
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A novel copper-chelating strategy for fluorescent proteins to image dynamic copper fluctuations on live cell surfaces†
Yoon-Aa
Choi
a,
Joo Oak
Keem
a,
Cha Yeon
Kim
b,
Hye Ryeon
Yoon
c,
Won Do
Heo
d,
Bong Hyun
Chung
*a and
Yongwon
Jung
*c
aBioNano Health Guard Research Center, 125 Gwahak-ro, Yuseong-gu, Daejeon, 305-806, Republic of Korea. E-mail: chungbh@kribb.re.kr; Fax: +82-42-860-4209; Tel: +82-42-860-4442
bGraduate School of Nanoscience and Technology, Korea Advanced Institute of Science and Technology (KAIST), Republic of Korea
cDepartment of Chemistry, KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon, 305-701, Republic of Korea. E-mail: ywjung@kaist.ac.kr; Fax: +82-42-350-2810; Tel: +82-42-350-2817
dDepartment of Biological Sciences, KAIST, Republic of Korea
First published on 19th November 2014
Abstract
Copper is indispensable in most aerobic organisms although it is toxic if unregulated as illustrated in many neurodegenerative diseases. To elucidate the mechanisms underlying copper release from cells, a membrane-targeting reporter which can compete with extracellular copper-binding molecules is highly desirable. However, engineering a reporter protein to provide both high sensitivity and selectivity for copper(II) has been challenging, likely due to a lack of proper copper(II)-chelating strategies within proteins. Here, we report a new genetically encoded fluorescent copper(II) reporter by employing a copper-binding tripeptide derived from human serum albumin (HSA), which is one of the major copper-binding proteins in extracellular environments. Optimized insertion of the tripeptide into the green fluorescent protein leads to rapid fluorescence quenching (up to >85% change) upon copper-binding, while other metal ions have no effect. Furthermore, the high binding affinity of the reporter enables reliable copper detection even in the presence of competing biomolecules such as HSA and amyloid beta peptides. We also demonstrate that our reporter proteins can be used to visualize dynamic copper fluctuations on living HeLa cell surfaces.
Introduction
Copper is an essential metal ion in most aerobic organisms, participating in many critical biological processes including energy generation, oxygen transport, cellular metabolism and signal transduction.1 However, when unregulated, copper can become toxic by generating reactive oxygen species (ROS) via Fenton-type reactions.2 The adverse effect of dysregulated copper homeostasis in humans is well illustrated in Menkes and Wilson's diseases, which are related to copper deficiency or overload, respectively.3 To address copper transport and trafficking in living systems, a variety of synthetic and genetically encoded probes have been developed with a focus mainly on visualizing intracellular copper,4 where Cu+ is the major form due to reduction upon cellular uptake and the reducing environment of the cytosol.5In contrast, extracellular copper trafficking has been insufficiently investigated due to a lack of appropriate tools for monitoring in living systems. In the more oxidizing extracellular media, Cu2+ is favored over Cu+.6 This oxidized form of copper has long been linked to many neurodegenerative diseases such as Alzheimer's disease, prion diseases, and Parkinson's disease through its participation in the aggregation of fibrogenic proteins.6 The protein–Cu2+ interactions are also directly involved in a catalytic cycle of ROS production, which exerts oxidative damage to cells.7 Moreover, exogenous copper plays a key role in neuronal signaling pathways by modulating the synaptic plasticity.8 Therefore, monitoring dynamic changes in the copper concentration in the extracellular environment will be critical for understanding the mechanisms of Cu2+ release and its physiological functions in the brain. In particular, imaging Cu2+ fluctuations on specifically targeted cell surfaces will greatly improve our insight into cellular responses to outside copper and copper-based cell-to-cell communication. Although a few cellular targeting strategies have been devised for synthetic metal reporters using organelle-targeting moieties,9 they cannot direct the probes to specific cells of interest. Furthermore, mislocalization of small-molecular probes inside cells might lead to controversy in data interpretation.10 In comparison, genetically encodable protein reporters have major advantages for the cell-specific and surface-specific detection of target molecules.
Several genetically encoded reporters have been applied for imaging copper in cellular context.11 However, they are limited by relatively small responses (15–50%),11b–e low copper selectivity,11a,11e or reversed responses in mammalian cells compared with the response in vitro and in living bacteria.11b Another challenge posed by imaging the extracellular pool of copper in particular is to provide the necessary binding affinity for Cu2+ sensors to compete with copper-binding molecules in the extracellular medium. While extracellular Cu2+ is present at micromolar concentrations (10–25 μM in blood plasma, 0.5–2.5 μM in cerebrospinal fluid (CSF), and 30 μM in the synaptic cleft),6 the metal ion is rather bound to highly abundant copper-binding proteins or small molecular ligands than present as a free ion.12 For example, human serum albumin (HSA), found at ∼600 μM in plasma, binds Cu2+ with a picomolar binding affinity.13 The HSA concentration in CSF is relatively low (∼3 μM) but represents 35–80% of the total CSF proteins.14
Developing protein-based Cu2+ sensors with strong but selective copper-binding properties will require potent Cu2+-chelating strategies within proteins. Previously reported sensors have employed simple histidines or a metal-chelating unnatural amino acid (3,4-dihydroxy-L-phenylalanine) to bind Cu2+ since the metal ion prefers to bind nitrogen or oxygen donors.11a,15 Unfortunately, reported binding affinities (KD 0.2–100 μM) of these sensors under physiologically relevant conditions have been still insufficient for imaging extracellular Cu2+, and it has not been demonstrated whether they could detect changes in copper concentration under conditions where competing copper-binding biomolecules are present.
Here, we report a new genetically encoded fluorescent reporter that binds Cu2+ with a KD of ∼8 nM while maintaining its selectivity for the metal ion. Green fluorescent protein (GFP) was engineered to contain a natural short but strong Cu2+-binding tripeptide, the amino terminal copper- and nickel-binding (ATCUN) motif, which is also the major copper-binding site of albumins in many species including humans.16 The location of this copper binding sequence in GFP was carefully optimized to provide maximum fluorescence quenching, strong Cu2+ binding, and stable protein folding in cells. The resulting reporter was sensitive enough to detect Cu2+ even in the presence of equimolar HSA in vitro. Furthermore, our reporter was stably displayed on cell surfaces and showed specific and reversible responses to extracellular Cu2+.
