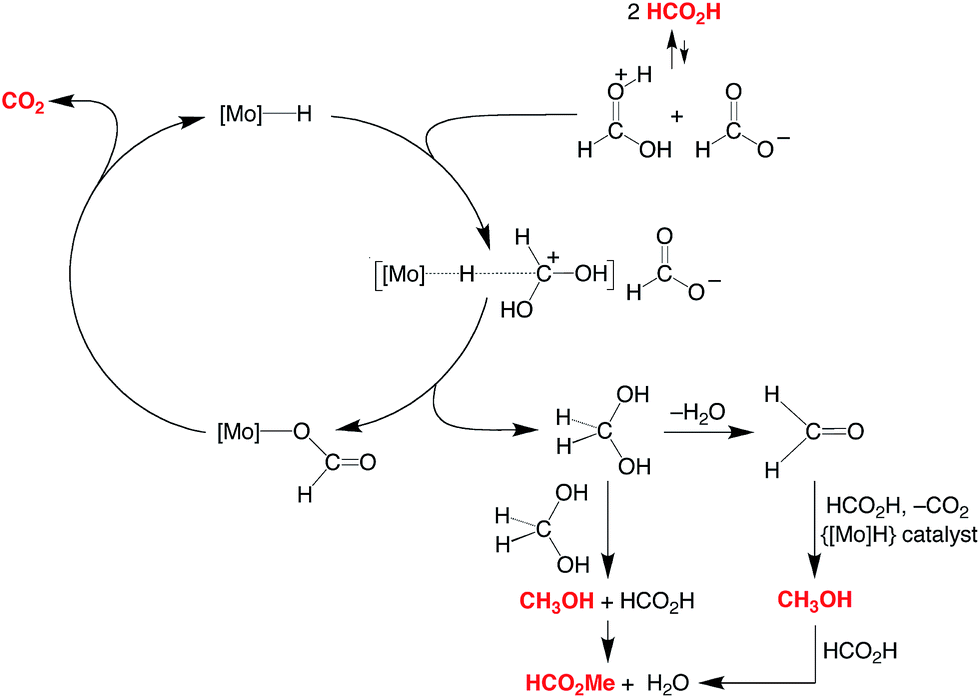 | ||
| Scheme 4 Essential features for the proposed mechanism of disproportionation of HCO2H by CpRMo(PMe3)3−x(CO)xH. | ||
Support for a direct proton and hydride transfer, rather than reaction with the H2 released via decarboxylation, is provided by the observation that performing the reaction under D2 (1 atm) results in no deuterium incorporation into the methanol or methyl formate.
On the basis of the mechanisms illustrated in Scheme 3 and Scheme 4 for the dehydrogenation and disproportionation of formic acid, the relative selectivity of the two processes (dehydrogenation versus disproportionation) is influenced by the preference of the metal center to transfer a hydride ligand to [HC(OH)2]+ (which is present in low concentration) relative to its tendency to undergo protonation and liberate H2 (Scheme 5). As such, the pKB and the hydricity of CpRMo(PMe3)3−x(CO)xH are expected to play an important role in influencing the selectivity. In this regard, while such values have been reported for a variety of metal hydride derivatives,41,52–54 data for all the compounds described here are not available. Nevertheless, the kinetic hydricity of CpMo(PMe3)(CO)2H is significantly greater than that of CpMo(CO)3H, by a factor of ca. 104,52 which is in accord with the notion that replacing a CO ligand by PMe3 would be expected to promote dissociation of hydride.55 However, it is also generally recognized that the basicity of a metal center increases upon replacing a CO ligand by PR3 (vide supra),41,42 and so it is nontrivial to predict a priori the relative influence of such substitution on the two pathways.
