
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiaryldichalcogenide radical cations†‡
Ole
Mallow
a,
Monther A.
Khanfar
bc,
Moritz
Malischewski
b,
Pamela
Finke
a,
Malte
Hesse
a,
Enno
Lork
a,
Timo
Augenstein
d,
Frank
Breher
d,
Jeffrey R.
Harmer
e,
Nadezhda V.
Vasilieva
f,
Andrey
Zibarev
fg,
Artem S.
Bogomyakov
h,
Konrad
Seppelt
b and
Jens
Beckmann
*a
aInstitut für Anorganische Chemie, Universität Bremen, Leobener Straße, 28359 Bremen, Germany. E-mail: j.beckmann@uni-bremen.de
bInstitut für Chemie und Biochemie, Freie Universität Berlin, Fabeckstraße 34/36, 14195 Berlin, Germany
cDepartment of Chemistry, The University of Jordan, Amman 11942, Jordan
dInstitut für Anorganische Chemie, Karlsruhe Institute of Technology, Engesserstr. 15, 76131 Karlsruhe, Germany
eCentre for Advanced Imaging, University of Queensland, St Lucia, Queensland 4072, Australia
fInstitute of Organic Chemistry, Russian Academy of Sciences, 630090 Novosibirsk, Russia
gDepartment of Physics, National Research University – Novosibirsk State University, 630090 Novosibirsk, Russia
hInternational Tomography Centre, Russian Academy of Sciences, 630090 Novosibirsk, Russia
First published on 21st October 2014
Abstract
One-electron oxidation of two series of diaryldichalcogenides (C6F5E)2 (13a–c) and (2,6-Mes2C6H3E)2 (16a–c) was studied (E = S, Se, Te). The reaction of 13a and 13b with AsF5 and SbF5 gave rise to the formation of thermally unstable radical cations [(C6F5S)2]˙+ (14a) and [(C6F5Se)2]˙+ (14b) that were isolated as [Sb2F11]− and [As2F11]− salts, respectively. The reaction of 13c with AsF5 afforded only the product of a Te–C bond cleavage, namely the previously known dication [Te4]2+ that was isolated as [AsF6]− salt. The reaction of (2,6-Mes2C6H3E)2 (16a–c) with [NO][SbF6] provided the corresponding radical cations [(2,6-Mes2C6H3E)2]˙+ (17a–c; E = S, Se, Te) in the form of thermally stable [SbF6]− salts in nearly quantitative yields. The electronic and structural properties of these radical cations were probed by X-ray diffraction analysis, EPR spectroscopy, and density functional theory calculations and other methods.
Introduction
The landmark paper by Gomberg on the stable free triphenylmethyl radical initiated numerous investigations on molecules containing unpaired electrons.1 Several kinds of persistent and stable radicals2 have been described ever since, and several more general classes of (poly)radicals have been developed in recent times.3 As stated recently, “much of the current interest in stable radicals probably arises […] from the fundamental structure and bonding issues that naturally arise with this class of compounds”.4 Most often, light carbon-based or heteroatom radicals have been studied. Investigations on heavier main group radicals are fewer in number, although the last decades witnessed spectacular discoveries in the chemistry of the heavier main group elements.5 The design of novel synthetic strategies, particularly the use of very bulky substituents, has led to the isolation of a wide range of compounds, including main group radicals, an area which has been reviewed recently.6 The interest in the one-electron oxidation of diorganodichalcogenides dates back to 1868 when it was observed that (PhS)2 (1a) dissolves in conc. H2SO4 to give intensively coloured solutions of radical cations (Scheme 1).7 Nowadays, it is understood that the formation of the latter begins with the one-electron oxidation of (PhS)2 (1a) providing the intermediate radical cation [(PhS)2]˙+ (2a), which upon loss of another electron gives rise to the intermediate dication [(PhS)2]2+ (3). Charge repulsion (“Coulomb explosion”) leads to the dissociation into two sulfenyl cations [PhS]+ (4), which undergo mutual electrophilic substitution of their phenyl rings in ortho-position to produce thianthrene (5). Another one-electron oxidation eventually yields the thianthrene radical cation (6) and unaccounted products (Scheme 1).8 The one-electron oxidation of (PhS)2 (1a) and its Se-congener (PhSe)2 (1b) in