Results and discussion
The ATCUN motif consists of three amino acids (Xaa-Xaa-His) with a free N-terminus, where Xaa is any amino acid that can provide an amide nitrogen. As depicted in the X-ray crystal structure, this motif binds Cu2+ by forming a square-planar complex with the α-NH2-terminal nitrogen, the two amide nitrogens, and the imidazole nitrogen in histidine.17 Although the peptide binds Cu2+ (KD ∼ 1.18 × 10−16 M)18 and Ni2+ (KD ∼ 10−16 M)19 with similarly high affinities, it is highly context dependent, such that the ATCUN in HSA was reported to bind Ni2+ with a nearly five orders of magnitude weaker binding affinity than Cu2+.20 Considering its small size and strong binding affinity for Cu2+, we hypothesized that the ATCUN motif can serve as a powerful Cu2+-binding site for protein sensors.To place the ATCUN motif in close proximity of the GFP chromophore, thereby inducing maximum fluorescence quenching by Cu2+ binding, the protein was circularly permuted to generate new N- and C-termini at the beginning of β-strand 7. Circular permutations on β-strand 7 have shown relatively minimal effects on the GFP fluorescence.21 The ATCUN motif (Gly-Gly-His) was then inserted into the N-terminal region in a manner in which the histidine at the third position replaced histidine 148 near the phenolic ring of the chromophore (Fig. 1 and Table 1). To provide an α-NH2-terminal nitrogen for ATCUN in the circularly permuted GFP, a free N-terminus at the first glycine of the motif was precisely generated by intein-based protein cleavage.22 N-terminal sequencing of the intein-cleaved protein confirmed proper insertion of the ATCUN motif (Fig. S1†). For fabrication of the sensor protein, we employed superfolder GFP (OPT)23 for higher stability in the assay buffer (100 mM HEPES, pH 7.0, 300 mM NaCl) and enhanced folding in E. coli as well as in mammalian cells (data not shown).
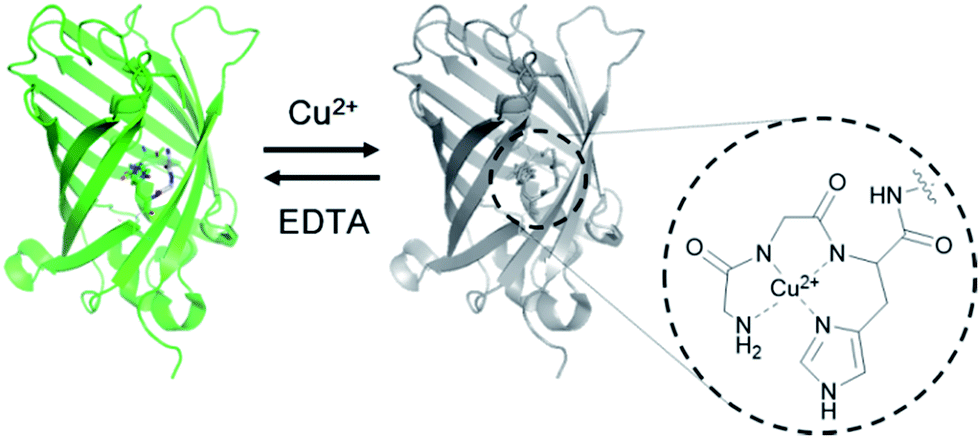 | ||
| Fig. 1 Design of a genetically encoded fluorescent Cu2+ reporter using the ATCUN motif. Structure of the Cu2+ complex of the ATCUN motif is shown. | ||
| Construct | +1 | H148 | −1 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| cpOPT | NH2− | E | L | S | H | N | V | Y | I | T | |
| GCS-1 | NH2− | G | G | H | N | V | Y | I | T | ||
| (−1)GCS-1 | NH2− | G | G | H | V | Y | I | T | |||
| GCS-2 | NH2− | G | G | H | H | N | V | Y | I | T | |
| G-GCS-2 | NH2− | G | G | G | H | H | N | V | Y | I | T |
| GCS-2-G | NH2− | G | G | H | G | N | V | Y | I | T |
Fluorescence spectra of the circularly permuted OPT (cpOPT) with the ATCUN insert were acquired by exciting at 480 nm and collecting the emission signals at 500–600 nm. Upon the addition of Cu2+, the fluorescence intensity of the sensor protein clearly decreased in a concentration-dependent manner (Fig. 2a). The decrease in the fluorescence signal (at 510 nm) was up to <5% of the apo-protein fluorescence (95% quenched). Compared with wild-type OPT (wtOPT) and cpOPT, the ATCUN-fused cpOPT (GCS-1) exhibited a considerably higher degree of quenching over a wide range of Cu2+ concentrations (Fig. 2b). Although cpOPT displayed noticeable fluorescence quenching at high Cu2+ concentrations (>10 μM), introduction of the ATCUN motif increased the Cu2+ sensitivity of the protein. In addition, longer incubation of the ATCUN-fused cpOPT at low Cu2+ concentrations further enhanced fluorescence quenching, which was likely due to the rather slow Cu2+ binding by ATCUN in cpOPT (Fig. 2c). We named the sensor protein as genetically-encoded copper(II) sensor (GCS)-1 (see Table 1 for a summary of N-terminal sequences in the GCS proteins).
