
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceOxygen evolution on well-characterized mass-selected Ru and RuO2 nanoparticles†
Elisa A.
Paoli
a,
Federico
Masini
a,
Rasmus
Frydendal
a,
Davide
Deiana
b,
Christian
Schlaup
a,
Mauro
Malizia
a,
Thomas W.
Hansen
b,
Sebastian
Horch
a,
Ifan E. L.
Stephens
*a and
Ib
Chorkendorff
*a
aCenter for Individual Nanoparticle Functionality (CINF), Department of Physics, Kgs. Lyngby DK-2800, Denmark. E-mail: ib.chorkendorff@fysik.dtu.dk
bCenter for Electron Nanoscopy (CEN), Kgs. Lyngby DK-2800, Denmark
First published on 26th September 2014
Abstract
Oxygen evolution was investigated on model, mass-selected RuO2 nanoparticles in acid, prepared by magnetron sputtering. Our investigations include electrochemical measurements, electron microscopy, scanning tunneling microscopy and X-ray photoelectron spectroscopy. We show that the stability and activity of nanoparticulate RuO2 is highly sensitive to its surface pretreatment. At 0.25 V overpotential, the catalysts show a mass activity of up to 0.6 A mg−1 and a turnover frequency of 0.65 s−1, one order of magnitude higher than the current state-of-the-art.
Introduction
Renewable sources of energy, such as wind or solar, are inherently intermittent. As they contribute an increasing amount to overall energy usage, more effective and sustainable methods are required to store the energy harvested. For instance, renewable energy could be used to split water electrochemically or photoelectrochemically to form hydrogen, as an energy carrier.1–5 Nonetheless, the efficiency of water splitting devices is largely defined by the sluggish kinetics of the oxygen evolution reaction (OER). As such, a judicious choice of the electrocatalyst material is imperative. The catalyst needs to be active, stable, and minimize the use of any scarce elements, so that it is scalable. Indeed, no water splitting device will make an impact to the global energy landscape, unless it can be scaled to the Terawatt (TW) level.6,7 This is a set of stringent requirements for the catalyst material. Indeed, there is a general call for more model studies of the catalysis of oxygen evolution, with the aim of improving the electrocatalysis of this reaction.2,8–13Of the different water splitting technologies, proton exchange membrane (PEM) electrolysers are arguably the most amenable towards small-scale delocalized storage of renewable electricity.4 Whereas traditional electrolysers operate in base, proton exchange membrane (PEM) electrolysers operate in acid. They hold some distinct advantages over traditional alkaline electrolysers, namely:14 (a) high efficiency at high current density, (b) the ability to manage fluctuating power inputs, (c) a solid electrolyte, and (d) a fast start up time. PEM electrolysers typically employ anode catalysts based on oxides of Ir, which are not only expensive, but also extremely scarce.6,15,16 Debe et al. showed that by employing an OER catalyst consisting of nanostructured whiskers of PtIrOx, they could achieve unprecedented low precious metal loadings of 0.3 mg cm−2 at the anode.17,18 We estimate that scaling up this device to a TW level of hydrogen storage capacity would require half a year of the annual global production of Pt and 10 years' Ir production (as described in the ESI†). Clearly, despite significant recent advances, current PEM electrolysis technology is not scalable to the TW level.1 One solution to this problem could be to replace the precious metals with more abundant elements. However, the harsh acidic and oxidizing conditions at the anode render most catalysts inactive or unstable, except for oxides of Ir or Ru,19 the latter also being subject to severe supply limitations.6
Consequently, a prerequisite for TW-scale PEM electrolyser technology is the development of a catalyst that has an appreciably higher mass activity (in A g−1 precious metal) than the current state-of-the-art. It turns out that RuO2 is more active than IrO2,11,20–22 but it is somewhat unstable towards dissolution towards RuO4.23–26 According to the theoretical “volcano” model developed by Rossmeisl, Nørskov and coworkers, RuO2 is the most active pure metal oxide catalyst for OER, because it exhibits the closest to optimal binding to the reaction intermediates.11 Further improvements can be achieved by combining ruthenium with nickel and cobalt;27–31 however, these bimetallic materials are highly unstable under acidic conditions.