the confined voids of the acidic pentasil zeolithe allowed the tentative characterization of the radical cations [(PhS)2]˙+ (2a) and [(PhSe)2]˙+ (2b) by EPR spectroscopy.9 Previous attempts to prepare a persistent dialkyldisulfide radical cation using (NeoS)2 (7; Neo = neopentyl) and nitrosyl triflate [NO][O3SCF3] afforded a dialkyldisulfide nitrosonium adduct [(NeoS)2·NO]+ (8) comprising a four-membered ring structure (Scheme 2).10 The related diamagnetic dicationic rings [(MeSe)4]2+ (9) and [(EtTe)4]2+ (10) were similarly obtained by the one-electron oxidation of heavier group 16 dialkyldichalcogenides (MeSe)2 (11) and (EtTe)2 (12) with [NO][O3SCF3].8h, 11 Compounds 9 and 10 can be regarded as dimers of persistent radical cations, which dimerise by π*–π* interactions.12 The dissociation energies of these rings were estimated to be in the order of magnitude of 50 kcal mol−1,10 which prompted us to investigate if persistent or even stable diaryldichalcogenide radical cations [(RE)2]˙+ can be prepared using fluorinated (R = C6F5) or bulky aromatic substituents (R = 2,6-Mes2C6H3).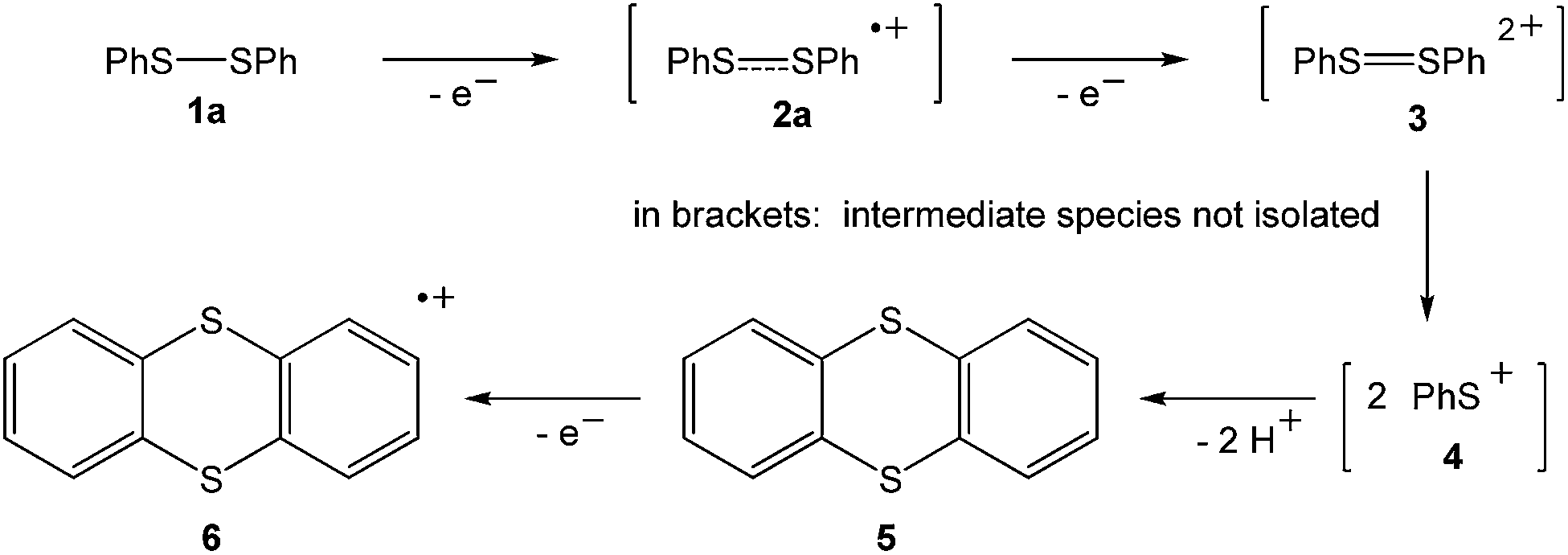 | ||
| Scheme 1 Oxidation of diphenyldisulfide in conc. sulfuric acid. | ||
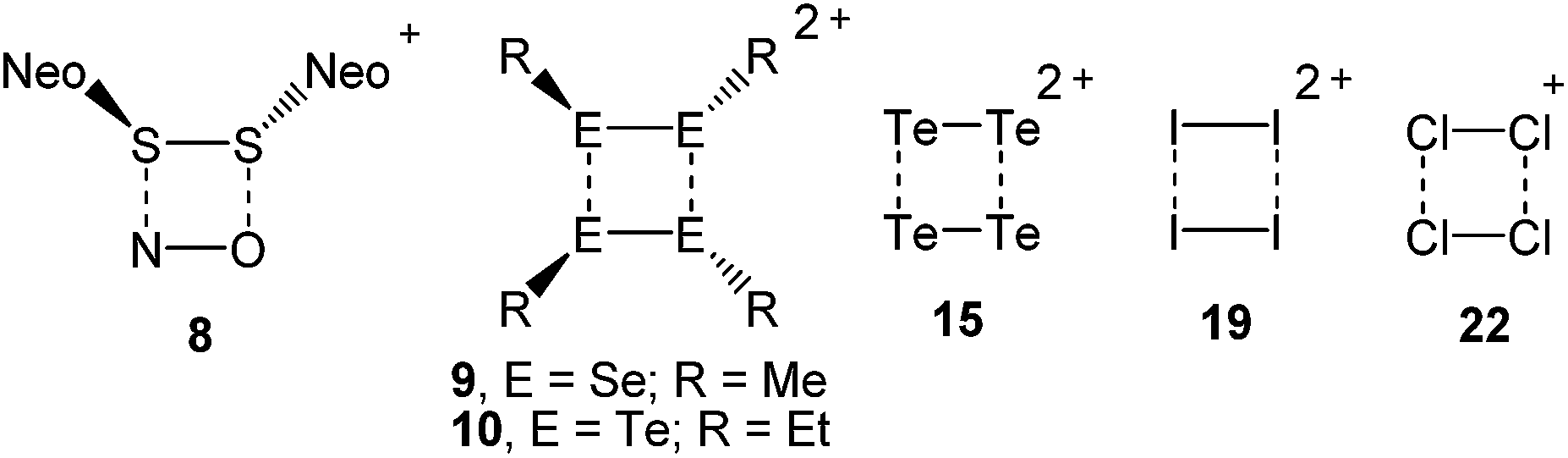 | ||
| Scheme 2 π*–π*-bonded four-membered rings. | ||
Results and discussion
In an initial foray, we investigated the one-electron oxidation of bis(pentafluorophenyl)dichalcogenides with antimony pentafluoride and arsenic pentafluoride. The reaction of (C6F5S)2 (13a) and (C6F5Se)2 (13b), respectively, with SbF5 or AsF5 gave an immediate colour change to dark blue and dark green upon contact, which suggested that paramagnetic species had formed (Scheme 3). Unfortunately, almost all attempts to isolate crystalline products by crystallisation from SO2ClF at low temperatures using co-solvents such as aHF, F114 and CFCl3 were impeded by decomposition. Only for the radical cations [(C6F5S)2]˙+ (14a; counterion [Sb2F11]−) and [(C6F5Se)2]˙+ (14b; counterion [As2F11]−) crystalline salts were obtained. However, even those crystals showed a limited thermal stability precluding any detailed spectroscopic characterisation. It is noted that the reaction of (C6F5S)2 (13a) with AsF5 in liquid SO2 was studied previously at room temperature and provided [(C6F5S)2SC6F5][AsF6].13 The reaction of (C6F5Te)2 (13c) with SbF5 or AsF5 under similar conditions gave no indication for the formation of radicals. The isolation of small amounts of the previously known [Te4]2+ (15; counterion [AsF6]−)14 provided evidence that Te–C bonds were cleaved.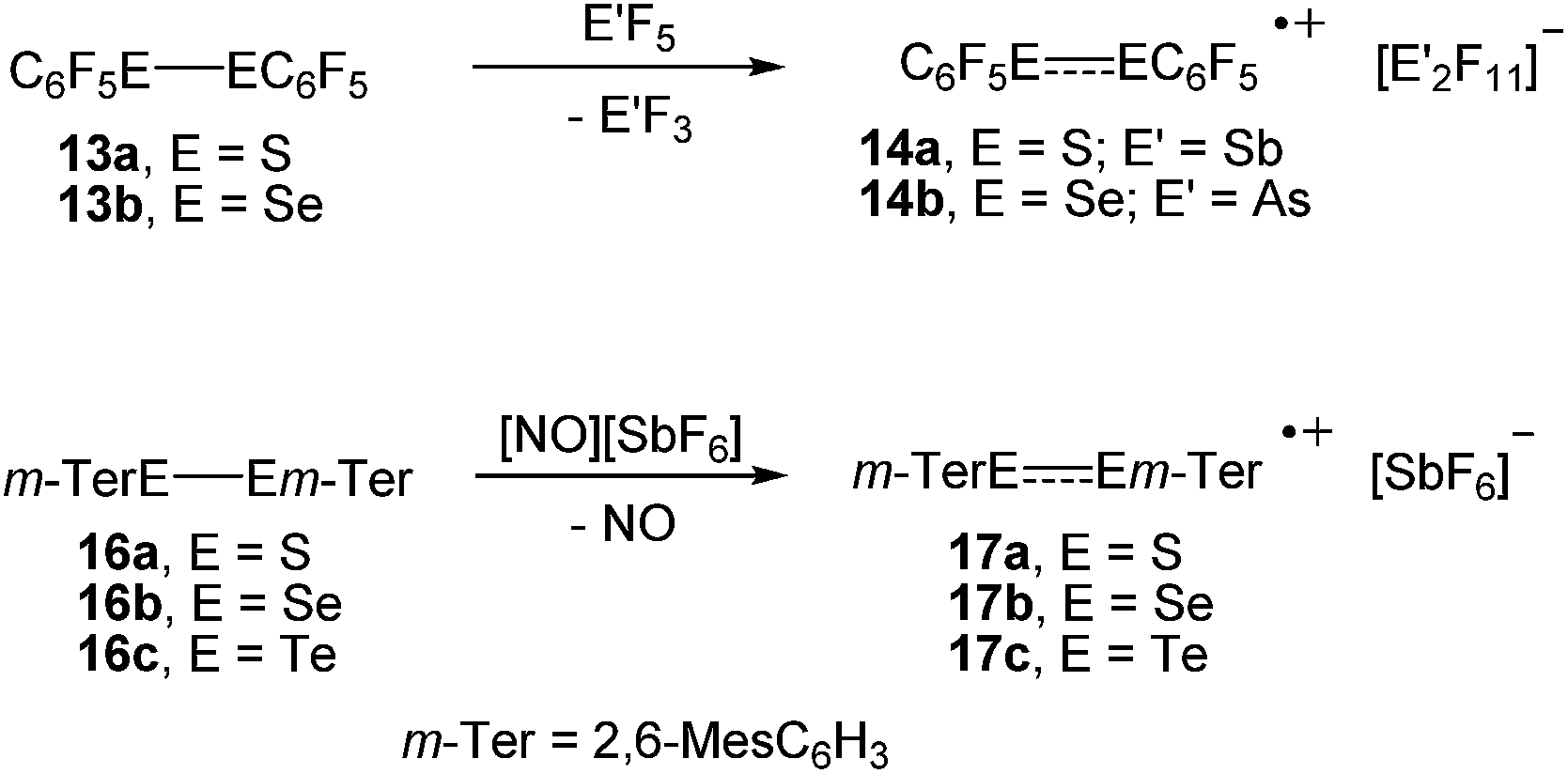 | ||
| Scheme 3 Synthesis of the radical cations 14a–b and 17a–c. | ||
The oxidation of the bulky bis(m-terphenyl)dichalcogenides were studied by cyclic voltammetry first. At a stationary Pt electrode, electrochemical oxidation of 16a–c in CH2Cl2/0.1 M [n-Bu4N][BF4] within the potential sweep range 0 < E < 1.5 V is characterized by a one-electron quasi-reversible peak (EOxp − ERedp = 0.07 − 0.08 V, EOxp − EOxp/2 = 0.06 V, IRedp/IOxp ≈ 0.8 − 0.9). The observed quasi-reversibility of the peak indicates relative stability of the radical cations. Significant differences in the peak currents for 16a–c can be tentatively attributed to the differences in their diffusion coefficients. The anodic peak potentials with respect to a saturated calomel electrode of (2,6-Mes2C6H3E)2 decrease from EOxp = 1.22 V (16a, E = S) over 1.09 V (16b, E = Se) to 0.79 V (16c, E = Te), respectively, which suggested nitrosonium salts to be suitable one-electron oxidizers.15 Indeed, the reaction of the bis(m-terphenyl)dichalcogenides (2,6-Mes2C6H3E)2 (16a, E = S; 16b, E = Se; 16c, E = Te) with [NO][SbF6] in propionitrile provided the corresponding radical cations [(2,6-Mes2C6H3E)2]˙+ (17a, E = S; 17b, E = Se; 17c, E = Te; counterion [SbF6]−) as dark blue crystals in very high yields, which showed no signs of decomposition for several months when isolated from the mother liquor and stored under argon (Scheme 3).