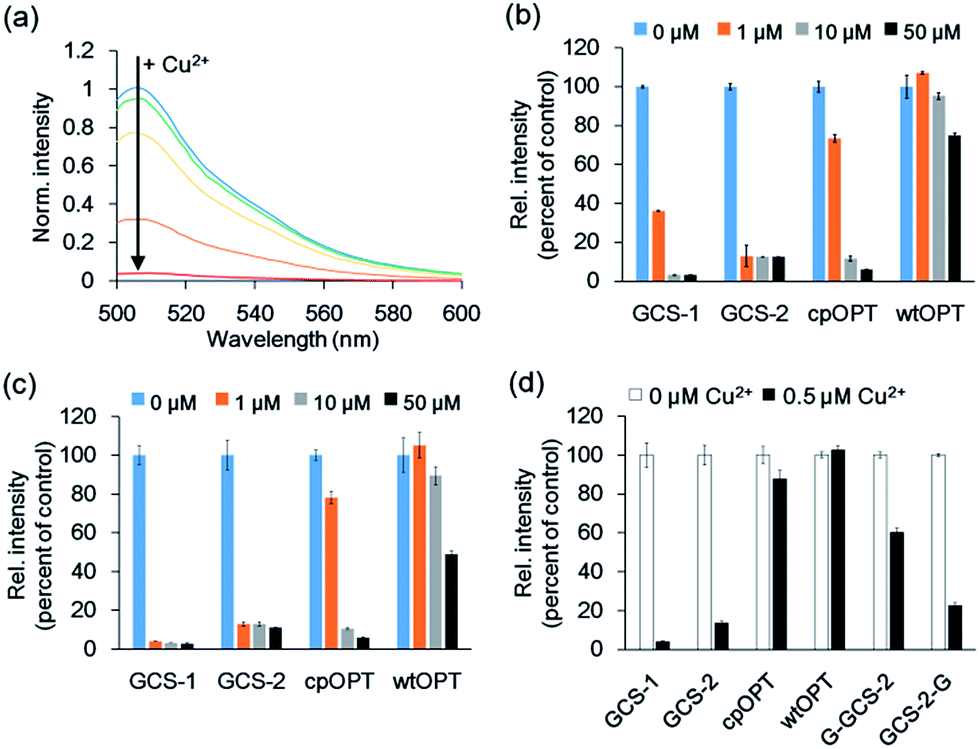 | ||
| Fig. 2 Fluorescence responses of GCS sensor proteins. (a) Fluorescence responses of 1 μM GCS-1 <5 min after the addition of 0, 0.4, 0.8, 1.6, and 3.2 μM Cu2+. (b) Fluorescence responses of 0.5 μM protein <5 min after the addition of 0, 1, 10, and 50 μM Cu2+. cpOPT: circularly permuted green fluorescent protein lacking the ATCUN motif; wtOPT: wild-type green fluorescent protein. (c) Fluorescence responses of 0.5 μM protein 1 h after the addition of 0, 1, 10, and 50 μM Cu2+. (d) Fluorescence responses of 0.1 μM protein 1 h after the addition of 0 and 0.5 μM Cu2+. The error bars correspond to the standard error of the mean of three independent measurements. | ||
To further optimize the Cu2+ responses, the location of the ATCUN motif in cpOPT was varied around H148 in GCS-1. ATCUN insertion at the −1 position of H148 ((−1)GCS-1, Table 1) resulted in a sensor protein with Cu2+ binding responses similar to those of GCS-1 but a poor protein yield (data not shown). In contrast, cpOPT with the ATCUN motif at the +1 position of H148 (GCS-2) showed rapidly saturated fluorescence quenching even at low concentrations of Cu2+ (Fig. 2b and c), possibly indicating a higher Cu2+ binding affinity. Although the degree of quenching was slightly lower for GCS-2 (<15% of apo-protein fluorescence, >85% quenched), the binding response of GCS-2 was significantly faster (within 10 s) than that of GCS-1 (>20 min, Fig. S2†). The improved response rate of GCS-2 might be due to an increased flexibility of the ATCUN motif in the protein. Unlike GCS-1, the ATCUN motif in GCS-2 does not include H148 which is known to interact with neighboring β-strands.23a Thereby the ATCUN tripeptide of GCS-2 can be more open for Cu2+ binding, exhibiting a faster association. On the other hand, H148 is also in a hydrogen-bonding network that stabilizes the chromophore.24 Conformational changes of H148 and subsequent effects on the chromophore by Cu2+ binding might be less significant in GCS-2 than in GCS-1. The absorbance spectrum of GCS-1 was altered more strongly by Cu2+ binding than that of GCS-2 (Fig. S3†), possibly indicating stronger influences on the chromophore and thereby a higher degree of quenching response in GCS-1.
To demonstrate that the ATCUN motif is indeed the major Cu2+ binding site in GCS-2, we constructed two GCS-2 variants, G-GCS-2 and GCS-2-G (Table 1). The ATCUN motif in G-GCS-2 was disrupted by adding a glycine residue to the N-terminus of GCS-2. Upon the addition of Cu2+, G-GCS-2 showed a severely diminished quenching response (Fig. 2d). On the other hand, the fluorescence intensity of GCS-2-G, in which the ATCUN motif is intact but H148 was mutated to glycine, was still reduced to <30% of the apo-protein fluorescence (>70% quenched) under the same conditions, supporting that the ATCUN motif is primarily responsible for Cu2+ binding (Fig. 2d). In addition, titration with Cu2+ revealed a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding stoichiometry between GCS-2 and Cu2+ (Fig. S4†), suggesting the presence of a single dominant metal binding site (such as the ATCUN tripeptide) in GCS-2. However, it should be noted that H148 of GCS-2 also plays a partial role in Cu2+ binding and the resulting fluorescence quenching since the Cu2+-mediated response of GCS-2-G was smaller than that of GCS-2.
:1 binding stoichiometry between GCS-2 and Cu2+ (Fig. S4†), suggesting the presence of a single dominant metal binding site (such as the ATCUN tripeptide) in GCS-2. However, it should be noted that H148 of GCS-2 also plays a partial role in Cu2+ binding and the resulting fluorescence quenching since the Cu2+-mediated response of GCS-2-G was smaller than that of GCS-2.