Most studies of the OER on RuO2, simply report the catalytic activity, but neglect corrosion; when the corrosion rate has been monitored, it has been significant, at around 10–40% of the total anodic current.13,24,26,32–35 Nonetheless, it turns out that RuO2 can be stabilized, both by mixing with other oxides such as Ir or Ti,23,36–39 or by providing it with appropriate oxidation treatment.34,40,41 Oxides grown anodically under ambient conditions from metallic precursors tend to be highly active, but also unstable.34,40 Moreover, according to a recent report by Strasser and coworkers, nanoparticles of 4–6 nm in diameter, with anodically-grown surface oxides, will show much lower stability than extended surfaces with the same pre-treatment.24,41 On the other hand, should extended surfaces of RuO2 be formed under thermal oxidation conditions, by pre-treating in O2 at temperatures above 350 °C, they will show much higher stability than the anodic oxides, but somewhat lower activity.34,40,41 Thus far, no studies have provided a comprehensive examination of the activity and stability of nanoparticulate RuO2 under well-defined conditions. As highlighted by Mayrhofer and coworkers in a recent review,8 systematic studies on the effect of particle size for the oxygen evolution reaction are missing, unlike the oxygen reduction reaction. Although size effects are reported on chemically synthesized RuO2 particles greater than 15 nm in diameter,30 it is of greater technological relevance to examine smaller particles, where the surface area is maximised.42,43 Moreover, size effects are typically likely to be more pronounced for particles smaller than 10 nm in diameter.43,44
Herein, we focus on the evaluation of oxygen evolution activity and corrosion of well-defined, mass-selected Ru nanoparticles, as a function of size. In particular, we aim to establish the extent to which the mass activity of nanoparticulate RuO2 can be maximized and at the same time monitor the catalyst stability. We adapt a methodology previously used in our laboratory to investigate the oxygen reduction reaction.43,45,46 The mass selected electrocatalysts are prepared in a magnetron sputter source, using an ultra-high-vacuum (UHV) compatible technique.47,48 This allows a high degree of control over critical parameters such as particle size, electrode coverage and density,49 and it avoids the inherent artefacts introduced by chemical synthesis from precursors and surfactants.50
Results and discussions
The catalysts were formed by vacuum deposition of mass-selected nanoparticles from a ruthenium target,51 deposited directly onto glassy carbon or Au electrodes. The mass ranged from 0.035 × 106 u to 2.9 × 106 u, which would correspond to a range in size of 2–9 nm, assuming perfectly spherical particles. We tested both (a) as-deposited particles (Ru NPs) which were not subjected to any further treatment before exposure to the electrochemical environment and (b) thermally oxidized particles (RuO2 NPs), which, prior to electrochemical measurements, were treated in a tube furnace at 400 °C under 1 bar oxygen for 1 minute. The loading was determined directly from the deposition current. The structure and composition were tested using X-ray Photoelectron Spectroscopy (XPS), Glancing Angle – X-Ray Diffraction (GA-XRD), High Angle Annular Dark Field – Scanning Transmission Electron Microscopy (HAADF-STEM) and Scanning Electron Microscopy (SEM). The electrocatalysts were tested in a Rotating Ring Disk Electrode set-up. Furthermore, Electrochemical Scanning Tunneling Microscopy (EC-STM) was used to directly observe the catalyst dissolution under reaction conditions.52 Inductively Coupled Plasma – Mass Spectrometry (ICP-MS) was used to analyze the electrolyte post-OER.The catalyst composition and crystallographic structure were probed using XPS and GA-XRD. The as-deposited particles (Fig. 1a) exhibit two pairs of doublets for the Ru 3d core level, with a separation of ∼1 eV; this indicates the presence of both metallic and oxidized Ru in the near-surface region. On other hand, the thermally oxidized particles (Fig. 1b) could only be fitted to a single doublet, confirming that a RuO2 layer of at least 1.5 nm has formed.
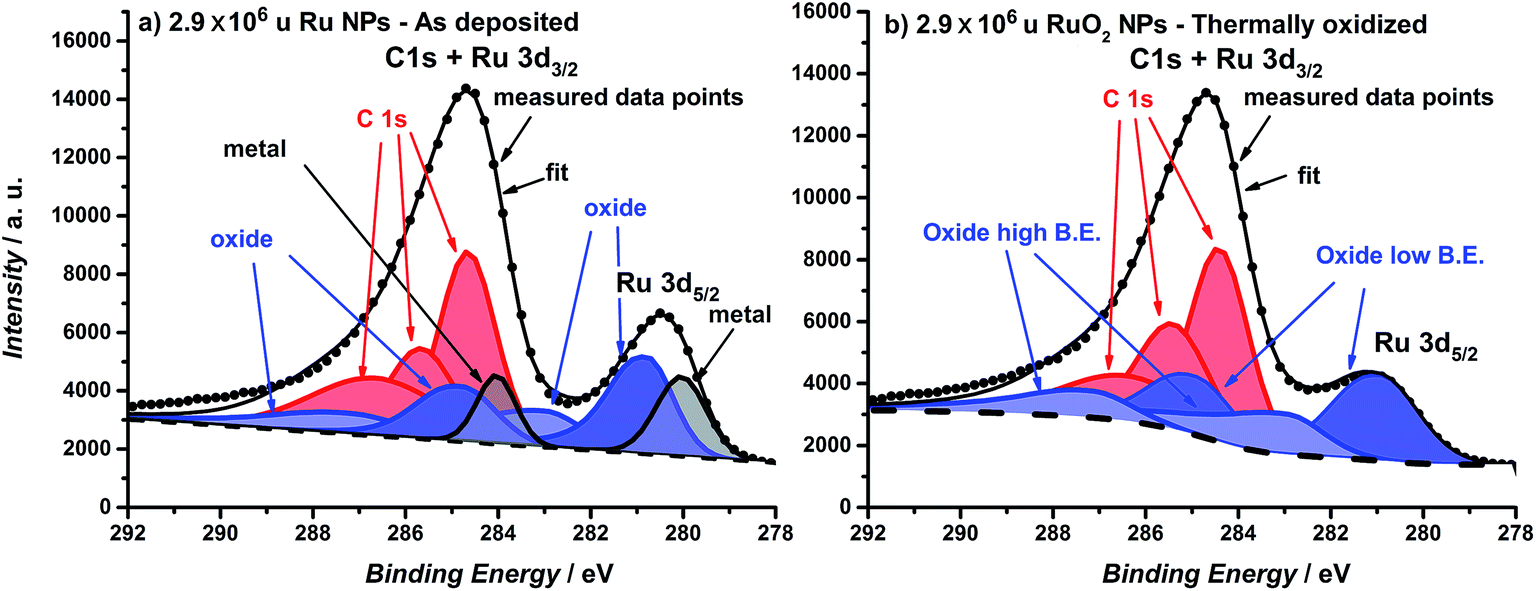 | ||
| Fig. 1 XPS spectra of the Ru 3d core level region of (a) as-deposited 2.9 × 106 u Ru nanoparticles and (b) thermally oxidized 2.9 × 106 u RuO2 nanoparticles. Ruthenium oxide is fitted with two species and carbon is fitted with three species. For further details about the fitting see the ESI.† | ||
GA-XRD measurements confirm the full oxidation of the particles (Fig. 2). The most intense peak from the reference pattern for metallic ruthenium can be identified in the as-deposited ruthenium catalyst. However, the peaks of the thermally oxidized ruthenium correspond closely to the RuO2 rutile phase. The remaining peaks correspond to the Au(111) substrate.