The molecular structures of [(C6F5E)2]˙+ (14a, E = S; 14b, E = Se) and [(2,6-Mes2C6H3E)2]˙+ (17a, E = S; 17b, E = Se; 17c, E = Te) are shown in Fig. 1. Selected bond parameters are collected in Table 1 together with those of the neutral parent compounds. The radical cations 14a, 14b, 17a and 17b containing S and Se atoms adopt nearly Cs symmetric conformations. The phenyl rings comprising C10–C15 are almost coplanar with the C10–E1–E2 plane pointing to delocalization of unpaired electron spin density across the aromatic π-system, whereas the phenyl rings including C20–C25 are perpendicular to the C20–E2–E1 plane (E = S, Se). The radical cation 16c containing Te atoms is centrosymmetric and possesses C2 symmetry. Consequently, only one crystallographically independent Te atom is present. In the radical cations [(C6F5S)2]˙+ (14a) and [(2,6-Mes2C6H3S)2]˙+ (17a) the delocalization is also reflected in the S–C bond lengths; S1–C10 (14a, 1.727(8) Å; 17a, 1.762(5) Å) is significantly shorter than S1–C20 (14a, 1.764(8); 17a, 1.799(5) Å) pointing to a quinoid structure of the coplanar phenyl ring (C10–C15). Indeed, the quinoid character of the coplanar phenyl rings of 14a (Q = 0.036 (26%)), 14b (Q = 0.012 (9%)), 17a (Q = 0.019 (14%)) and 17b (Q = 0.019 (14%)) is substantially higher than that of the perpendicular phenyl rings of 14a (Q = 0.024 (17%)), 14b (Q = 0.001 (<1%)), 17a (Q = 0.001 (<1%)) and 17b (Q = 0.002 (1%)).16 The quinoid character of 17c (Q = 0.037 (27%)) adopts the highest value and nearly approaches that of the hexafluorobenzene radical cation [C6F6]˙+ (Q = 0.042 (30%)).17 Comparison of the parent compounds with the corresponding radical cations reveals a shortening of the E–E bonds by 0.008 Å for 13a/14a, 0.030 Å for 13b/14b, 0.075 Å for 16a/17a, 0.050 Å for 16b/17b and 0.049 Å for 16c/17c (E = Te), respectively. The E–E bond shortening unambiguously suggests that the bond order increased as electron density from π*-orbitals of the chalcogens has been depleted upon oxidation. The increase of the E–E bond orders should be also reflected by an increase of the E–E stretching vibrations, however, all attempts to obtain reasonable Raman spectra failed due to the intense colour of the compounds. The most striking structural difference upon going from the parent compounds to the radical cations is the dramatic increase of the C–E–E–C torsion angles from 84.6(2)° to 175.8(4)° for 13a/14a, 127.2(1) to 174.6(3)° for 16a/17a (E = S), 75.3(1) to 178.1(1)° for 13b/14b, 128.2(3) to 172.6(5)° for 16b/17b (E = Se) and 123.1(1) to 155.5(3)° for 16c/17c (E = Te), respectively. The positive charges of 17a–c seem to be compensated by intramolecular Menshutkin interactions between chalcogen atoms and mesityl groups of the m-terphenyl substituents (E–Zπca. 3 Å; Zπ = centroid of the phenyl ring).21 The neutral parent compounds exhibit interactions which are substantially longer (E–Zπca. 3.4 Å). Presumably for the same reason, 14a and 14b possesses a short intramolecular S⋯F (2.712(6) Å) and Se⋯F (2.770(2) Å) contacts. These structural changes upon oxidation were satisfactorily reproduced by DFT calculations on two series of parent compounds, namely (PhE)2 (1a–c) and (C6F5E)2 (13a–c), and radical cations, namely [(PhE)2]˙+ (2a–c) and [(C6F5E)2]˙+ (14a–c) for E = S, Se, Te. The adiabatic ionization energies of 1a–c (6.98–7.38 eV) are lower than those of 13a–c (7.64–8.16 eV) and follow the same trend as the cathodic peak potentials of series (2,6-Mes2C6H3E)2 (16a–c) for E = S, Se, Te. In contrast to the neutral (PhE)2 (1a–c) that all have a C2 (H2O2 type) structure with dihedral angles around 90°, all radical cations are calculated having much larger dihedral angles. The phenyl-substituted radical cations [(PhE)2]˙+ (2a–c), can be considered as essentially freely rotating around the E–E bonds, with a minimum energy at dihedral angles around 160° and a C2-symmetric structure. The experimentally found Cs symmetric structure is less than 1 kcal mol−1 higher in energy and corresponds to a calculated transition state (one imaginary frequency (>10 cm−1)). The size of the butterfly shaped m-terphenyl groups easily explains why the radical cations [(2,6-Mes2C6H3E)2]˙+ (17a–b; E = S, Se) have Cs symmetry, simply for reasons of sterical crowding. In the pentafluorophenyl substituted radical cations [(C6F5E)2]˙+ (14a–c) with E = S and Se, the experimentally observed Cs symmetry is the ground state, and the C2 symmetric structure is about 1.5 kcal mol−l higher in energy. Interestingly, [(C6F5Te)2]˙+ (14c) behaves differently. Its ground state is C2h symmetric, completely flat, with a dihedral angle of 180°. The Cs symmetric structure is again only <0.5 kcal mol−l higher in energy. It is unclear whether this peculiar structure is the reason why it has not been possible to isolate it. The HOMO of (C6F5Se)2 (13b) is an admixture of components situated at the Se atoms and the π-system of the pentafluorophenyl groups, whereas in the SOMO of the corresponding radical cation [(C6F5Se)2]˙+ (14b), the unpaired electron is strongly distributed over the coplanar phenyl group (Fig. 2).
 | ||
| Fig. 1 Molecular structures of the radical cations 14a,b and 17a–c; thermal ellipsoids are set at 30% probability. | ||
| (RE)2[(RE)2]˙+ | 13a (ref. 18)/14a, E = S, R = C6F5 | 13b (ref. 18)/14b, E = Se, R = C6F5 | 16a/17a, E = S, R = m-Ter | 16b (ref. 19)/17b, E = Se, R = m-Ter | 16c (ref. 20)/17c, E = Te, R = m-Ter |
|---|---|---|---|---|---|
| a Quinoid character is defined as Q(C10–C15) = (dC10–C11 + dC12–C13 + dC13–C14 + dC15–C10)/4 − (dC11–C12 + dC14–C15)/2 and Q(C20–C25) = (dC20–C21 + dC22–C23 + dC23–C24 + dC25–C20)/4 − (dC21–C22 + dC24–C25)/2 and is 0 for a perfectly delocalized hexagonal benzene structure and 0.138 for a perfect quinoid structure where dC10–C11 = dC12–C13 = dC13–C14 = dC15–C10 = 1.455 Å and dC20–C21 = dC22–C23 = 1.317 Å. b Element distance to the centroid of the phenyl ring E–Zπ. | |||||
| E–E | 2.022(2) | 2.319(4) | 2.073(1) | 2.339(2) | 2.711(1) |
| 2.014(3) | 2.289(1) | 1.998(2) | 2.289(7) | 2.662(1) | |
| C–E | 1.785(6) | 1.90(2) | 1.790(2) | 1.926(6) | 2.144(3) |
| 1.796(6) | 1.92(1) | 1.787(2) | 1.926(7) | 2.151(3) | |
| 1.727(8) | 1.876(2) | 1.762(5) | 1.94(1) | 2.131(8) | |
| 1.764(8) | 1.904(2) | 1.799(5) | 1.947(8) | ||
| C–E–E | 104.5(2) | 98.7(5) | 104.8(1) | 102.2(3) | 103.3(1) |
| 106.2(2) | 98.9(6) | 104.4(1) | 102.3(3) | 103.0(1) | |
| 96.2(3) | 93.71(7) | 95.4(2) | 95.9(3) | 98.5(2) | |
| 107.5(3) | 103.9(1) | 110.4(2) | 105.6(3) | ||
| C–E–E–C | 84.6(2) | 75.3(1) | 127.2(1) | 128.2(3) | 123.1(1) |
| 175.8(4) | 178.1(1) | 174.6(3) | 172.6(5) | 155.5(3) | |
| Q (C10–C15) | 0.036 | 0.012 | 0.019 | 0.019 | 0.037 |
| (26%) | (9%) | (14%) | (14%) | (27%) | |
| Q (C20–C25) | 0.024 | 0.001 | 0.001 | 0.002 | — |
| (17%) | (<1%) | (<1%) | (1%) | ||
| E–Zπb | — | — | 3.439(1) | 3.452(2) | 3.377(1) |
| — | — | 2.975(2) | 3.01(1) | 3.481(1) | |
| 3.161(2) | |||||
| E⋯F | 2.712(6) | 2.770(2) | — | — | — |
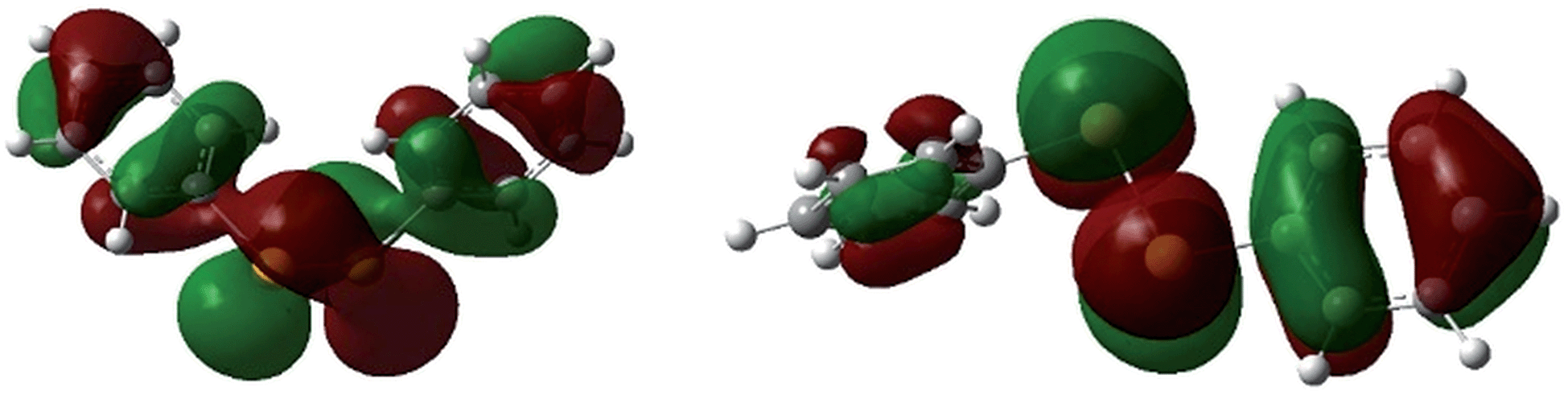 | ||
| Fig. 2 HOMO (left) of C2 symmetric (C6F5Se)2 (13b) and SOMO (right) of Cs symmetric [(C6F5Se)2]˙+ (14b). | ||
The Wiberg bond indices (WBIs) of the E–E bonds increase from about 1 for the parent compounds 1a–c and 13a–c to values between 1.216 and 1.315 for the [(PhE)2]˙+ (2a–c; E = S, Se, Te) series and between 1.176 and 1.282 for the [(C6F5E)2]˙+ (14a–c; E = S, Se, Te) series of radical cations. In the absence of electron delocalization across the coplanar phenyl rings the WBIs of the radical cations would have been expected to be close to 1.5.