The metal selectivity of both GCS proteins was subsequently investigated by monitoring changes in fluorescence intensity in the GCS proteins when other biologically relevant metal ions (Cr3+, Mn2+, Fe3+, Co2+, Ni2+, Zn2+, Ag+, Cd2+, Hg2+, Pb2+, K+, Ca2+, and Mg2+) were present. Most of these metal ions had no effect on the fluorescence quenching by Cu2+ (Fig. 3a and b). Although Ni2+ caused a small fluorescence change for GCS-2, subsequent Cu2+ addition readily induced fluorescence quenching (Fig. 3b). The ATCUN motif in our GCS sensor proteins likely binds to Ni2+ more weakly than to Cu2+, which is similar to the ATCUN motif in HSA.20 The effects of Cu+ were examined in a buffer containing a 50-fold molar excess of L-ascorbic acid. L-ascorbic acid had no influence on the fluorescence intensities of the GCS proteins (Fig. S5†) or Cu2+ binding of the ATCUN motifs.25 Cu+ addition showed no change in fluorescence for both GCS proteins (Fig. 3c). The reversibility of the fluorescence quenching of the sensor protein was also tested, and fluorescence signals from the Cu2+-bound GCS proteins were recovered upon addition of metal-chelating ethylenediaminetetraacetic acid (EDTA, Fig. 3d).
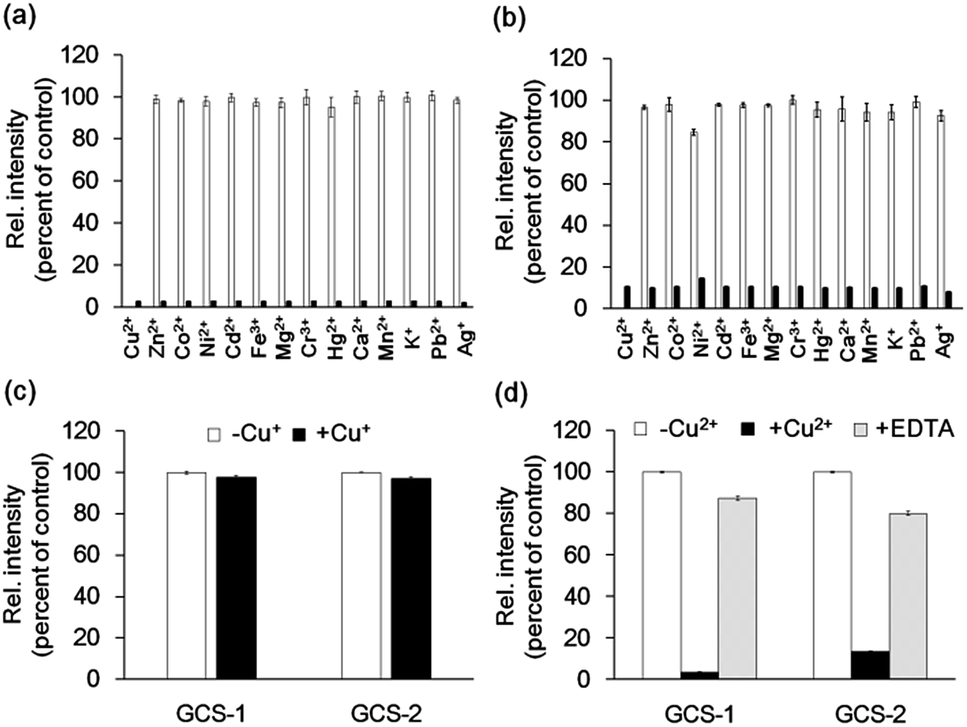 | ||
| Fig. 3 Cu2+ selectivity and reversibility of GCS proteins. (a and b) Fluorescence responses of 1 μM GCS-1 (a) and GCS-2 (b) to various metal ions. White bars represent fluorescence responses with 5 μM of the respective metal ions. Black bars represent fluorescence responses after the addition of 5 μM Cu2+. (c) Fluorescence responses of 1 μM GCS protein with 5 μM Cu+ including 250 μM of L-ascorbic acid to prevent oxidation of Cu+ to Cu2+. Fluorescence spectra were collected within 5 min after Cu+ addition. (d) Fluorescence responses of 1 μM GCS proteins with 5 μM Cu2+ followed by the addition of 50 μM EDTA. All responses were normalized against the fluorescence intensity of 1 μM GCS protein lacking a metal ion. The error bars correspond to the standard error of the mean of three independent measurements. | ||
The Cu2+-binding affinities of the GCS proteins were determined by measuring fluorescence changes upon titration with copper.15b All assays were performed at relatively high buffer and salt concentrations (100 mM HEPES, pH 7.0, 300 mM NaCl), which might reflect various extracellular media.26 The dissociation constant for GCS-2 was calculated to be 7.93 ± 3.20 nM (Fig. 4a), which is the strongest Cu2+ binding among the currently available protein-based Cu2+ sensors (Table S1†). The use of a natural copper(II)-specific tripeptide provides a strong chelating strategy for robust Cu2+ capture near the GFP chromophore, presenting high sensitivity for the metal ion while not compromising the metal selectivity of the GCS proteins. GCS-1 was found to bind Cu2+ more weakly (94.04 ± 19.33 nM) as discussed above (Fig. S6†).