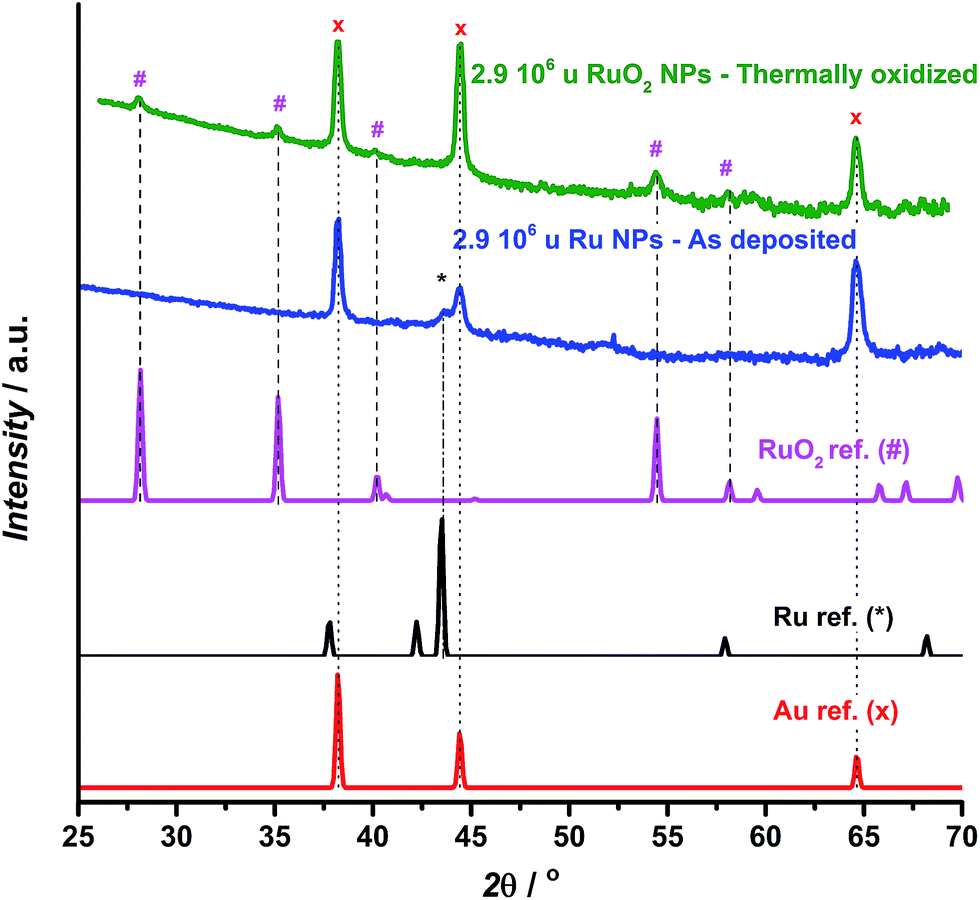 | ||
| Fig. 2 GA-XRD for as deposited (blue) and thermally oxidized (green) 2.9 × 106 u ruthenium nanoparticles deposited on a Au(111) single crystalline substrate, together with reference patterns for Au,53 Ru54 and RuO2.55 | ||
The projected particle area distribution for four different thermally oxidized samples, together with the corresponding HAADF-STEM images, are shown in Fig. 3a–d. Smaller particles show a more regular, spherical shape, whereas the larger particles are rough and form tetrahedrons. These observations correspond well with earlier studies from our laboratory on metallic Ru.49–51
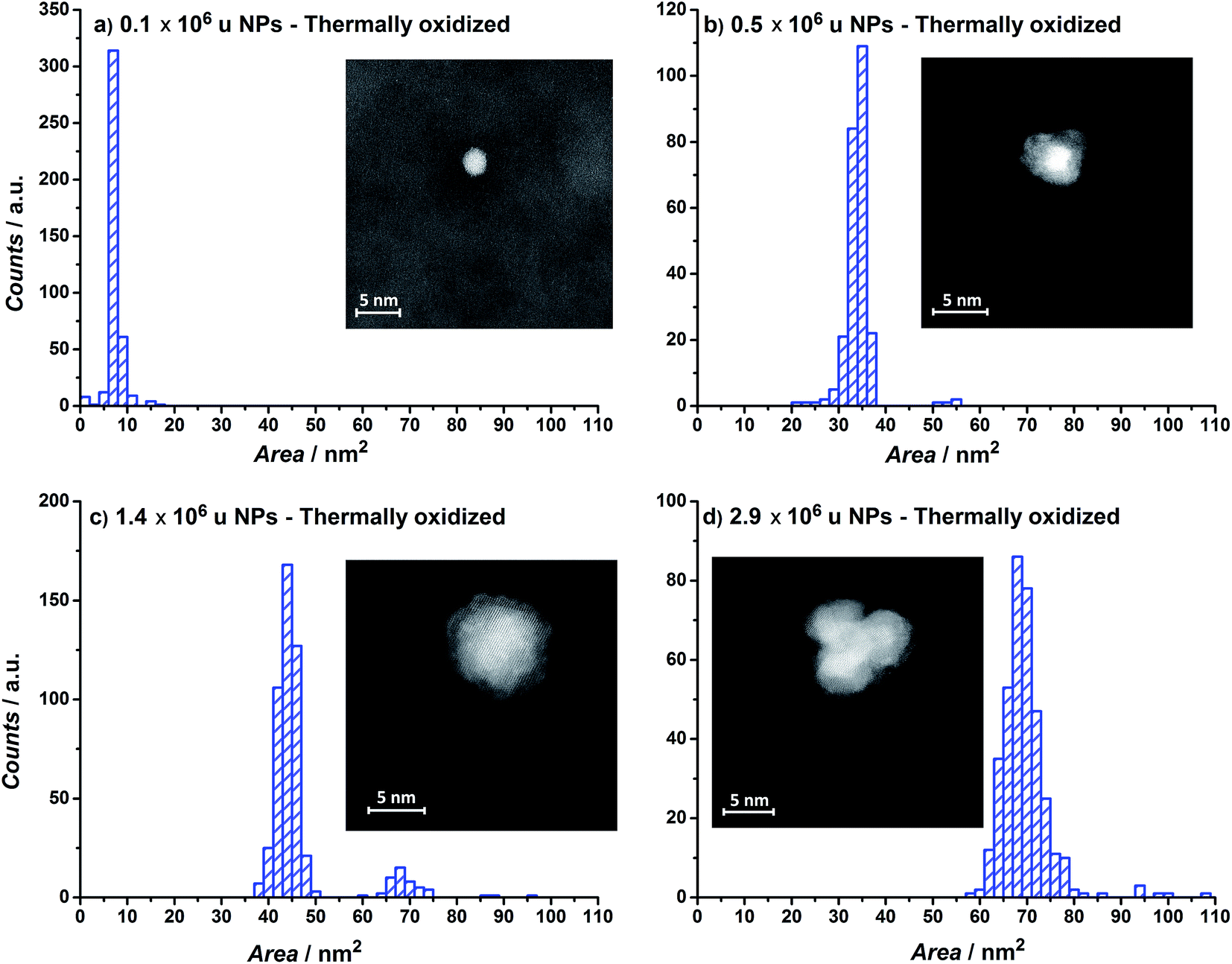 | ||
| Fig. 3 Nanoparticle area distributions and HAADF-Scanning Transmission Electron Microscopy images of thermally annealed RuO2/Si3N4 samples for particle mass of (a) 0.1 × 106, (b) 0.5 × 106, (c) 1.4 × 106 and (d) 2.9 × 106 u. | ||
The OER activity of glassy carbon supported thermally oxidized RuO2 nanoparticles was measured voltammetrically in N2-saturated 0.05 M H2SO4. The OER mass activity of the thermally oxidized RuO2 particles is plotted in Fig. 4a as a function of particle mass. There is some scatter in the data, which we attribute to uncertainties in the total mass deposited. This is despite significant endeavours to minimize the error in the measurement, by carefully choosing deposition parameters and monitoring the coverage of particles using XPS, as described in the ESI.† Incidentally, we obtained a similar catalytic activity on a polycrystalline gold electrode, indicating no influence from the substrate. We can tentatively identify a maximum in activity for the particles with a diameter of 3–5 nm. This could suggest the terrace is the active site for oxygen evolution, analogous to similar reports for the oxygen reduction reaction.43,44,56 Smaller particles will have a decreased number of terraces sites, and hence will exhibit a lower specific activity.