The stability of 17a–c in solution dramatically depends on the solvents and in CH2Cl2 on the nature of the chalcogen. Ink-blue solutions of 17a and 17b in CH2Cl2 are stable for months, whereas purple-blue 17c decomposes within a few days. The blue colour arises from very broad absorptions in the near IR region, which stretch into the visible range. Only 17c shows an absorption maximum (CH2Cl2) at λmax = 583 nm in the visible range that is only slightly shifted compared to that of 16c (553 nm) and responsible for the purple tinge. The absorption is tentatively assigned to a n(Te) → σ* transition.22 Dark blue solutions of 17a–c in acetonitrile and propionitrile are stable only for a few days. Notably, after some time the parent compounds 16a–c slowly form back and eventually precipitate. Electrospray mass spectra (MeCN, positive mode) of 17a–c show prominent mass clusters at m/z = 690.4, 786.3 and 882.2, which were unambiguously assigned to the radical cations on the basis of the correct isotopic patterns. The molecular conductivities (MeCN, c = 5 × 10−7 mol L−1) of Λ = 1800, 600 and 540 Ω−1 cm2 mol−1 confirm a high concentration of electrolytes in solutions of 17a–c. The one-electron oxidation upon going from (2,6-Mes2C6H3E)2 (16b, E = Se; 16c, E = Te) to the radical cation [(2,6-Mes2C6H3E)2]˙+ (17b, E = Se; 17c, E = Te) is well reflected by multinuclear NMR spectroscopy. The 77Se NMR spectra show signals at δ = 426.2 (CDCl3) for 16b and at δ = 1362.3 (CDCl3, 223 K) and 1362.4 (MeCN) for 17b, respectively. The 125Te NMR spectra exhibit signals at δ = 322.2 (CDCl3) for 16c and at δ = 1703.8 (CDCl3) and 1698.7 (MeCN) for 17c, respectively. These values are in good agreement with those calculated for (PhSe)2 (1b, δ = 451.5), [(PhSe)2]˙+ (2b, δ = 1237.2 and 1493.0; average 1365.1), (PhTe)2 (1c, δ = 185.1) and [(PhTe)2]˙+ (1c, δ = 1346.8 and 1778.9; average 1562.9). 1H and 13C NMR spectra gave expectedly broad signals. The paramagnetism of the radical cations was unambiguously confirmed by EPR spectroscopy and SQUID magnetometry. X- and Q-band field-sweep EPR spectra measured in frozen CH2Cl2/THF solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) show that all three species have g values that deviate significantly from the free-electron value. All radicals exhibit a rhombic g-matrix with the same g-value ordering, inferring the electronic structure of the three radicals is very similar. The g anisotropy for the radical cations [(2,6-Mes2C6H3E)2]˙+ increases in the order S (17a) < Se (17b) < Te (17c), as expected from the increasing spin–orbit coupling constant and for radicals where the spin density is predominately located on the central dichalcogen moiety. The EPR spectra of the [(2,6-Mes2C6H3E)2]˙+ radical cations feature a single proton hyperfine coupling resolved at g1 and g2 and this coupling was fully characterized by 1H Davies ENDOR spectra recorded at Q-band (Fig. 3, see ESI† for details). Simulation of the data yielded the hyperfine matrix (Table 2, see ESI† for details), which reveals a small but significant isotropic component of |aiso| = 7.1 MHz (aiso = (a1 + a2 + a3)/3), proving a small amount of spin density delocalises onto one of the m-terphenyl ligands.23 The findings were supported by spin density calculation (B3LYP/def2-TZVP) showing noticeable negative spin density on the para-H atom of one m-terphenyl ring (Fig. 4). The EPR spectra of the [(2,6-Mes2C6H3Se)2]˙+ (17b) at X- (Fig. 3 top) and Q-band (Fig. 3 bottom) exhibit two distinct Se hyperfine couplings along g1 (77Se, S = 1/2, 7.6% abundance), consistent with a radical containing a Se–Se moiety with a small asymmetry in the electronic structure. Along g2 and g3, 77Se hyperfine splittings were partially resolved and enabled an estimation of the remaining principal values of the two hyperfine interactions. Q-band Davies ENDOR was used to investigate the proton hyperfine interactions (see ESI† for details) which revealed (again) one largest resolved proton hyperfine interaction. The latter is, however, smaller than in the case of 17a, with |aiso| = 4.2 MHz. This result is consistent with the small asymmetry in the spin density inferred from the two inequivalent 77Se hyperfine couplings. The EPR spectra of [(2,6-Mes2C6H3Te)2]˙+ (17c) established principal values for the rhombic g-matrix (Fig. 3 and Table 2). Because of the low natural abundance of 125Te (S = 1/2, 7% abundance), the broad EPR spectrum and small background signals of the data only allowed estimates for the hyperfine couplings and were not of sufficient quality to resolve a potential inequivalence in the two Te nuclei. Along g1, the hyperfine coupling is resolved with |A(125Te)| = 1000 MHz; along g2 and g3, the 125Te hyperfine couplings were partially resolved and allowed the estimates shown in Table 2. Davies ENDOR was used to investigate the proton hyperfine interactions and showed that the largest proton hyperfine interaction has further decreased in comparison to 17a and 17b. Along g2, the coupling is |A| = 3.8 MHz, whereas for 17b: |A| = 6.4 MHz, and for 17a: |A| = 11.0 MHz. The experimental EPR data are very well modelled by DFT calculations (Fig. 4 and ESI† for details). The principal g-values are well reproduced as well as the two inequivalent hyperfine couplings for 77Se. The DFT data are able to unambiguously assign the largest proton coupling to the para-H atom of the central phenyl group of the one of the m-terphenyl substituents (Fig. 4). The trend to smaller 1H hyperfine values observed experimentally in the series 17a, 17b, and 17c follows the change in the orientation of two m-terphenyl substituents with respect to the central dichalcogen moiety: for 17a, the phenyl group π system of one m-terphenyl substituent is well orientated to allow overlap with the π-type orbitals carrying the unpaired electron on the S–S moiety and thus facilitates spin density delocalization. In contrast, the phenyl group of the second m-terphenyl substituent is approximately at 90° to this orientation and thus there is poor overlap with the S π-orbitals carrying the unpaired electron and less spin delocalisation. For 17c, the π-orbitals of both phenyl rings of the m-terphenyl
substituent have essentially the same orientation with respect to the π-type orbitals of the Te–Te moiety carrying the unpaired electron but with relatively poor orbital overlap, resulting in an equivalent but relatively small delocalisation of the spin densities onto the two m-terphenyl substituents. As expected, 17b has a structure intermediate between 17a and 17c and this is reflected in the experimental 1H hyperfine coupling assigned to one para-H of the m-terphenyl group. The summed chalcogen-based spin densities from DFT calculations are 0.698, 0.734 and 0.778 for 17a, 17b, and 17c, respectively. These findings in conjunction with the EPR data leave no doubt that all three radical cations are characterized best as chalcogen-centred (i.e. ca. 70–80% spin density on the chalcogen atoms). The effective magnetic moments (μeff) of 17a–c at 300 K are 1.61, 1.48 and 1.41 μB, respectively, and reasonably close to the theoretically expected value of 1.744 μB, 1.7880 μB, 1.8625 μB, respectively, for a system of uncoupled paramagnetic centers with spins S = 1/2; the μeff decreases slightly with lowering temperature, that implies weak antiferromagnetic interactions in the solid state (see ESI† for details).