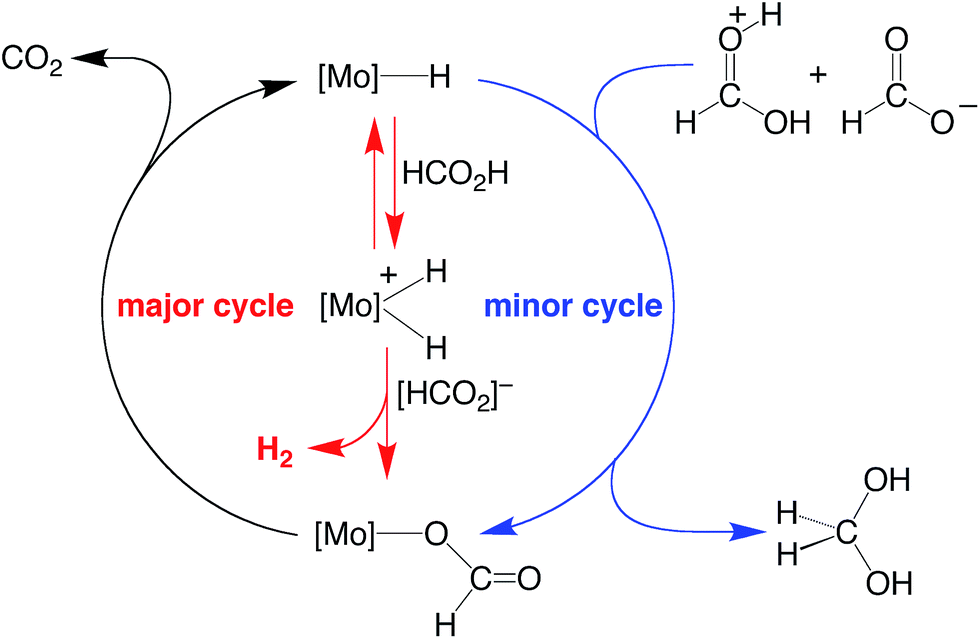 | ||
| Scheme 5 Possible origin of the selectivity of disproportionation versus dehydrogenation of formic acid. | ||
The ability of these molybdenum compounds to catalyze the disproportionation of formic acid prompted the possibility that a system could be developed to use formic acid as a reagent in the transfer hydrogenation of other substrates. In this regard, transfer hydrogenation,56,57 often employing isopropanol as the reductant, offers considerable advantages over direct reduction by hydrogen because it obviates the need to use H2.56 However, disadvantages of using isopropanol are that (i) isopropanol is obtained commercially by the hydration of propene,58 and (ii) the reactions are often reversible, such that the isopropanol needs to be used as a solvent to drive the equilibrium.56 In contrast, formic acid is available from renewable sources, and the release of CO2 as a byproduct renders the reaction effectively irreversible.56 However, catalysts that are commonly used to effect transfer hydrogenation using formic acid are largely restricted to the platinum group metals, namely Ru, Rh, Pd and Ir,59–61 with few reports employing other metals,62 and none employing Mo. It is, therefore, noteworthy that CpMo(CO)3H is also capable of effecting transfer hydrogenation of a variety of carbonyl compounds, namely RCHO (R = Me, Pri), RC(O)Me (R = Me, Pri, But, Ph) and Ph2CO, using formic acid as the reductant,63 as illustrated in Scheme 6. For example, Ph2CO is reduced by HCO2H to Ph2CH(OH) with a selectivity of 29% relative to decarboxylation and disproportionation, while PriCHO is reduced to BuiOH and HCO2Bui (3:1) with a selectivity of 32%.64–67
 | ||
| Scheme 6 Ionic hydrogenation of carbonyl compounds. | ||
Conclusions
In summary, the cyclopentadienyl molybdenum hydride compounds, CpRMo(PMe3)3−x(CO)xH (CpR = Cp, Cp*; x = 0, 1, 2 or 3), are catalysts for the dehydrogenation of formic acid. The activity of these catalysts is not a monotonic function of the number of CO or PMe3 ligands and is greatest for CpRMo(PMe3)2(CO)H. Specifically, while CpMo(CO)3H has a low activity because the metal center is not susceptible towards protonation, CpMo(PMe3)3H has a low activity because the protonated derivative [CpMo(PMe3)3H2]+ is relatively stable with respect to elimination of H2. As such, the hybrid carbonyl/phosphine derivatives, CpRMo(PMe3)2(CO)H, have the greatest reactivity. In addition to catalyzing the dehydrogenation of formic acid, CpRMo(PMe3)3−x(CO)xH also catalyzes its disproportionation to methanol and CO2via a transfer hydrogenation reaction. Similarly, CpMo(CO)3H also catalyzes the reduction of aldehydes and ketones by formic acid via a mechanism that involves ionic hydrogenation. These investigations demonstrate that molybdenum hydride compounds have much potential with respect to transfer hydrogenation reactions involving formic acid.Acknowledgements