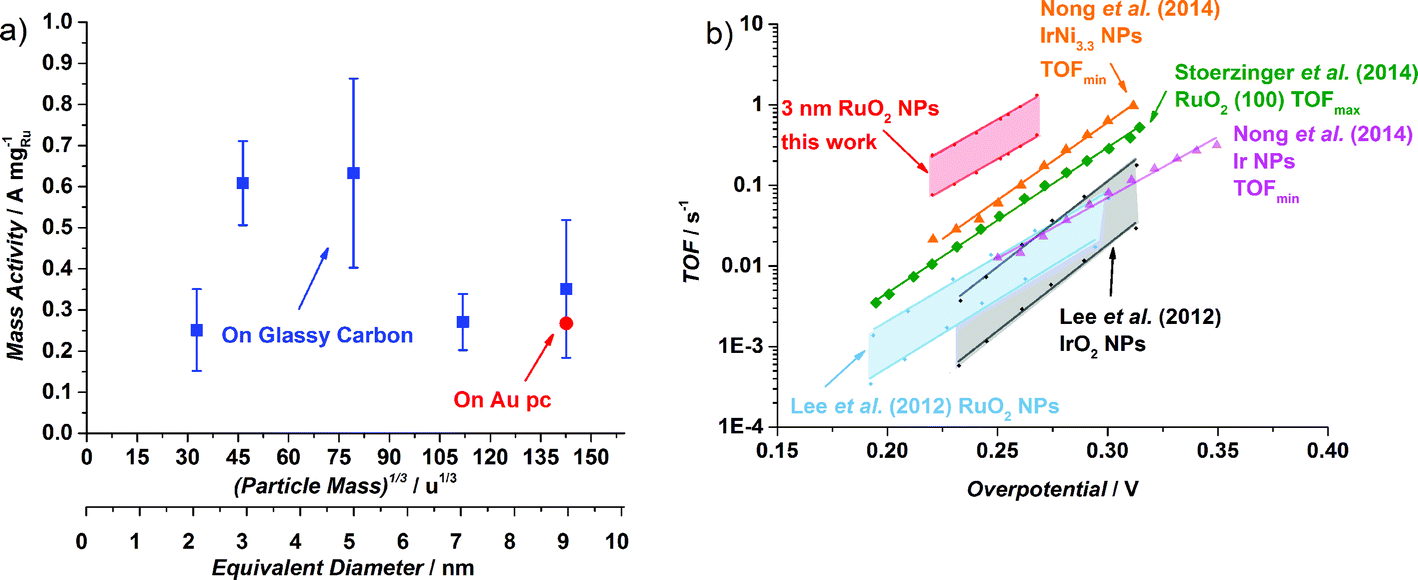 | ||
| Fig. 4 (a) OER mass activities at 1.48 V (vs. RHE) of different thermal oxidized RuO2 particle masses (0.035 × 106 u, 0.1 × 106 u, 0.5 × 106 u, 1.4 × 106 u and 2.9 × 106 u) on a Glassy Carbon (GC) disk, from the first ohmic and capacitance corrected CV. The Ru mass was evaluated from the deposition current. The error bars are based on four independent measurements. The activity for 2.9 × 106 u nanoparticles on a Au polycrystalline disk is also shown. The particle size is shown as both the (particle mass)1/3 and the equivalent diameter that the particles would be, should they be perfectly spherical. (b) Turnover frequency comparison of different catalysts in acid. When possible, both minimum and maximum TOF were estimated. Data adapted from this work for thermally oxidized 0.1 × 106 u NPs; adapted from ref. 57 for RuO2 and IrO2 NPs; from ref. 22 for RuO2 (100); and from ref. 59 for Ir NPs and Ir Ni3.3 NPs. | ||
It is challenging to assess the specific activity of the catalyst as a function of particle size, as we do not have an accurate measure of the surface area for all particle sizes, in particular the larger nanoparticles, which have a non-spherical morphology. Nevertheless, since the smaller particles do in fact exhibit a spherical morphology, we can estimate the surface area and hence the specific activity. It turns out that the specific activity of the 0.1 × 106 u RuO2 nanoparticles at 1.48 V (vs. RHE) is 0.32 mA cmRu−2 which is over an order of magnitude more active than both films and nanoparticles reported in the literature.22,57 (For further details see the ESI†). Moreover, based on the specific activity and mass activity, we can also estimate the turnover frequency (TOF), a fundamental parameter which represents the number of oxygen molecules produced per second. We can estimate the lower bound, i.e. TOFmin, by assuming that each Ru atom is active for the OER. On the other hand, we can estimate the upper bound, TOFmax, by assuming only the surface atoms are active (in reality, we view this TOFmax as a conservative estimate; the surface will be covered with a range of different catalytic sites, whereas the activity will be dominated by the most active). In Fig. 4b we show the estimated TOF of the 0.1 × 106 u RuO2 nanoparticles from this work, in comparison with the most active catalysts in the literature (we have only included catalysts with a well-defined mass or surface area). For further details on how the TOF was estimated, see the ESI.† There is a factor of three difference between TOFmin and TOFmax; although this is significant, it is lower than that reported for other similar systems.58 The figure demonstrates that the TOF frequency of the particles reported herein is the highest in the literature for the OER under acidic conditions. We speculate that this might be related to the preparation technique, which allowed us to obtain well-defined and monodisperse nanoparticles, without the inherent impurities that could be introduced from a chemical synthesis method.