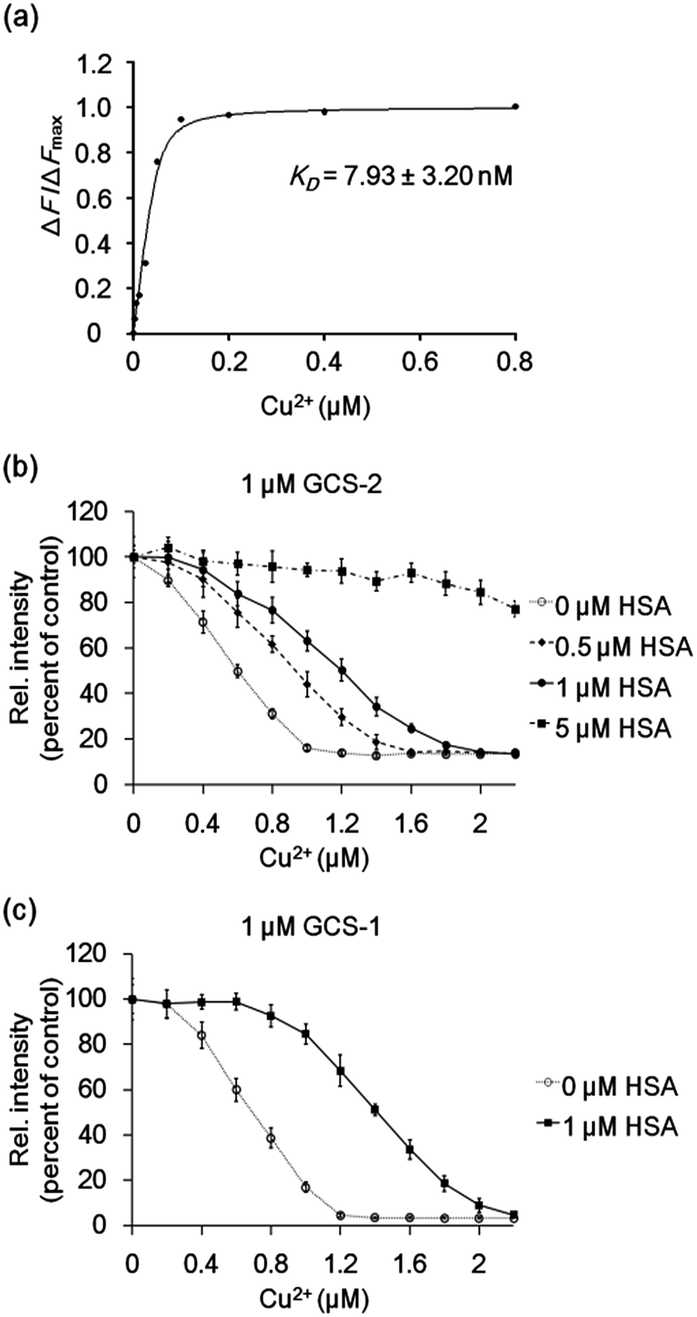 | ||
| Fig. 4 (a) Representative plot of ΔF/ΔFmax of 50 nM GCS-2 with various Cu2+ concentrations. The binding constant KD was derived from three independent experiments. ΔF: change in measured fluorescence, ΔFmax: maximum fluorescence change. (b) GCS-2 fluorescence changes with varying concentration of Cu2+ in the absence and presence of 0.5, 1, and 5 μM HSA. (c) GCS-1 fluorescence changes with varying concentration of Cu2+ in the absence and presence of 1 μM HSA. The error bars correspond to the standard error of the mean of three independent measurements. | ||
To test whether the binding affinity of the GCS proteins for Cu2+ is high enough to monitor the metal ion even in the presence of other copper-binding molecules, the GCS responses to Cu2+ were measured in the presence of various amounts of HSA. GCS-2 showed Cu2+ binding properties comparable to HSA, where >40% of the added Cu2+ was bound to GCS-2 in the presence of equimolar HSA (Fig. 4b). Considering that the reported dissociation constants for HSA are in the picomolar range,13 Cu2+ binding to GCS-2 should be weaker by at least three orders of magnitude. This discrepancy is likely due to the different experimental methods used for KD measurements, components/concentration of buffers, pH, or ionic strengths.27 Nevertheless, under our conditions, an optimized ATCUN tripeptide in GCS-2 showed nearly as strong binding as the ATCUN motif in HSA. In contrast, the fluorescence intensity of GCS-1 began to decrease only after the concentration of Cu2+ was high enough to fill in most of the ATCUN sites in HSA (Fig. 4c). Therefore, GCS-1 was not able to effectively compete with HSA. We also performed competing experiments with an amyloid beta peptide (Aβ40), another copper-binding molecule (KD 0.47–30 μM) which plays a key role in the pathogenesis of Alzheimer's disease.28 Aβ40, which has a rather weak Cu2+-binding ability, was outcompeted by both GCS proteins (Fig. S7†). Taken together, our data show that GCS-2 (KD ∼ 8 nM) is a highly sensitive reporter that can efficiently compete with other high-affinity copper-binding molecules in extracellular media. In comparison, GCS-1 (KD ∼ 94 nM) might be useful for applications in which low-affinity reporter proteins are required.
Finally, we used the GCS proteins to detect dynamic Cu2+ fluctuations on the surfaces of live mammalian cells. A signal peptide for the secretory pathway and the transmembrane domain of the platelet-derived growth factor receptor were fused to GCS-2 for display on the extracellular plasma membrane. The signal peptide is cleaved during sensor protein synthesis in the endoplasmic reticulum, generating the ATCUN motif precisely at the N-terminus for effective Cu2+ binding. HeLa cells expressing the GCS-2 construct were monitored with a total internal reflection fluorescence (TIRF) microscope for selective visualization of the cell surface fluorescence. Upon Cu2+ addition (50 μM), the GCS-2 fluorescence intensity on the cell surface was rapidly decreased within ∼1.5 min, and it was saturated to 47% (average from 13 cells in the same well, 53% quenched) of the initial signal after 3 min (Fig. 5). EDTA was then added to the cells, and the GCS-2 fluorescence intensity was recovered to 97% within 4 min, which is in agreement with the in vitro results. GCS-1 that was expressed on HeLa cell surfaces provided fluorescence changes similar to those of GCS-2 against extracellular Cu2+ (data not shown).