We thank the US Department of Energy, Office of Basic Energy Sciences (DE-FG02-93ER14339) for support of this research. M. C. N. acknowledges the National Science Foundation for a Graduate Research Fellowship under Grant no. DGE 11-44155. Dr Deven P. Estes and Jonathan L. Kuo are thanked for technical assistance with high pressure experiments.Notes and references
- M. Z. Jacobson, Energy Environ. Sci., 2009, 2, 148–173 CAS.
- (a) M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC; (b) S. Enthaler, J. von Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 RSC; (c) S. Enthaler, ChemSusChem, 2008, 1, 801–804 CrossRef CAS PubMed; (d) F. Joó, ChemSusChem, 2008, 1, 805–808 CrossRef PubMed.
- S. Satyapal, J. Petrovic, C. Read, G. Thomas and G. Ordaz, Catal. Today, 2007, 120, 246–256 CrossRef CAS PubMed.
- L. Schlapbach and A. Züttel, Nature, 2001, 414, 353–358 CrossRef CAS PubMed.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS PubMed.
- W.-H. Wang, Y. Himeda, J. T. Muckerman and E. Fujita, Adv. Inorg. Chem., 2014, 66, 189–222 CrossRef CAS.
- (a) B. Loges, A. Boddien, F. Gärtner, H. Junge and M. Beller, Top. Catal., 2010, 53, 902–914 CrossRef CAS; (b) G. Laurenczy, Chimia, 2011, 65, 663–666 CrossRef CAS.
- For example, formic acid can be obtained from methyl formate and synthesis gas. See: (a) W. Reutemann and H. Kieczka, Ullmann's Encyclopedia of Industrial Chemistry, 2011, pp. 13–33 Search PubMed; (b) I. Wender, Annu. Rev. Energy, 1986, 11, 295–314 Search PubMed.
- The synthesis of formic acid from biomass has also emerged as another promising source. See: A. Kruse and A. Gawlik, Ind. Eng. Chem. Res., 2003, 42, 267–279 CrossRef CAS.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- A. Behr and K. Nowakowski, Adv. Inorg. Chem., 2014, 66, 223–258 CrossRef CAS.
- (a) M. Peters, B. Köhler, W. Kuckshinrichs, W. Leitner, P. Markewitz and T. E. Müller, ChemSusChem, 2011, 4, 1216–1240 CrossRef CAS PubMed; (b) M. He, Y. Sun and B. Han, Angew. Chem., Int. Ed., 2013, 52, 9620–9633 CrossRef CAS PubMed; (c) A. M. Appel, J. E. Bercaw, A. B. Bocarsly, H. Dobbek, D. L. Dubois, M. Dupuis, J. G. Ferry, E. Fujita, R. Hille, P. J. A. Kenis, C. A. Kerfeld, R. H. Morris, C. H. F. Peden, A. R. Portis, S. W. Ragsdale, T. B. Rauchfuss, J. N. H. Reek, L. C. Seefeldt, R. K. Thauer and G. L. Waldrop, Chem. Rev., 2013, 113, 6621–6658 CrossRef CAS PubMed.