:1) show that all three species have g values that deviate significantly from the free-electron value. All radicals exhibit a rhombic g-matrix with the same g-value ordering, inferring the electronic structure of the three radicals is very similar. The g anisotropy for the radical cations [(2,6-Mes2C6H3E)2]˙+ increases in the order S (17a) < Se (17b) < Te (17c), as expected from the increasing spin–orbit coupling constant and for radicals where the spin density is predominately located on the central dichalcogen moiety. The EPR spectra of the [(2,6-Mes2C6H3E)2]˙+ radical cations feature a single proton hyperfine coupling resolved at g1 and g2 and this coupling was fully characterized by 1H Davies ENDOR spectra recorded at Q-band (Fig. 3, see ESI† for details). Simulation of the data yielded the hyperfine matrix (Table 2, see ESI† for details), which reveals a small but significant isotropic component of |aiso| = 7.1 MHz (aiso = (a1 + a2 + a3)/3), proving a small amount of spin density delocalises onto one of the m-terphenyl ligands.23 The findings were supported by spin density calculation (B3LYP/def2-TZVP) showing noticeable negative spin density on the para-H atom of one m-terphenyl ring (Fig. 4). The EPR spectra of the [(2,6-Mes2C6H3Se)2]˙+ (17b) at X- (Fig. 3 top) and Q-band (Fig. 3 bottom) exhibit two distinct Se hyperfine couplings along g1 (77Se, S = 1/2, 7.6% abundance), consistent with a radical containing a Se–Se moiety with a small asymmetry in the electronic structure. Along g2 and g3, 77Se hyperfine splittings were partially resolved and enabled an estimation of the remaining principal values of the two hyperfine interactions. Q-band Davies ENDOR was used to investigate the proton hyperfine interactions (see ESI† for details) which revealed (again) one largest resolved proton hyperfine interaction. The latter is, however, smaller than in the case of 17a, with |aiso| = 4.2 MHz. This result is consistent with the small asymmetry in the spin density inferred from the two inequivalent 77Se hyperfine couplings. The EPR spectra of [(2,6-Mes2C6H3Te)2]˙+ (17c) established principal values for the rhombic g-matrix (Fig. 3 and Table 2). Because of the low natural abundance of 125Te (S = 1/2, 7% abundance), the broad EPR spectrum and small background signals of the data only allowed estimates for the hyperfine couplings and were not of sufficient quality to resolve a potential inequivalence in the two Te nuclei. Along g1, the hyperfine coupling is resolved with |A(125Te)| = 1000 MHz; along g2 and g3, the 125Te hyperfine couplings were partially resolved and allowed the estimates shown in Table 2. Davies ENDOR was used to investigate the proton hyperfine interactions and showed that the largest proton hyperfine interaction has further decreased in comparison to 17a and 17b. Along g2, the coupling is |A| = 3.8 MHz, whereas for 17b: |A| = 6.4 MHz, and for 17a: |A| = 11.0 MHz. The experimental EPR data are very well modelled by DFT calculations (Fig. 4 and ESI† for details). The principal g-values are well reproduced as well as the two inequivalent hyperfine couplings for 77Se. The DFT data are able to unambiguously assign the largest proton coupling to the para-H atom of the central phenyl group of the one of the m-terphenyl substituents (Fig. 4). The trend to smaller 1H hyperfine values observed experimentally in the series 17a, 17b, and 17c follows the change in the orientation of two m-terphenyl substituents with respect to the central dichalcogen moiety: for 17a, the phenyl group π system of one m-terphenyl substituent is well orientated to allow overlap with the π-type orbitals carrying the unpaired electron on the S–S moiety and thus facilitates spin density delocalization. In contrast, the phenyl group of the second m-terphenyl substituent is approximately at 90° to this orientation and thus there is poor overlap with the S π-orbitals carrying the unpaired electron and less spin delocalisation. For 17c, the π-orbitals of both phenyl rings of the m-terphenyl
substituent have essentially the same orientation with respect to the π-type orbitals of the Te–Te moiety carrying the unpaired electron but with relatively poor orbital overlap, resulting in an equivalent but relatively small delocalisation of the spin densities onto the two m-terphenyl substituents. As expected, 17b has a structure intermediate between 17a and 17c and this is reflected in the experimental 1H hyperfine coupling assigned to one para-H of the m-terphenyl group. The summed chalcogen-based spin densities from DFT calculations are 0.698, 0.734 and 0.778 for 17a, 17b, and 17c, respectively. These findings in conjunction with the EPR data leave no doubt that all three radical cations are characterized best as chalcogen-centred (i.e. ca. 70–80% spin density on the chalcogen atoms). The effective magnetic moments (μeff) of 17a–c at 300 K are 1.61, 1.48 and 1.41 μB, respectively, and reasonably close to the theoretically expected value of 1.744 μB, 1.7880 μB, 1.8625 μB, respectively, for a system of uncoupled paramagnetic centers with spins S = 1/2; the μeff decreases slightly with lowering temperature, that implies weak antiferromagnetic interactions in the solid state (see ESI† for details).
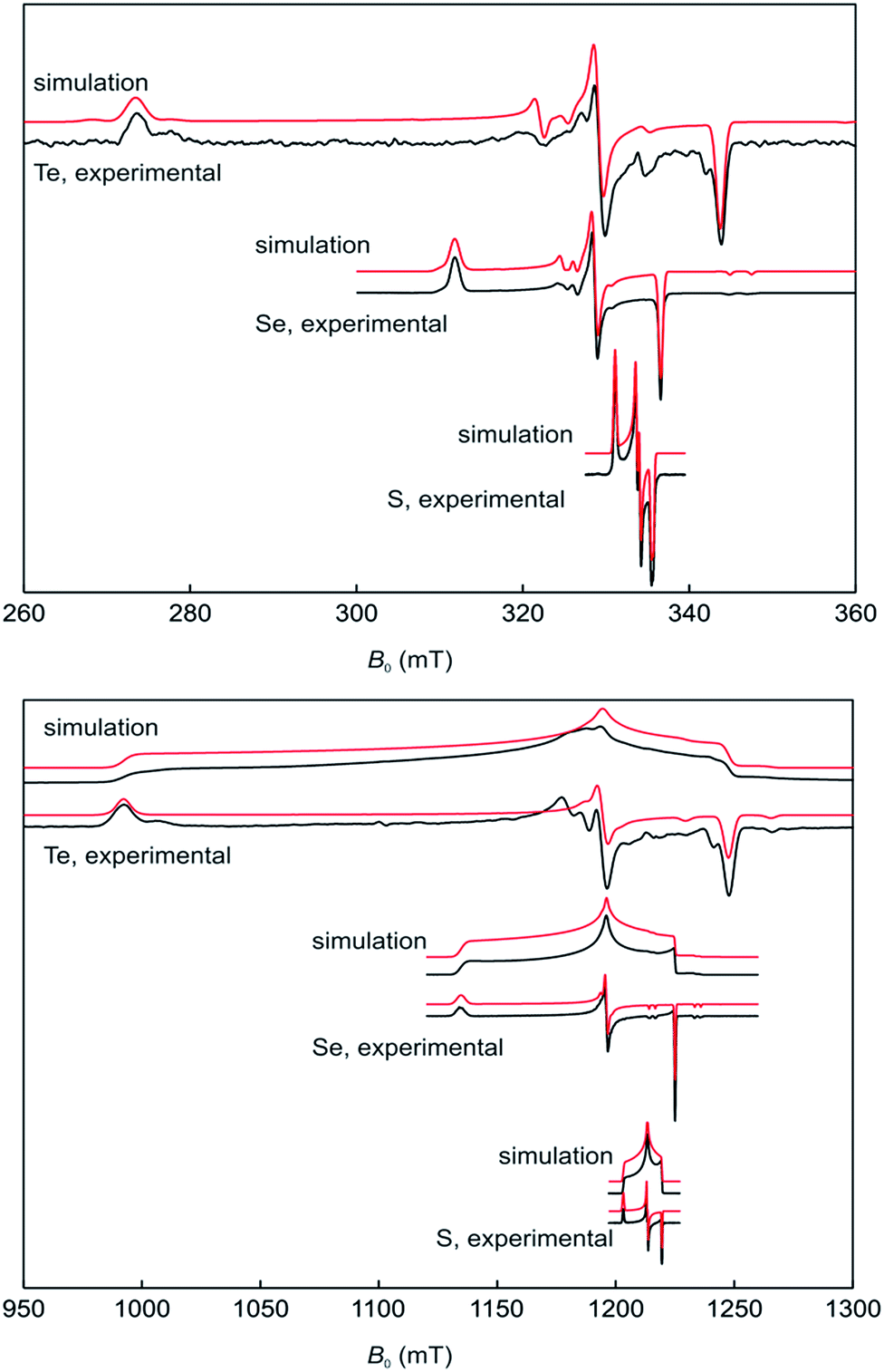 | ||
| Fig. 3 Field-sweep EPR spectra for 17a (E = S) 17b (E = Se) and 17c (E = Te) measured in frozen CH2Cl2/THF solution (1:1), along with the corresponding simulations. | ||
| Parameter | Principal values |
|---|---|
| 17a | |
| g | 2.0014, 2.0115, 2.0285 (2.0022, 2.0107, 2.0213) |
| A(1H) | −3.0, −7.2, −11.0 (−1.8, −5.8, −8.5) |
|
|
| 17b | |
| g | 1.9956, 2.0438, 2.1543 (2.0021, 2.0452, 2.1165) |
| A(77Se) | −50, −100, 465 (−106, −116, 306) |
| A(77Se) | −80, −115, 610 (−135, −140, 407) |
| A(1H) | −1.8, −4.3, −6.4 (−1.2, −4.2, −6.1) |
|
|
| 17c | |
| g | 1.9542, 2.0411, 2.4566 (2.0021, 2.0876, 2.3688) |