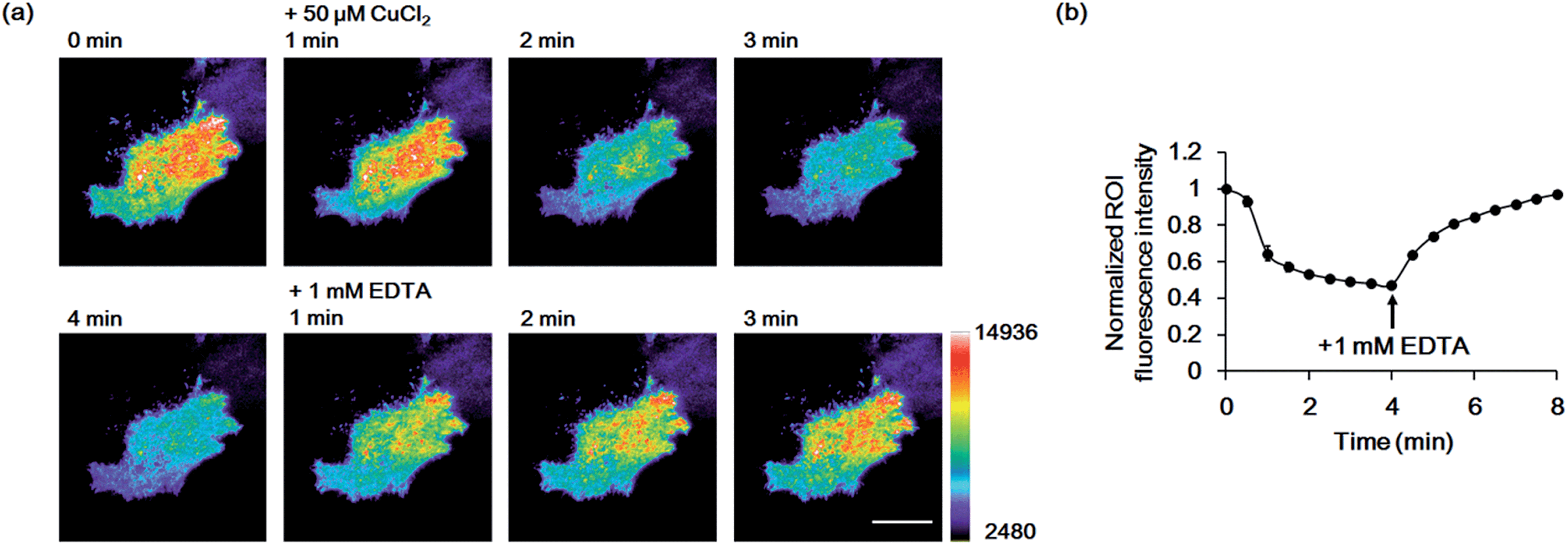 | ||
| Fig. 5 Imaging of Cu2+ on live HeLa cell surfaces by GCS-2. (a) Pseudocolored images from 1 min prior to Cu2+ addition to 1, 2, 3 and 4 min after the addition of 50 μM Cu2+, and 1, 2 and 3 min after the addition of 1 mM EDTA. Images were collected using total internal reflection fluorescence (TIRF) microscopy. The color bar represents the fluorescence intensity in the GFP channel (excitation at 488 nm). Scale bar: 20 μm. (b) Plot of the average fluorescence response of 13 cells vs. time after 50 μM Cu2+ addition. All fluorescence intensity values were subtracted by background signals and normalized by the fluorescence before Cu2+ addition. The time of 1 mM EDTA addition is shown. The error bars correspond to the standard error of the mean value of 13 individual cells. | ||
To confirm that the decrease in fluorescence of GCS-2 was specific for Cu2+ binding, Zn2+, another crucial metal ion in the brain, was added to the cells (50 μM) before Cu2+ treatment. Interestingly, the fluorescence intensity of several cells of the 13 monitored cells was slightly increased (<115%) by Zn2+ (Fig. 6). This could result from structural stabilization of the β-barrel of the GCS-2 protein under relatively high zinc concentrations as recently reported for a red fluorescent mKate2 mutant.11a Zinc(II)-mediated changes in protein distribution in the plasma membrane might also affect the fluorescence intensity of GCS-2 as observed using TIRF imaging. Nevertheless, all cells exhibited rapid fluorescence quenching upon Cu2+ addition (Fig. 6). The copper(II)-specific fluorescence decrease for GCS-2 was further confirmed by its co-expression with mCherry, a red fluorescent protein, on the cell surfaces. Addition of 10 μM CuCl2 decreased the fluorescence intensity of GCS-2 but not that of mCherry, which again shows the applicability of GCS-2 for specific Cu2+ imaging on the surface of live cells (Fig. 7). Moreover, the fluorescence signals of cpOPT proteins on cell surfaces were not altered by 10 μM Cu2+ (Fig. S8†).