- S.-W. Ting, C. Hu, J. K. Pulleri and K.-Y. Chan, Ind. Eng. Chem. Res., 2012, 51, 4861–4867 CrossRef CAS and references therein.
- M. Vogt, A. Nerush, Y. Diskin-Posner, Y. Ben-David and D. Milstein, Chem. Sci., 2014, 5, 2043–2051 RSC.
- (a) R. S. Coffey, Chem. Commun., 1967, 923–924 Search PubMed; (b) Y. Gao, J. Kuncheria, G. P. A. Yap and R. J. Puddephatt, Chem. Commun., 1998, 2365–2366 RSC; (c) M. L. Man, Z. Zhou, S. M. Ng and C. P. Lau, Dalton Trans., 2003, 2, 3727–3735 RSC; (d) A. Boddien, B. Loges, H. Junge and M. Beller, ChemSusChem, 2008, 1, 751–758 CrossRef CAS PubMed; (e) C. Fellay, N. Yan, P. J. Dyson and G. Laurenczy, Chem. Eur. J., 2009, 15, 3752–3760 CrossRef CAS PubMed; (f) S. Fukuzumi, T. Kobayashi and T. Suenobu, J. Am. Chem. Soc., 2010, 132, 1496–1497 CrossRef CAS PubMed; (g) S. Savourey, G. Lefèvre, J.-C. Berthet, P. Thuéry, C. Genre and T. Cantat, Angew. Chem., Int. Ed., 2014, 53, 10466–10470 CrossRef CAS PubMed.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, ChemSusChem, 2008, 1, 827–834 CrossRef CAS PubMed.
- (a) Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC; (b) Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360–7367 RSC; (c) A. J. M. Miller, D. M. Heinekey, J. M. Mayer and K. I. Goldberg, Angew. Chem., Int. Ed., 2013, 52, 3981–3984 CrossRef CAS PubMed; (d) Y. Manaka, W.-H. Wang, Y. Suna, H. Kambayashi, J. T. Muckerman, E. Fujita and Y. Himeda, Catal. Sci. Technol., 2014, 4, 34–37 RSC.
- J. Broggi, V. Jurčík, O. Songis, A. Poater, L. Cavallo, A. M. Z. Slawin and C. S. J. Cazin, J. Am. Chem. Soc., 2013, 135, 4588–4591 CrossRef CAS PubMed.
- (a) C. Bianchini, M. Peruzzini, A. Polo, A. Vacca and F. Zanobini, Gazz. Chim. Ital., 1991, 121, 543–549 CAS; (b) A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science., 2011, 333, 1733–1736 CrossRef CAS PubMed; (c) T. Zell, B. Butschke, Y. Ben-David and D. Milstein, Chem. Eur. J., 2013, 19, 8068–8072 CrossRef CAS PubMed; (d) C. Federsel, A. Boddien, R. Jackstell, R. Jennerjahn, P. J. Dyson, R. Scopelliti, G. Laurenczy and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 9777–9780 CrossRef CAS PubMed; (e) A. Boddien, B. Loges, F. Gärtner, C. Torborg, K. Fumino, H. Junge, R. Ludwig and M. Beller, J. Am. Chem. Soc., 2010, 132, 8924–8934 CrossRef CAS PubMed; (f) A. Boddien, F. Gärtner, D. Mellmann, P. Sponholz, H. Junge, G. Laurenczy and M. Beller, Chimia, 2011, 65, 214–218 CrossRef CAS.
- J. H. Shin, D. G. Churchill and G. Parkin, J. Organomet. Chem., 2002, 642, 9–15 CrossRef CAS.
- E. V. Kudrik, S. V. Makarov, E. S. Ageeva and I. A. Dereven'kov, Macroheterocycles, 2009, 2, 69–70 CAS.