| A(127Te) | 300, 350, −1000 (350, 370, −720) (352, 372, −724) |
| A(1H) | −, −, −3.8 (−0.6, −2.6, −3.8) |
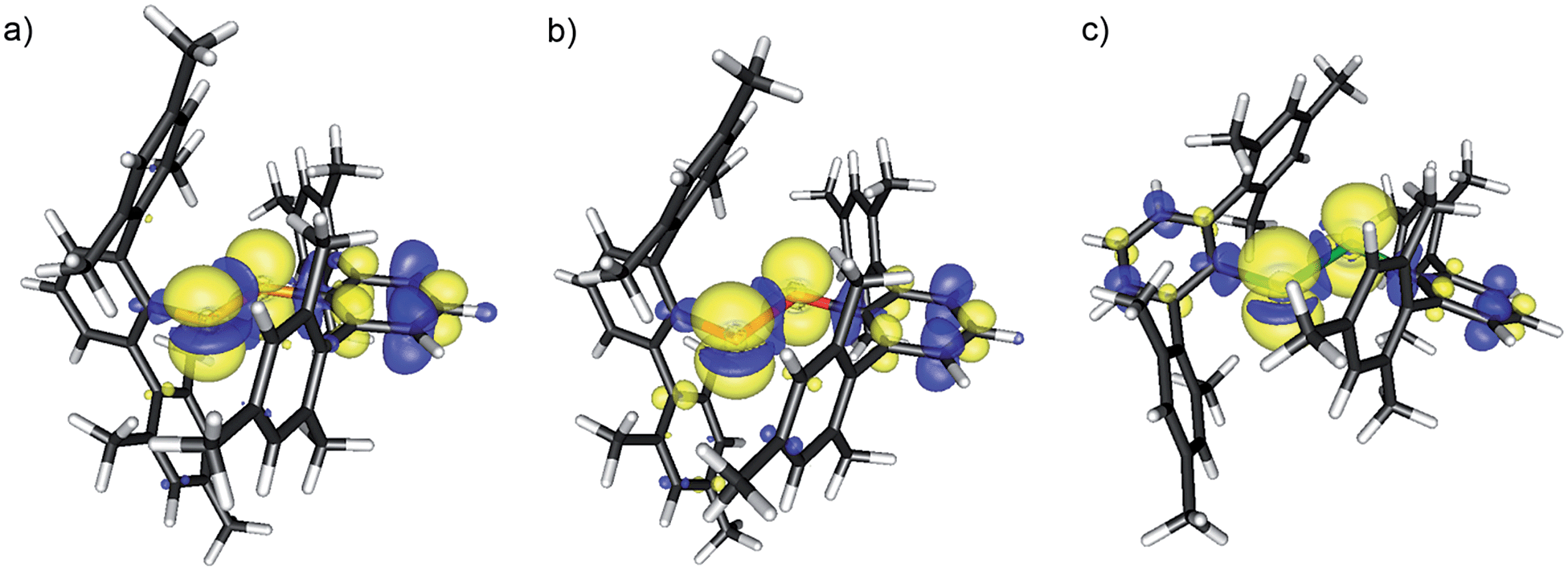 | ||
| Fig. 4 Calculated spin densities for 17a–c at contour levels of 0.0025 (yellow) and −0.0005 (blue). Note the small but noticeable negative spin density on the para-H atom of one terphenyl ring for 17a and 17b. | ||
Conclusion
The stable group 16 radical cations [(RE)2]˙+ (14a–b, 17a–c; E = S, Se, Te) were prepared and fully characterized by various methods for the first time. Note that these radical cations are isoelectronic with the previously described group 15 radical anions [(RE)2]˙− (E = P, As, Sb; R = 2,6-[(Me3Si)2CH]2-4-[(Me3Si)3C]-C6H2).24 Bearing in mind that the aryltellurenyl fragment RTe is isolobal with atomic iodine I, the paramagnetic [(RTe)2]˙+ (17c) and the diamagnetic [(RTe)4]2+ (10, R = Et)11 also resemble the paramagnetic [I2]˙+ (18)25 and the diamagnetic I42+ (19).26 In the same notion, the radical cations [(RSe)2]˙+ (14b, R = C6F5; 17b, R = 2,6-Mes2C6H3) and [(RS)2]˙+ (14a, R = C6F5; 17a, R = 2,6-Mes2C6H3) are closely related to the known [Br2]˙+ (20)27 and the elusive [Cl2]˙+ (21), which forms [Cl4]˙+ (22) with Cl2 (Scheme 2).28 The reported findings leave no doubt that the radical cations 14a-b, 17a-c contain chalcogen-centred odd-electron π-bonds.29 It is noteworthy that recently also odd-electron S–S and Se–Se σ-bonded 1,8-bis(arylchalcogenyl)naphthalene were investigated.30 In view of the fact that many odd-electron species exist as (reactive) intermediates in various chemical reactions and play an important role in bond formation and cleavage processes, alongside many applications that have been reported or envisaged for stable radicals, the successful isolation of the title compounds provide a suitable entry point for an in-depth exploration of these and related species. Accordingly, the isolation and characterization of other examples for odd-electron bonding, which is of both fundamental and practical interest, is currently under way in our laboratories.Experimental
Synthesis of (C6F5S)2Sb2F11 ([14a][Sb2F11])
SbF5 (220 mg, 1.0 mmol) was added to solid 13a (40 mg, 0.1 mmol) in a PFA tube. 13a directly changed the colour to dark blue when SbF5 reached the solid. The mixture has been kept 3 h at room temperature until the dissolving of 13a in SbF5 completed. The reaction mixture was then evacuated and SO2ClF (1.0 mL) was condensed into the PFA tube at −196 °C. The mixture was allowed to slowly warm up to −80 °C using a dry ice/ethanol bath. After the melting of the solvent (1 h) completed, the reaction mixture was allowed to slowly warm up to room temperature (4 h) to ensure the complete dissolution of the reaction mixture. Afterwards the PFA tube was sealed under reduced temperature and pressure. Dark blue needles of [14a][Sb2F11] formed at −30 °C after several careful cycles of cooling and warming between −50 °C and to −78 °C.Synthesis of (C6F5Se)2As2F11 ([14b][As2F11])
SO2ClF (0.20 mL) was slowly condensed onto solid 13b (25 mg, 0.05 mmol) in a PFA tube at −196 °C. The mixture was slowly warmed up to room temperature for complete dissolution. Then the mixture was cooled again to −196 °C and AsF5 (0.04 mL, 0.5 mmol) was condensed into the PFA tube. The solidified mixture directly changed the colour to dark violet. Afterwards the mixture was slowly warmed up to −78 °C where the mixture melts and partial dissolution of the solid gave a deep green colour. The reaction was allowed to further warm up until reaching −30 °C after 3 h, when additional SO2ClF (1.0 mL) was added. Afterwards the PFA tube was sealed under reduced temperature and pressure. Black needles of [14b][As2F11] formed after careful cooling to −78 °C over two days.Attempts to oxidize (C6F5Te)2 (13c)
At −196 °C, SO2 (0.20 mL) was condensed onto 13c (30 mg, 0.05 mmol) in a PFA tube. After warming up to room temperature, the reaction mixture was cooled down to −196 °C a second time to add AsF5 (0.04 mL, 0.5 mmol). The colour of the reaction changed to dark violet during the process of condensation. Afterwards the mixture was slowly warmed up to −78 °C, to remove the excess of AsF5 under reduced pressure. After a third cooling process (−196 °C), further SO2 (1.0 mL) was added and the reaction mixture was allowed to slowly warm up to −78 °C to give a deep violet solution. For a complete melting process of the solvent, the mixture was further warmed up to −30 °C over a period of 3 h. Afterwards F114 (0.3 mL) has been condensed onto the solidified reaction mixture at −196 °C and the PFA tube sealed afterwards. [Te4][AsF6]2 crystallized as dark violet plates after a careful cooling process from room temperature to −30 °C over two days.Synthesis of 2,6-Mes2C6H3SH
A solution of 2,6-dimesityliodobenzene (6.50 g, 14.7 mmol) in n-hexane (150 mL) was treated with a 2.5 M solution of n-BuLi (6.60 mL, 14.7 mmol) at room temperature and stirred overnight. The volume of the suspension has been reduced to 20 mL under reduced pressure and the white solid has been filtered off. The product, 2,6-dimesitylphenyl lithium (3.33 g, 10.4 mmol), was dissolved in THF (50 mL), cooled to −78 °C and sulfur powder (0.40 g, 12.5 mmol) was cautiously added. The red suspension was stirred overnight and allowed to warm up to room temperature during that period. Hydrochloric acid (40 mL, 10%) was added to the suspension and the stirring has been extended for another 2 h. The organic phase was extracted with CHCl3 (3 × 50 mL), filtrated and the solvent removed under reduced pressure. The crude product was recrystallized from CH2Cl2 to yield pale yellow crystals of 2,6-Mes2C6H3SH (3.02 g, 8.72 mmol, 84%; Mp. 192–194 °C).1H-NMR (CDCl3): δ = 7.27 (t, 1H, 3J = 8.1 Hz), 7.06 (d, 2H, 3J = 7.5 Hz), 7.02 (s, 4H), 3.07 (s, 1H, SH), 2.38 (s, 6H), 2.07 (s, 12H) ppm. 13C{1H}-NMR (CDCl3): δ = 138.7, 137.3, 136.2, 132.7, 128.4, 128.2, 128.0, 125.1, 21.2, 20.0 ppm.