 | ||
| Fig. 6 Fluorescence response of GCS-2 to 50 μM Zn2+ on live HeLa cell surfaces followed by addition of 50 μM Cu2+. (a) Images of 1 min prior to Zn2+ addition, 3 min after the addition of Zn2+, and 1 and 3 min after the addition of Cu2+. Scale bar: 10 μm. (b) Plot of the average response of 13 cells vs. time after 50 μM Zn2+ addition. All fluorescence intensity values were subtracted by background signals and normalized by the fluorescence before Zn2+ addition. The time of 50 μM Cu2+ addition is shown. The error bars correspond to the standard error of the mean value of 13 individual cells. | ||
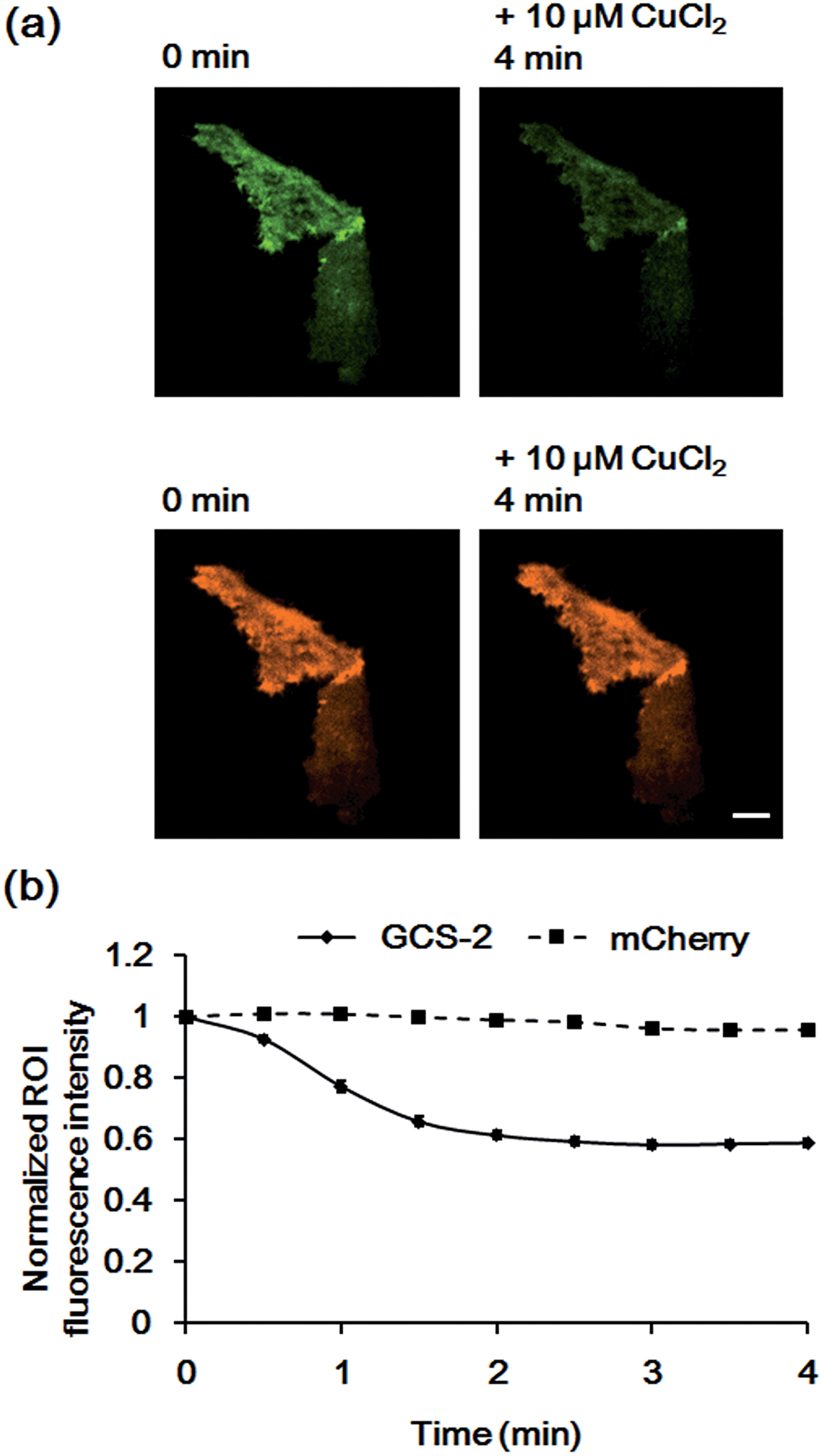 | ||
| Fig. 7 Fluorescence responses of GCS-2 and mCherry co-expressed in HeLa cells to 10 μM Cu2+. (a) Images of GCS-2 (top) and mCherry (bottom) of 1 min prior to Cu2+ addition and 4 min after the addition of Cu2+. Scale bar: 10 μm (b) plots of the average fluorescence responses of GCS-2 and mCherry of 8 cells vs. time after 10 μM Cu2+ addition. All fluorescence intensity values were subtracted by background signals and normalized by the fluorescence before Cu2+ addition. The error bars correspond to the standard error of the mean value of eight individual cells. | ||
Conclusions
The GCS proteins demonstrate that the natural and small Cu2+ binding ATCUN motif can be successfully utilized as a strong and specific copper chelating site for genetically encoded Cu2+ reporters. The tight Cu2+ binding of GCS-2 allows for reliable Cu2+ monitoring in the presence of HSA, one of the major Cu2+ binding biomolecules in extracellular environments. The sensor proteins also demonstrate high selectivity for Cu2+ compared with other biologically relevant metal ions. Imaging Cu2+ on the surface of live cells with GCS-2 showed a quick and reversible response, which demonstrates the possibility of monitoring oscillating Cu2+ levels in living systems. The copper-chelating strategy described herein has multiple advantages. Taken from one of the major players in extracellular copper trafficking, the ATCUN motif may enable non-invasive detection of dynamic changes in copper concentration in biological systems. The motif is composed of only three amino acids, creating minimal disruption in the fluorescent protein’s structure. To expand the applicability of GCS sensors to intracellular Cu2+ imaging, strategies to generate ATCUN motifs and protect these motifs from various post-translational modifications29 inside cells should be developed. Nonetheless, we envision that GCS proteins can be employed to monitor Cu2+ release on neuronal cells. Specifically, copper release in the synaptic cleft which is induced by chemical or biological cues could be detected by GCS proteins expressed on pre- or postsynaptic cell surfaces. Further studies will be aimed at improving the membrane targeting efficiency and applications of red fluorescent proteins that enable Cu2+ imaging in live animals.Acknowledgements
This work was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Science, ICT & Future Planning (MSIP) (2011-0020322) and BioNano Health Guard Research Center funded by MSIP as Global Frontier Project (H-GUARD_2014M3A6B2060512 & 2013M3A6B2078950).Notes and references
- B. E. Kim, T. Nevitt and D. J. Thiele, Nat. Chem. Biol., 2008, 4, 176–185 CrossRef CAS PubMed.
- B. Halliwell and J. M. Gutteridge, Biochem. J., 1984, 219, 1–14 CAS.
- E. Madsen and J. D. Gitlin, Annu. Rev. Neurosci., 2007, 30, 317–337 CrossRef CAS PubMed.
- (a) K. P. Carter, A. M. Young and A. E. Palmer, Chem. Rev., 2014, 114, 4564–4601 CrossRef CAS PubMed; (b) D. W. Domaille, E. L. Que and C. J. Chang, Nat. Chem. Biol., 2008, 4, 168–175 CrossRef CAS PubMed.
- E. M. Rees and D. J. Thiele, J. Biol. Chem., 2007, 282, 21629–21638 CrossRef CAS PubMed.