- T. W. Myers and L. A. Berben, Chem. Sci., 2014, 5, 2771–2777 RSC.
- The only other Mo-containing compound that catalyzes the decarboxylation of formic acid contains Ru. See ref. 15c.
- For other decomposition pathways of formic acid, see: (a) P. Mars, J. J. F. Scholten and P. Zwietering, Adv. Catal., 1963, 14, 35–113 CAS; (b) Q. Luo, M. Beller and H. Jiao, J. Theor. Comput. Chem., 2013, 12, 1330001 CrossRef.
- J. C. Fettinger, H.-B. Kraatz, R. Poli, E. A. Quadrelli and R. C. Torralba, Organometallics, 1998, 17, 5767–5775 CrossRef CAS.
- (a) M. Brookhart, K. Cox, F. G. N. Cloke, J. C. Green, M. L. H. Green and P. M. Hare, J. Chem. Soc., Dalton Trans., 1985, 423–433 RSC; (b) F. Abugideiri, M. A. Kelland and R. Poli, Organometallics, 1992, 11, 1303–1311 CrossRef CAS.
- H. G. Alt, H. E. Engelhardt, W. Kläui and A. Müller, J. Organomet. Chem., 1987, 331, 317–327 CrossRef CAS.
- P. Kalck, R. Pince, R. Poilblanc and J. Roussel, J. Organomet. Chem., 1970, 24, 445–452 CrossRef CAS.
- (a) R. B. King, in Organometallic Syntheses, Transition Metal Compounds, ed. J. J. Eisch and R. B. King, Academic Press, Inc.: New York, 1965, vol. 1, ch. H.1, pp. 156–158 Search PubMed; (b) R. P. L. Burchell, P. Sirsch, A. Decken and G. S. McGrady, Dalton Trans., 2009, 30, 5851–5857 RSC; (c) E. O. Fischer, Inorg. Synth., 1963, 7, 136–139 CrossRef CAS.
- The formation of CO was not observed.
- CpMo(PMe3)2(CO)H TOF = 0.77 h−1; Cp*Mo(PMe3)2(CO)H, TOF = 1.8 h−1 (values at 50% conversion and room temperature).
- Likewise, Cp*Mo(PMe3)3H also converts to Cp*Mo(PMe3)2(CO)H in the presence of formic acid. See ref. 20.
- [CpMo(PMe3)3H2][HCO2] is best isolated by performing the reaction in THF or pentane, rather than benzene.
- For the structure of [CpW(PMe3)(CO)2H2][OTf], see: R. M. Bullock, J.-S. Song and D. J. Szalda, Organometallics, 1996, 15, 2504–2516 CrossRef CAS.
- It is worth noting that Poli has demonstrated that protonation of Cp*Mo(PMe3)2(CO)H by HBF4 yields a complex that exists as the dihydrogen compound, [Cp*Mo(PMe3)2(CO)(η2-H2)]+, in THF and as the dihydride, [Cp*Mo(PMe3)2(CO)H2]+, in CH2Cl2, both of which dissociate H2 at low temperature (230 K).a The former is characterized by a broad singlet at δ –5.14 in the hydride region, while the latter is characterized by a triplet at δ –3.92 (JP–H = 55 Hz), which is comparable to that observed for [CpMo(PMe3)2(CO)H2]+. Bullock has also shown that protonation of CpMo(CO)(dppe)H by TfOH at low temperature generates a dihydride species [CpMo(CO)(dppe)H2][OTf] which releases H2 to form CpMo(CO)(dppe) at room temperature.b See: (a) P. A. Dub, N. V. Belkova, O. A. Filippov, J. C. Daran, L. M. Epstein, A. Lledos, E. S. Shubina and R. Poli, Chem. Eur. J., 2010, 16, 189–201 CrossRef CAS PubMed; (b) T.-Y. Cheng, D. J. Szalda, J. Zhang and R. M. Bullock, Inorg. Chem., 2006, 45, 4712–4720 CrossRef CAS PubMed.