Although it is a prerequisite for efficient catalysts to have high activity, it is also essential that the catalyst stability is tested, particularly due to the corrosive conditions under which OER catalysts operate.26,60 The cyclic voltammogram of the largest nanoparticles, 2.9 × 106 u, is plotted in Fig. 5a. The as-deposited Ru particles possess a higher activity than the thermally oxidized RuO2 particles, in agreement with earlier reports on extended surfaces.61 Even so, the as-deposited nanoparticles corrode immediately to form RuO4. According to ring current measurements,32,35 the anodic dissolution on our Ru nanoparticles constituted 15% of the total current, a similar degree of stability to commercial Ru/C catalysts24 or polycrystalline Ru.13 On the other hand, the thermally oxidized RuO2 nanoparticles showed a negligible ring current, suggesting that the activity should be solely due to the OER. This notion was confirmed by both ICP-MS and the direct measurement of oxygen using gas chromatography, as described in the ESI.†
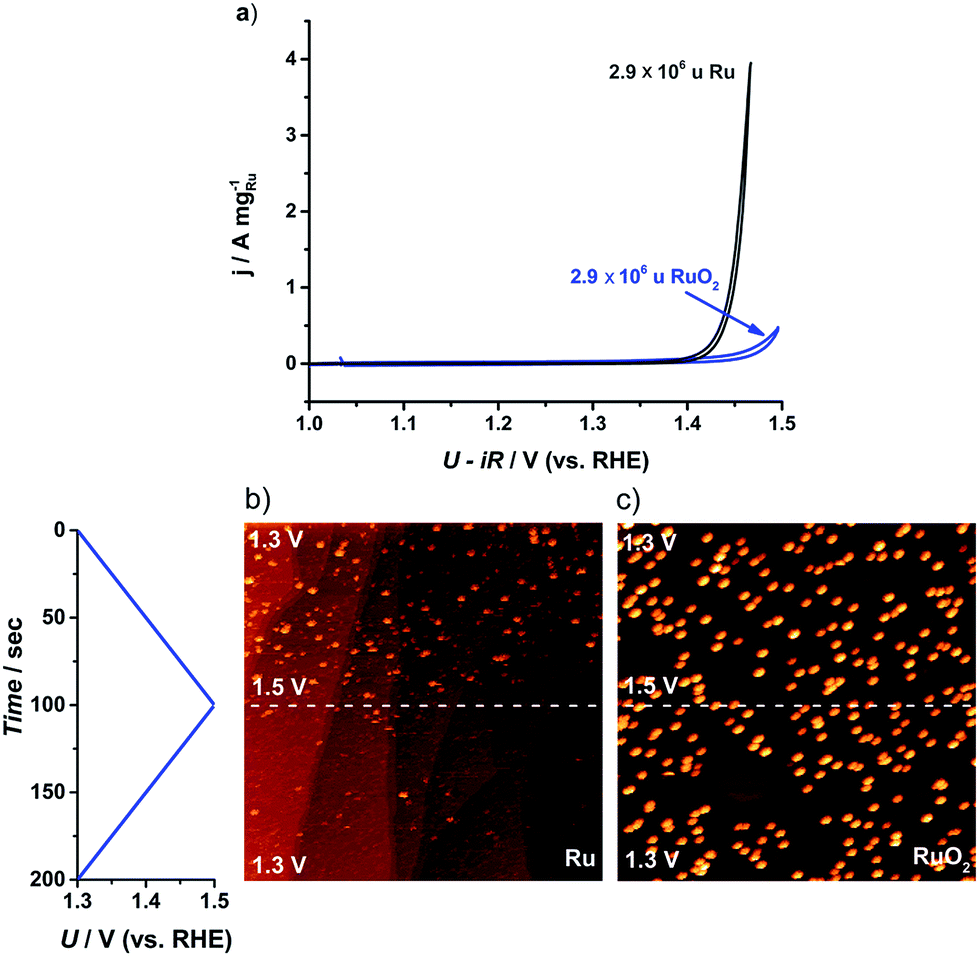 | ||
| Fig. 5 (a) Cyclic voltammogram (CV) for the first cycle of the as-deposited Ru and thermally oxidized RuO2 2.9 × 106 u nanoparticles on GC. CV recorded in N2 saturated 0.05 M H2SO4 between 1.0 and 1.5 V (vs. RHE) at 20 mV s−1 and 1600 rpm. (b) and (c) Potentiodynamic STM images of Ru dissolution in Ar-saturated 0.05 M H2SO4 solution. The potential was scanned between 1.3 and 1.5 V (vs. RHE) at 2 mV s−1: (b) 0.001 monolayer of as-deposited 0.1 × 106 u Ru nanoparticles, (520 nm),2UB = −299 mV, IT = 1 nA; (c) 0.001 monolayer of thermally treated 0.1 × 106 u RuO2 nanoparticles, (516 nm),2UB = −245 mV, IT = 1 nA. | ||
In order to directly observe the catalyst dissolution, we employed electrochemical scanning tunnelling microscopy (EC-STM). The STM images, shown in Fig. 5b and c, of Ru dissolution were taken in Ar-saturated 0.05 M H2SO4. The potential was scanned from 1.3 V to 1.5 V vs. RHE. The as-deposited and thermally oxidized nanoparticles show distinct behaviour from each other, under OER conditions. The as-deposited Ru (Fig. 5b) disappeared from view, as the potential was raised, due to dissolution, while RuO2 (Fig. 5c) exhibited no evidence of corrosion in the same potential range. These microscopic data correlate closely with the data from RRDE experiments and ICP-MS, described in the ESI.†
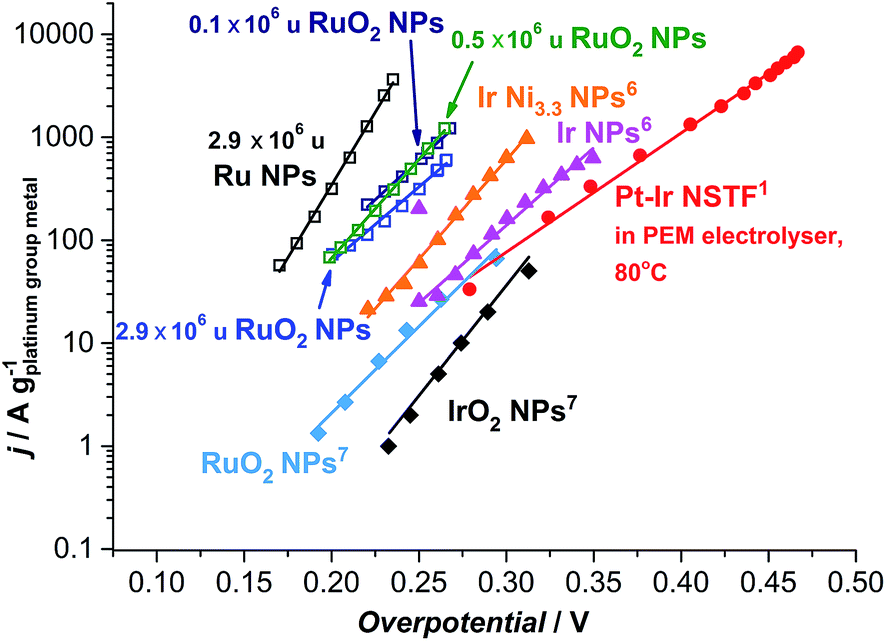 | ||
| Fig. 6 State-of-the-art for the oxygen evolution reaction in acidic media. Data adapted from this work for as-deposited and thermally oxidized 2.9 × 106 u nanoparticles (NPs) and thermally oxidized 0.1 and 0.5 × 106 u NPs; adapted from ref. 57 for RuO2 and IrO2 NPs; from ref. 59 for Ir NPs and Ir Ni3.3 NPs; and ref. 17 for Pt–Ir nanostructured thin films (NSTF) at 80 °C. The solid lines serve to guide the eye. | ||