- E. L. Que, D. W. Domaille and C. J. Chang, Chem. Rev., 2008, 108, 1517–1549 CrossRef CAS PubMed.
- E. Gaggelli, H. Kozlowski, D. Valensin and G. Valensin, Chem. Rev., 2006, 106, 1995–2044 CrossRef CAS PubMed.
- E. D. Gaier, B. A. Eipper and R. E. Mains, J. Neurosci. Res., 2013, 91, 2–19 CAS.
- (a) R. J. Radford, W. Chyan and S. J. Lippard, Chem. Sci., 2014, 5, 4512–4516 RSC; (b) Y. H. Lee, N. Park, Y. B. Park, Y. J. Hwang, C. Kang and J. S. Kim, Chem. Commun., 2014, 50, 3197–3200 RSC; (c) R. J. Radford, W. Chyan and S. J. Lippard, Chem. Sci., 2013, 4, 3080–3084 RSC; (d) S. Iyoshi, M. Taki and Y. Yamamoto, Org. Lett., 2011, 13, 4558–4561 CrossRef CAS PubMed; (e) D. Li, S. Chen, E. A. Bellomo, A. I. Tarasov, C. Kaut, G. A. Rutter and W. H. Li, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 21063–21068 CrossRef CAS PubMed; (f) S. C. Dodani, S. C. Leary, P. A. Cobine, D. R. Winge and C. J. Chang, J. Am. Chem. Soc., 2011, 133, 8606–8616 CrossRef CAS PubMed.
- K. A. Price, J. L. Hickey, Z. G. Xiao, A. G. Wedd, S. A. James, J. R. Liddell, P. J. Crouch, A. R. White and P. S. Donnelly, Chem. Sci., 2012, 3, 2748–2759 RSC.
- (a) X. Yu, M. P. Strub, T. J. Barnard, N. Noinaj, G. Piszczek, S. K. Buchanan and J. W. Taraska, PLoS One, 2014, 9, e95808 Search PubMed; (b) J. Liu, J. Karpus, S. V. Wegner, P. R. Chen and C. He, J. Am. Chem. Soc., 2013, 135, 3144–3149 CrossRef CAS PubMed; (c) M. S. Koay, B. M. Janssen and M. Merkx, Dalton Trans., 2013, 42, 3230–3232 RSC; (d) J. Liang, M. Qin, R. Xu, X. Gao, Y. Shen, Q. Xu, Y. Cao and W. Wang, Chem. Commun., 2012, 48, 3890–3892 RSC; (e) S. V. Wegner, H. Arslan, M. Sunbul, J. Yin and C. He, J. Am. Chem. Soc., 2010, 132, 2567–2569 CrossRef CAS PubMed.
- J. H. Kaplan and S. Lutsenko, J. Biol. Chem., 2009, 18, 25461–25465 CrossRef PubMed.
- M. Rozga, M. Sokolowska, A. M. Protas and W. Bal, J. Biol. Inorg. Chem., 2007, 12, 913–918 CrossRef CAS PubMed.
- H. Reiber, Clin. Chim. Acta, 2001, 310, 173–186 CrossRef CAS.
- (a) E. É. Bálint, J. Petres, M. Szabó, C. K. Orbán, L. Szilágyi and B. Ábrahám, J. Fluoresc., 2013, 23, 273–281 CrossRef PubMed; (b) N. Ayyadurai, N. S. Prabhu, K. Deepankumar, S.-G. Lee, H.-H. Jeong, C.-S. Lee and H. Yun, Angew. Chem., Int. Ed., 2011, 50, 6534–6537 CrossRef CAS PubMed; (c) T. A. Richmond, T. T. Takahashi, R. Shimkhada and J. Bernsdorf, Biochem. Biophys. Res. Commun., 2000, 268, 462–465 CrossRef CAS PubMed.
- C. Harford and B. Sarkar, Acc. Chem. Res., 1997, 30, 123–130 CrossRef CAS.
- N. Camerman, A. Camerman and B. Sarkar, Can. J. Chem., 1976, 54, 1309–1316 CrossRef CAS.
- S. J. Lau, T. P. Kruck and B. Sarkar, J. Biol. Chem., 1974, 249, 5878–5884 CAS.
- E. C. Long, Acc. Chem. Res., 1999, 32, 827–836 CrossRef CAS.
- W. Bal, M. Sokolowska, E. Kurowska and P. Faller, Biochim. Biophys. Acta, 2013, 1830, 5444–5455 CrossRef CAS PubMed.
- G. S. Baird, D. A. Zacharias and R. Y. Tsien, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11241–11246 CrossRef CAS.
- S. Chong, K. S. Williams, C. Wotkowicz and M. Q. Xu, J. Biol. Chem., 1998, 273, 10567–10577 CrossRef CAS PubMed.
- (a) J. D. Pedelacq, S. Cabantous, T. Tran, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2006, 24, 79–88 CrossRef CAS PubMed; (b) S. Cabantous, T. C. Terwilliger and G. S. Waldo, Nat. Biotechnol., 2005, 23, 102–107 CrossRef CAS PubMed.
- M. A. Elsliger, R. M. Wachter, G. T. Hanson, K. Kallio and S. J. Remington, Biochemistry, 1999, 38, 5296–5301 CrossRef CAS PubMed.
- Y. Jin and J. A. Cowan, J. Am. Chem. Soc., 2005, 127, 8408–8415 CrossRef CAS PubMed.
- G. G. Somjen, in Ions in the Brain: Normal Function, Seizures, and Stroke, Oxford University Press, Inc, New York, 2004, p. 16 Search PubMed.
- Y. Zhang, S. Akilesh and D. E. Wilcox, Inorg. Chem., 2000, 39, 3057–3064 CrossRef CAS.
- V. Tougu, A. Karafin and P. Palumaa, J. Neurochem., 2008, 104, 1249–1259 CrossRef CAS PubMed.
- T. Meinnel and C. Giglione, Proteomics, 2008, 8, 626–649 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03027c |
| This journal is © The Royal Society of Chemistry 2015 |