- Weak Brønsted acids bind to the carbonyl ligands of Cp*Mo(PMe3)2(CO)H, while strong Brønsted acids interact preferentially with the hydride ligand to yield a dihydrogen adduct. See: P. A. Dub, O. A. Filippov, N. V. Belkova, J.-C. Daran, L. M. Epstein, R. Poli and E. S. Shubina, Dalton Trans., 2010, 39, 2008–2015 Search PubMed.
- However, although decarboxylation of CpMo(PMe3)2(CO)(κ1-O2CH) is thermodynamically downhill, treatment of CpMo(PMe3)2(CO)H with a high pressure of CO2 (59.9 atm) demonstrates that CpMo(PMe3)2(CO)(κ1-O2CH) can be generated as a component of an equilibrium mixture (K = 0.01 atm−1).
- E. A. Quadrelli, H.-B. Kraatz and R. Poli, Inorg. Chem., 1996, 35, 5154–5162 Search PubMed.
- For protonation of CpMo(PPh3)(CO)2H with TfOH, see: (a) R. M. Bullock and J.-S. Song, J. Am. Chem. Soc., 1994, 116, 8602–8612 Search PubMed; (b) K.-T. Smith, J. R. Norton and M. Tilset, Organometallics, 1996, 15, 4515–4520 Search PubMed.
- For other reports of CpMo(CO)3OTf, see: (a) M. Appel, K. Schloter, J. Heidrich and W. Beck, J. Organomet. Chem., 1987, 322, 77–88 Search PubMed; (b) E. Lindner, M. Henes, W. Wielandt, K. Eichele, M. Steimann, G. A. Luinstra and H.-H. Gortz, J. Organomet. Chem., 2003, 681, 12–23 Search PubMed.
- R. H. Morris, J. Am. Chem. Soc., 2014, 136, 1948–1959 Search PubMed.
- (a) E. J. Moore, J. M. Sullivan and J. R. Norton, J. Am. Chem. Soc., 1986, 108, 2257–2263 Search PubMed; (b) G. Jia and R. H. Morris, J. Am. Chem. Soc., 1991, 113, 875–883 Search PubMed.
- G. A. Olah; A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, Weinheim, 2009 Search PubMed.
- For an early report of the heterogenous catalytic disproportionation of formic acid, see: P. Sabatier and A. Mailhe, Compt. Rend. Acad. Sci., 1911, 152, 1212–1215 Search PubMed.
- Selectivity for formation of methanol/methyl formate is defined as the percentage of formic acid used to produce methanol/methyl formate, based on the stoichiometry of the disproportionation reaction: 3 × {[MeOH]formed + [HCO2Me]formed}/{[HCO2H]consumed − [HCO2Me]formed} × 100.
- The selectivity of the ruthenium system may, however, be increased to 50% by addition of methane sulfonic acid. See ref. 15g.
- S. Namorado, M. A. Antunes, L. F. Veiros, J. R. Ascenso, M. T. Duarte and A. M. Martins, Organometallics, 2008, 27, 4589–4599 Search PubMed.
- R. M. Bullock, Chem. Eur. J., 2004, 10, 2366–2374 Search PubMed.
- (a) R. M. Bullock and M. H. Voges, J. Am. Chem. Soc., 2000, 122, 12594–12595 Search PubMed; (b) M. H. Voges and R. M. Bullock, J. Chem. Soc., Dalton Trans., 2002, 759–770 Search PubMed; (c) B. F. M. Kimmich, P. J. Fagan, E. Hauptman, W. J. Marshall and R. M. Bullock, Organometallics, 2005, 24, 6220–6229 Search PubMed; (d) J.-S. Song, D. J. Szalda, R. M. Bullock, C. J. C. Lawrie, M. A. Rodkin and J. R. Norton, Angew. Chem., Int. Ed., 1992, 31, 1233–1235 Search PubMed; (e) M. A. Antunes, S. Namorado, C. G. de Azevedo, M. A. Lemos, M. T. Duarte, J. R. Ascenso and A. M. Martins, J. Organomet. Chem., 2010, 695, 1328–1336 Search PubMed.