Synthesis of (2,6-Mes2C6H3S)2 (16a)
A solution of 2,6-Mes2C6H3SH (2.00 g, 5.77 mmol) in toluene (80 mL) was treated with a solution of ethyl nitrite in ethanol (∼15%, 25 mL, 40 mmol). The solution was kept at room temperature for 4 h before stirring it at 76 °C over a period of 72 h. The resulting yellow solid was filtered off and dried under vacuum. The crude product was recrystallized from CH2Cl2 to afford yellow crystals of 16a (1.80 g, 2.60 mmol, 90%; Mp. >230 °C).1H-NMR (CDCl3): δ = 7.19 (t, 2H, 3J = 7.4 Hz), 6.88 (d, 4H, 3J = 7.5 Hz), 6.80 (8H), 2.35 (12H), 1.70 (24H) ppm. 13C{1H}-NMR (CDCl3): δ = 142.9, 137.4, 136.6, 136.5, 136.1, 129.2, 127.7, 127.1, 21.1, 20.3 ppm. Anal. calcd for C48H50S2 (691.06): C, 83.43; H, 7.29. Found C, 83.29; H, 6.91.
Synthesis of 2,6-Mes2C6H3SeH
A solution of 2,6-dimesityliodobenzene (6.50 g, 14.7 mmol) in n-hexane (150 mL) was treated with a 2.5 M solution of n-BuLi (6.60 mL, 14.7 mmol) at room temperature and stirred overnight. The volume of the suspension has been reduced to 20 mL under reduced pressure and the white solid has been filtered off. The product, 2,6-dimesitylphenyl lithium (3.33 g, 10.4 mmol), was dissolved in THF (50 mL), cooled to −78 °C and selenium powder (0.990 g, 12.3 mmol) was cautiously added. The black suspension was stirred overnight and allowed to warm up to room temperature during that period. Hydrochloric acid (40 mL, 5%) was added to the suspension and the stirring was extended for another 2 h. The organic phase has been extracted with CHCl3 (3 × 50 mL), filtrated and the solvent removed under reduced pressure. The remaining solid was dissolved in THF (50 mL) and slowly added to a suspension of LiAlH4 (0.79 g, 20.8 mmol) in THF (10 mL) at 0 °C. The suspension was stirred for 1 h, cautiously poured onto ice-water and extracted with CHCl3 (3 × 50 mL). The crude product was recrystallized from n-hexane to afford yellow crystals of 2,6-Mes2C6H3SeH (1.76 g, 4.47 mmol, 43%; Mp. = 222–224 °C).1H-NMR (CDCl3): δ = 7.19 (t, 1H, 3J = 7.5 Hz), 7.09 (d, 2H, 3J = 7.4 Hz), 7.04 (s, 4H), 2.41 (s, 6H), 2.10 (s, 12H), 1.11 (s, 1H, SeH), 1J (77Se–1H = 63.4 Hz) ppm. 13C{1H}-NMR (CDCl3): δ = 141.1, 138.9, 137.3, 136.0, 129.1, 128.4, 128.0, 126.0, 21.2, 20.0 ppm. 77Se-NMR (CDCl3): δ = 71.8 (d, 1J (1H–77Se = 63.4 Hz) ppm.
Synthesis of (2,6-Mes2C6H3Se)2 (16b)
A solution of 2,6-Mes2C6H3SeH (1.72 g, 4.37 mmol) was dissolved in toluene (100 mL) and treated with a solution of ethyl nitrite in ethanol (∼15%, 16.2 g, 32.3 mmol). The solution was kept at room temperature for 4 h before stirring it at 82 °C over a period of 48 h. The remaining solvent was removed under reduced pressure. The product was crystallized from CHCl3 to afford deep red prisms of 16b (580 mg, 1.49 mmol, 34%; Mp. >230 °C).1H-NMR (CDCl3): δ = 7.19 (t, 2H, 3J = 7.5 Hz), 6.82 (d, 4H, 3J = 7.5 Hz), 6.74 (8H), 2.32 (12H), 1.72 (24H) ppm. 13C{1H}-NMR (CDCl3): δ = 144.1, 138.2, 136.4, 136.2, 132.8, 129.0, 127.9, 127.3, 20.9, 20.4 ppm. 77Se-NMR (CDCl3): δ = 426.2 ppm.
Synthesis of (2,6-Mes2C6H3S)2SbF6 ([17a][SbF6])
Solid 16a (193 mg, 0.28 mmol) was added to a stirred solution of [NO][SbF6] in propionitrile (15 mL). After 18 h of stirring, the solvent was removed under reduced pressure affording a deep blue solid that was recrystallized from CH2Cl2/n-hexane (1:1) at room temperature to yield [17a][SbF6] (250 mg, 0.027 mmol, 96%).
ESI MS (CH3CN, positive mode): m/z = 690.4 [C48H50S2]˙+ for 17a. Molar conductivity (CH3CN, c = 5 × 10−7 mol L−1): Λ = 1800 Ω−1 cm2 mol−1. SQUID: μeff (300 K) = 1.61 μB. Anal. calcd for C48H50F6S2Sb (926.80): C, 62.21; H, 5.44. Found C, 62.57; H, 5.62.
Synthesis of (2,6-Mes2C6H3Se)2SbF6 ([17b][SbF6])
Solid 16b (220 mg, 0.28 mmol) was added to a stirred solution of [NO][SbF6] in propionitrile (15 mL). After 12 h of stirring, the solvent was removed under reduced pressure affording a deep blue solid that was recrystallized from CH2Cl2/n-hexane (1:1) at room temperature to yield [17b][SbF6] (280 mg, 0.27 mmol, 98%).
77Se-NMR (CDCl3, r.t.): δ = no signal. 77Se-NMR (CDCl3, 223 K): δ = 1362.3 ppm. 77Se-NMR (CD3CN): δ = 1362.4 ppm. ESI MS (CH3CN, positive mode): m/z = 786.3 [C48H50Se2]˙+ for 17b. UV-vis (CH2Cl2, c = 1 × 10−3 mol L−1): λmax = 710 nm. Molar conductivity (CH3CN, c = 5 × 10−7 mol L−1): Λ = 600 Ω−1 cm2 mol−1. SQUID: μeff (300 K) = 1.48 μB. Anal. calcd for C48H50F6Se2Sb (1105.59): C, 56.49; H, 4.94. Found C, 56.57; H, 5.12.
Synthesis of (2,6-Mes2C6H3Te)2SbF6 ([17c][SbF6])
Solid 16c (247 mg, 0.28 mmol) was added to a stirred solution of [NO][SbF6] in propionitrile (15 mL). After 8 h of stirring, the solvent was removed under reduced pressure affording a purple blue almost black solid that was recrystallized from propionitrile at room temperature to yield [17c][SbF6] (300 mg, 0.27 mmol, 95%).125Te-NMR (CD3CN): δ = 1698.7 ppm. 125Te-NMR (CDCl3): δ = 1703.8 ppm. ESI MS (CH3CN, positive mode): m/z = 882.2 [C48H50Te2]˙+ for 17c. UV-vis (CH2Cl2, c = 1 × 10−3 mol L−1): λmax = 583 nm. Molar conductivity (CH3CN, c = 5 × 10−7 mol L−1): Λ = 540 Ω−1 cm2 mol−1. SQUID: μeff (300 K) = 1.41 μB. Anal. calcd for C51H55F6NTe2Sb (1172.94): C, 52.22; H, 4.73; N, 1.19. Found C, 52.38; H, 4.62; N, 1.24.
Acknowledgements
The Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged for financial support. JRH thanks the ARC (FT120100421) for financial support and the Australian National Fabrication Facility for use of equipment. MM thanks the Fonds der Chemischen Industrie (VCI) for a scholarship.Notes and references
- (a) M. Gomberg, Ber. Dtsch. Chem. Ges., 1900, 33, 3150 CrossRef CAS; (b) M. Gomberg, J. Am. Chem. Soc., 1900, 22, 757 CrossRef.
- Stable radicals can be isolated and handled as a pure compound, whereas radicals that are sufficiently long-lived to be observed using conventional spectroscopic methods but cannot be isolated are classified as persistent. D. Griller and K. U. Ingold, Acc. Chem. Res., 1976, 9, 13 CrossRef CAS.
- (a) S. González-Gallardo and F. Breher, Main Group Biradicaloids, Comprehensive Inorganic Chemistry II, 2013, vol. 1, pp. 413–455 Search PubMed; (b) F. Breher, Coord. Chem. Rev., 2007, 251, 1007 CrossRef CAS; (c) M. Abe, Chem. Rev., 2013, 113, 7011 CrossRef CAS PubMed.