Most strikingly, the particles tested here show a mass activity of at least one order of magnitude higher than the state-of-the-art for precious metal based OER catalysts in acid, as shown in Fig. 6.17,57 The thermally oxidized 0.5 and 0.1 × 106 u RuO2 particles exhibit a 45-fold increase, relative to RuO2 nanoparticles prepared by a chemical route,57 a 30-fold improvement compared to the PtIrOx in an electrolyser at 80 °C,17 and a 9-fold enhancement relative to Ir Ni3.3 core–shell nanoparticles.59 Assuming that the activity enhancement persists to higher overpotentials, we demonstrate that the precious metal oxide loading in a PEM electrolyser and in other technologies using oxygen evolution could be decreased by an order of magnitude. Our data provides an impetus for synthetic chemists to produce such active catalysts, using a more scalable method.
Conclusion
In summary, we have investigated the activity and stability of as-deposited and thermally oxidized Ru nanoparticles. By using mass selected nanoparticles, our work provides a model investigation of the OER on RuO2 and the effect of particle size. The particles reported here exhibit a one order of magnitude improvement in activity relative to the state-of-the-art and a tentative maximum at around 3–5 nm. An appropriate oxidation treatment can provide moderate stability to RuO2, and only slightly compromise on its high activity. Moreover, the well-defined shape and small size of the nanoparticles reported herein allow us to make an accurate estimate of the turnover frequency; it turns out it is appreciably higher than previous reports from the literature for oxygen evolution on any catalyst in acid. Should further improvements be made to the stability of RuO2 nanoparticles (for instance through the addition of Ir or Ti23,36–39), PEM electrolysis, as well as other technologies which are limited by oxygen evolution, could eventually become scalable to the TW level.Author contributions
E.A.P. performed the measurements of the electrocatalytic activity and stability, the GA-XRD experiments, and wrote the first draft and designed the figures; R.F. co-performed the electrochemical measurements and GA-XRD; F.M. prepared the nanoparticles, performed XPS and SEM; D.D performed STEM; C.S. performed EC-STM; M.M performed the gas chromatography experiments; T.W.H. supervised the STEM; S.H. supervised the EC-STM experiments; I.E.L.S supervised the electrochemical measurements and co-wrote the first draft; I.C. supervised the vacuum experiments and conceived the idea. All authors discussed and analysed the data and commented on the manuscript.Acknowledgements
The authors gratefully acknowledge financial support from the Danish Ministry of Science's UNIK initiative, Catalysis for Sustainable Energy and the Danish Council for Strategic Research’s project MEDLYS (10-093906). The Center for Individual Nanoparticle Functionality is supported by the Danish National Research Foundation (DNRF54).References
- E. Fabbri, A. Habereder, K. Waltar, R. Kotz and T. Schmidt, Catal. Sci. Technol., 2014 10.1039/C4CY00669K , accepted.
- H. Dau, C. Limberg, T. Reier, M. Risch, S. Roggan and P. Strasser, ChemCatChem, 2010, 2, 724–761 CrossRef CAS.
- M. G. Walter, E. L. Warren, J. R. McKone, S. W. Boettcher, Q. Mi, E. a. Santori and N. S. Lewis, Chem. Rev., 2010, 110, 6446–6473 CrossRef CAS PubMed.
- S. Kotrel and S. Bräuninger, in Handbook of Heterogeneous Catalysis, ed. G. Ert, Wiley-CPH, 2008, pp. 1936–1958 Search PubMed.