- K ap = 2.2 × 10−7. See: (a) T. C. Wehman and A. I. Popov, J. Phys. Chem., 1968, 72, 4031–4036 Search PubMed; (b) S. Rondinini, P. Longhi, P. R. Mussini and T. Mussini, Pure Appl. Chem., 1987, 59, 1693–1702 Search PubMed.
- (a) C. Wakai, S. Morooka, N. Matubayasi and M. Nakahara, Chem. Lett., 2004, 33, 302–303 Search PubMed; (b) S. Morooka, C. Wakai, N. Matubayasi and M. Nakahara, J. Phys. Chem. A, 2005, 109, 6610–6619 Search PubMed; (c) S. Morooka, N. Matubayasi and M. Nakahara, J. Phys. Chem. A, 2007, 111, 2697–2705 Search PubMed.
- T. Y. Cheng, B. S. Brunschwig and R. M. Bullock, J. Am. Chem. Soc., 1998, 120, 13121–13137 Search PubMed.
- M. Horn, L. H. Schappele, G. Lang-Wittkowski, H. Mayr and A. R. Ofial, Chem. Eur. J., 2013, 19, 249–263 Search PubMed.
- W. W. Ellis, J. W. Raebiger, C. J. Curtis, J. W. Bruno and D. L. DuBois, J. Am. Chem. Soc., 2004, 126, 2738–2743 Search PubMed.
- W. W. Ellis, R. Ciancanelli, S. M. Miller, J. W. Raebiger, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2003, 125, 12230–12236 Search PubMed.
- (a) J. Ito and H. Nishiyama, Tetrahedron Lett., 2014, 55, 3133–3146 Search PubMed; (b) S. Gladiali and E. Alberico, Chem. Soc. Rev., 2006, 35, 226–236 Search PubMed; (c) S. Gladiali and E. Alberico, in Transition Metals for Organic Synthesis, ed. M. Beller and C. Bolm, Wiley-VCH, Weinheim, 2nd edn, 2004, vol. 2, pp. 145–166 Search PubMed; (d) R. Malacea, R. Poli and E. Manoury, Coord. Chem. Rev., 2010, 254, 729–752 Search PubMed; (e) R. H. Morris, Chem. Soc. Rev., 2009, 38, 2282–2291 Search PubMed; (f) J. S. M. Samec, J.-E. Bäckvall, P. G. Andersson and P. Brandt, Chem. Soc. Rev., 2006, 35, 237–248 Search PubMed; (g) R. Noyori and S. Hashiguchi, Acc. Chem. Res., 1997, 30, 97–102 Search PubMed.
- (a) R. A. W. Johnstone, A. H. Wilby and I. D. Entwistle, Chem. Rev., 1985, 85, 129–170 Search PubMed; (b) S. Rajagopal, M. K. Anwer and A. F. Spatola, in Peptides: Design, Synthesis, and Biological Activity, Birkhäuser, Boston, 1994, pp. 11–26 Search PubMed; (c) B. Štefane and F. Požgan, Catal. Rev., 2014, 56, 82–174 Search PubMed; (d) Z. Shen, Y. Zhang, F. Jin, X. Zhou, A. Kishita and K. Tohji, Ind. Eng. Chem. Res., 2010, 49, 6255–6259 Search PubMed.
- J. E. Logsdon and R. A. Loke, Isopropyl alcohol, Kirk-Othmer Encycl. Chem. Technol., 2000 DOI:10.1002/0471238961.0919151612150719.a01.