- R. G. Hicks, Org. Biomol. Chem., 2007, 5, 1321 CAS . However, apart from these fundamentally important questions, many applications of stable radicals have been reported or envisaged such as spin labelling, spin trapping, EPR imaging, radical polymerization, or catalysis, just to mention a few. In particular, the development of new materials with technologically relevant properties, such as magnetism or conductivity is in the current focus of interest. A very appealing idea in this context is to use stable radicals as both a charge carrier and magnetic coupler. Selected reviews: (a) Stable Radicals – Fundamentals and Applied Aspects of Odd-Electron Compounds, ed. R. G. Hicks, John Wiley & Sons Ltd, 2010 Search PubMed; (b) A. Rajca, Chem. Rev., 1994, 94, 871 CrossRef CAS; (c) J. S. Miller, Adv. Mater., 2002, 14, 1105 CrossRef CAS; (d) Special issue on, Molecular Conductors: Chem. Rev., 2004, 104 Search PubMed; (e) J. M. Rawson, A. Alberola and A. Whalley, J. Mater. Chem., 2006, 16, 2560 RSC.
- (a) P. P. Power, Nature, 2010, 463, 171 CrossRef CAS PubMed; (b) T. Chivers and J. Konu, Comments Inorg. Chem., 2009, 30, 131 CrossRef CAS.
- P. P. Power, Chem. Rev., 2003, 103, 789 CrossRef CAS PubMed.
- (a) G. Stenhouse, Proc. R. Soc. London, 1868, 17, 62 CrossRef; (b) G. Stenhouse, Justus Liebigs Ann. Chem., 1869, 149, 247 CrossRef.
- (a) K. Fies and W. Volk, Chem. Ber., 1909, 42, 1170 CrossRef; (b) K. Fries, H. Koch and H. Stuckebrock, Justus Liebigs Ann. Chem., 1929, 468, 162 CrossRef CAS; (c) J. Giordan and H. Bock, Chem. Ber., 1982, 115, 2548 CrossRef CAS; (d) H. Bock, A. Rauschenbach, C. Näther, M. Kleine and Z. Havlas, Chem. Ber., 1994, 127, 2043 CrossRef CAS; (e) P. Rangappa and H. J. Shine, J. Sulfur Chem., 2006, 27, 617 CrossRef CAS; (f) J. Beck, T. Bredow and R. T. Tjahjanto, Z. Naturforsch., B: J. Chem. Sci., 2009, 64, 145 CAS; (g) R. T. Tjahjanto, M. F. Peintinger, T. Bredow and J. Beck, Eur. J. Inorg. Chem., 2012, 3625 CrossRef CAS; (h) H. Poeschner and K. Seppelt, Angew. Chem., 2013, 125, 13072 ( Angew. Chem., Int. Ed. , 2013 , 52 , 12838 ) CrossRef.
- (a) P. S. Lakkaraju, D. Zhou and H. D. Roth, J. Chem. Soc., Perkin Trans. 2, 1998, 1119 RSC; (b) P. S. Lakkaraju, K. Shen, H. D. Roth and H. García, J. Phys. Chem. A, 1999, 103, 7381 CrossRef CAS; (c) V. Martí, L. Fernández, V. Fornés, H. García and H. D. Roth, J. Chem. Soc., Perkin Trans. 2, 1999, 145 RSC.
- B. Mueller, T. T. Takaluoma, R. S. Laitinen and K. Seppelt, Eur. J. Inorg. Chem., 2011, 4970 CrossRef CAS.
- (a) J. Passmore, E. K. Richardson and P. Taylor, J. Chem. Soc., Dalton Trans., 1976, 1006 RSC; (b) B. Mueller, H. Poleschner and K. Seppelt, Dalton Trans., 2008, 4424 RSC.
- Numerous classical inorganic molecules such (NO)2, [S2O4]2, [S2I4]2+and [Te6]4+ show π*–π* interactions: N. Burford, J. Passmore and J. C. P. Sanders, in From Atoms to Polymers, Isoelectronic Analogies, ed. J. F. Liebman and A. Greenberg, Wiley-VCH, Weinheim, 1989, pp. 53–108 Search PubMed.
- S. Brownridge, T. S. Cameron, J. Passmore, G. Schatte and G. W. Sutherland, Can. J. Chem., 1998, 76, 1050 CrossRef CAS.
- (a) R. C. Burns, R. J. Gillespie, W. C. Luk and D. R. Slim, Inorg. Chem., 1979, 18, 3084 Search PubMed; (b) R. Faggiani, R. J. Gillespie and J. E. Vekris, Chem. Commun., 1988, 902 RSC; (c) J. Beck, F. Stecken, A. Reich and H. Foehlsing, Z. Anorg. Allg. Chem., 2003, 629, 1073 CrossRef CAS.
- If preferred the potentials might be referenced to the external ferrocene standard Fc/Fc+ (E1/2 + 0.46 V). N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877 CrossRef CAS PubMed.
- J. Beckmann, D. Dakternieks, A. Duthie and C. Mitchell, Aust. J. Chem., 2005, 58, 119 CrossRef CAS.
- H. Shorafa, D. Mollenhauer, B. Paulus and K. Seppelt, Angew. Chem., 2009, 121, 5959 ( Angew. Chem Int. Ed. , 2009 , 48 , 5845 ) CrossRef.
- C. M. Woodard, D. S. Brown, J. D. Lee and A. G. Massey, J. Organomet. Chem., 1976, 121, 333 CrossRef CAS.
- J. J. Ellison, K. Ruhland-Senge, H. H. Hope and P. P. Power, Inorg. Chem., 1995, 34, 49 CrossRef CAS.
- J. Beckmann, M. Hesse, H. Poleschner and K. Seppelt, Angew. Chem., 2007, 119, 8425 ( Angew. Chem., Int. Ed. , 2007 , 46 , 8277 ) CrossRef.
- (a) J. Zuckerman-Schpector and J. Haiduc, CrystEngComm, 2002, 4, 178 RSC; (b) H. Schmidbaur and A. Schnier, Organometallics, 2008, 27, 2361 CrossRef CAS.
- (a) R. I. Vinokurova, L. A. Sorokina, I. V. Olifirenko, L. M. Kataeva and E. G. Kataev, Zh. Obshch. Khim., 1983, 53, 599 CAS; (b) H. Kunkely and A. Vogler, Inorg. Chim. Acta, 2009, 362, 281 CrossRef CAS.
- The spin density is asymmetric since the largest hyperfine couplings from the second Ar ligand is significantly smaller, Amax < 3.5 MHz.
- T. Sasamori, E. Mieda, N. Nagahora, K. Sato, D. Shiomi, T. Takui, Y. Hosoi, Y. Furukawa, N. Takagi, S. Nagase and N. Tokitoh, J. Am. Chem. Soc., 2006, 128, 12582 CrossRef CAS PubMed.
- C. G. Davies, R. J. Gillespie, P. R. Ireland and J. M. Sowa, Can. J. Chem., 1974, 52, 2048 CrossRef CAS.
- (a) R. J. Gillespie, J. B. Milne and M. J. Morton, Inorg. Chem., 1968, 7, 2221 CrossRef CAS; (b) R. J. Gillespie, R. Kapoor, R. Fggiani, C. J. L. Lock, M. Murchie and J. Passmore, J. Chem. Soc., Chem. Commun., 1983, 8 RSC; (c) R. Faggiani, R. J. Gillespie, R. Kapoor, C. J. L. Lock and J. E. Vekris, Inorg. Chem., 1988, 27, 4350 CrossRef CAS.
- A. J. Edwards and G. R. Stone, J. Chem. Soc. A, 1971, 2318 RSC.
- S. Seidel and K. Seppelt, Angew. Chem., 2000, 112(21), 4072 ( Angew. Chem., Int. Ed. , 2000 , 39 , 3923 ) CrossRef.
- H. Grützmacher and F. Breher, Angew. Chem., 2002, 114, 4178 ( Angew. Chem., Int. Ed. , 2002 , 41 , 4006 ) CrossRef.
- (a) F. R. Knight, R. A. M. Randall, T. L. Roemmele, R. T. Boeré, B. E. Bode, L. Crawford, M. Bühl, A. M. Z. Slawin and J. D. Woollins, ChemPhysChem, 2013, 14, 3199 CrossRef CAS PubMed; (b) S. Zhang, X. Wang, Y. Su, Y. Qiu, Z. Zhang and X. Wang, Nat. Commun., 2014, 5, 4127 CAS; (c) S. Zhang, X. Wang, Y. Sui and X. Wang, J. Am. Chem. Soc., 2014, 136, 14666 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental considerations, cyclic voltammograms of 16a–c; UV-vis spectra of 16a–c and 17a–c; ESI-MS spectra of 17a–c; experimental and DFT EPR parameters of 17a–c; SQUID magnetic moments of 17a–c; crystal and refinement data of [14a][Sb2F11], [14b][As2F11], 16a, [17a][SbF6], [17b][SbF6]·CH2Cl2, [17c][SbF6]·NCCH2CH3; molecular structure of 16a; results from the conformational analysis of 1a–c, 2a–c, 13a–c and 14a–c; ionisation energies of 1a–c and 13a–c; Wiberg bond indices (WBIs), NBO and Mulliken charges of 1a–c, 2a–c, 13a–c and 14a–c. CCDC 999869–999873 and 1028017. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02964j |
| ‡ Dedicated to Professor Hubert Schmidbaur on the occasion of his 80th birthday. |
| This journal is © The Royal Society of Chemistry 2015 |