- (a) J. Václavík, P. Sot, B. Vilhanová, J. Pecháček, M. Kuzma and P. Kačer, Molecules, 2013, 18, 6804–6828 Search PubMed; (b) X. Wu, C. Wang and J. Xiao, Platinum Met. Rev., 2010, 54, 3–19 Search PubMed; (c) A. Robertson, T. Matsumoto and S. Ogo, Dalton Trans., 2011, 40, 10304–10310 Search PubMed; (d) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 Search PubMed; (e) C.-Y. Huang, K.-Y. Kuan, Y.-H. Liu, S.-M. Peng and S.-T. Liu, Organometallics, 2014, 33, 2831–2836 Search PubMed; (f) V. Fábos, L. T. Mika and I. T. Horváth, Organometallics, 2014, 33, 181–187 Search PubMed; (g) T. Thananatthanachon and T. B. Rauchfuss, ChemSusChem, 2010, 3, 1139–1141 Search PubMed; (h) D. S. Matharu, D. J. Morris, G. J. Clarkson and M. Wills, Chem. Commun., 2006, 3232–3234 Search PubMed; (i) Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara, H. Arakawa and K. Kasuga, J. Mol. Catal. A: Chem., 2003, 195, 95–100 Search PubMed; (j) T. Mizugaki, Y. Kanayama, K. Ebitani and K. Kaneda, J. Org. Chem., 1998, 63, 2378–2381 Search PubMed; (k) A. Fujii, S. Hashiguchi, N. Uematsu, T. Ikariya and R. Noyori, J. Am. Chem. Soc., 1996, 118, 2521–2522 Search PubMed; (l) Y. Watanabe, T. Ohta and Y. Tsuji, Bull. Chem. Soc. Jpn., 1982, 55, 2441–2444 Search PubMed; (m) C. A. Mebi, R. P. Nair and B. J. Frost, Organometallics, 2007, 26, 429–438 Search PubMed; (n) S. Bolano, L. Gonsalvi, F. Zanobini, F. Vizza, V. Bertolasi, A. Romerosa and M. Peruzzini, J. Mol. Catal. A: Chem., 2004, 224, 61–70 Search PubMed.
- J. Yu and J. B. Spencer, Chem. Eur. J., 1999, 5, 2237–2240 Search PubMed.
- A. Nova, D. J. Taylor, A. J. Blacker, S. B. Duckett, R. N. Perutz and O. Eisenstein, Organometallics, 2014, 33, 3433–3442 Search PubMed.
- See for example: (a) T. Nakano, J. Ando, Y. Ishii and M. Ogawa, Technol. Rep. Kansai Univ., 1987, 29, 69–76 Search PubMed; (b) G. Wienhöfer, F. A. Westerhaus, R. V. Jagadeesh, K. Junge, H. Junge and M. Beller, Chem. Commun., 2012, 48, 4827–4829 Search PubMed; (c) G. Wienhöfer, I. Sorribes, A. Boddien, F. Westerhaus, K. Junge, H. Junge, R. Llusar and M. Beller, J. Am. Chem. Soc., 2011, 133, 12875–12879 Search PubMed.
- It is pertinent to note that compounds of this class have been used as catalysts for ionic hydrogenation employing H2. See, for example, ref. 48.
- Selectivities are the average of two runs.
- Selectivity for transfer hydrogenation is defined as {[ROH]formed + [HCO2R]formed}/{[HCO2H]consumed − [HCO2R]formed − [HCO2Me]formed} × 100.
- Formate esters have also been observed in other systems. See, for example, ref. 59l and 62a.
- Note that it is more difficult to reduce esters by ionic hydrogenation than it is to reduce aldehydes and ketones by ionic hydrogenation. See, for example: J. S. Song, D. J. Szalda and R. M. Bullock, Organometallics, 2001, 20, 3337–3346 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1028432. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03128h |
| This journal is © The Royal Society of Chemistry 2015 |