- K. Maeda and K. Domen, J. Phys. Chem. Lett., 2010, 1, 2655–2661 CrossRef CAS.
- P. C. K. Vesborg and T. F. Jaramillo, RSC Adv., 2012, 2, 7933–7947 RSC.
- R. E. Smalley, Mater. Matters, 2004, 30, 412–417 Search PubMed.
- I. Katsounaros, S. Cherevko, A. R. Zeradjanin and K. J. J. Mayrhofer, Angew. Chem., Int. Ed., 2013, 52, 2–22 Search PubMed.
- O. Diaz-Morales, F. Calle-Vallejo, C. de Munck and M. T. M. Koper, Chem. Sci., 2013, 4, 2334–2343 RSC.
- J. Suntivich, K. J. May, H. A. Gasteiger, J. B. Goodenough and Y. Shao-Horn, Science, 2011, 334, 1383–1385 CrossRef CAS PubMed.
- I. C. Man, H.-Y. Su, F. Calle-Vallejo, H. A. Hansen, J. I. Martínez, N. G. Inoglu, J. Kitchin, T. F. Jaramillo, J. K. Nørskov and J. Rossmeisl, ChemCatChem, 2011, 3, 1159–1165 CrossRef CAS.
- J. Rossmeisl, A. Logadottir and J. K. Nørskov, Chem. Phys., 2005, 319, 178–184 CrossRef CAS PubMed.
- N. Danilovic, R. Subbaraman, K.-C. Chang, S. H. Chang, Y. J. Kang, J. D. Snyder, A. P. Paulikas, D. Strmcnik, Y.-T. Kim, D. J. Myers, V. R. Stamenkovic and N. M. Markovic, J. Phys. Chem. Lett., 2014, 5, 2474–2478 CrossRef CAS.
- K. E. Ayers, E. B. Anderson, C. Capuano, B. Carter, L. Dalton, G. Hanlon, J. Manco and M. Niedzwiecki, ECS Trans., 2010, 33, 3–15 CAS.
- A. Marshall, B. Børresen, G. Hagen, M. Tsypkin and R. Tunold, Energy, 2007, 32, 431–436 CrossRef CAS PubMed.
- a. S. Aricò, S. Siracusano, N. Briguglio, V. Baglio, a. Blasi and V. Antonucci, J. Appl. Electrochem., 2012, 43, 107–118 CrossRef PubMed.
- M. K. Debe, S. M. Hendricks, G. D. Vernstrom, M. Meyers, M. Brostrom, M. Stephens, Q. Chan, J. Willey, M. Hamden, C. K. Mittelsteadt, C. B. Capuano, K. E. Ayers and E. B. Anderson, J. Electrochem. Soc., 2012, 159, K165–K176 CrossRef CAS PubMed.
- S. Grigoriev, V. Porembsky and V. Fateev, Int. J. Hydrogen Energy, 2006, 31, 171–175 CrossRef CAS PubMed.
- M. Pourbaix, Atlas of Electrochemical Equilibria in Aqueous Solutions, Houston, TX, 1974 Search PubMed.
- J. Rossmeisl, Z.-W. Qu, H. Zhu, G.-J. Kroes and J. K. Nørskov, J. Electroanal. Chem., 2007, 607, 83–89 CrossRef CAS PubMed.
- S. Trasatti, Electrochim. Acta, 1984, 29, 1503–1512 CrossRef CAS.
- K. A. Stoerzinger, L. Qiao, M. D. Biegalski and Y. Shao-horn, J. Phys. Chem. Lett., 2014, 5, 1636–1641 CrossRef CAS.
- R. Kötz and S. Stucki, Electrochim. Acta, 1986, 31, 1311–1316 CrossRef.
- T. Reier, M. Oezaslan and P. Strasser, ACS Catal., 2012, 2, 1765–1772 CrossRef CAS.
- K. E. Ayers, L. T. Dalton and E. B. Anderson, ECS Trans., 2012, 41, 27–38 CAS.
- S. Cherevko, A. R. Zeradjanin, A. A. Topalov, N. Kulyk and J. J. Mayrhofer, ChemCatChem, 2014, 6, 2219–2223 CAS.
- N. B. Halck, V. Petrykin, P. Krtil and J. Rossmeisl, Phys. Chem. Chem. Phys., 2014, 16, 13682–13688 RSC.
- R. Forgie, G. Bugosh, K. C. Neyerlin, Z. Liu and P. Strasser, Electrochem. Solid-State Lett., 2010, 13, B36–B39 CrossRef CAS PubMed.
- K. C. Neyerlin, G. Bugosh, R. Forgie, Z. Liu and P. Strasser, J. Electrochem. Soc., 2009, 156, B363–B369 CrossRef CAS PubMed.
- K. Macounová, J. Jirkovský, M. V. Makarova, J. Franc and P. Krtil, J. Solid State Electrochem., 2009, 13, 959–965 CrossRef.
- V. Petrykin, K. Macounová, M. Okube, S. Mukerjee and P. Krtil, Catal. Today, 2013, 202, 63–69 CrossRef CAS PubMed.
- M. Vukovic, J. Chem. Soc., Faraday Trans., 1990, 86, 3743–3746 RSC.
- S. H. Chang, N. Danilovic, K.-C. Chang, R. Subbaraman, A. P. Paulikas, D. D. Fong, M. J. Highland, P. M. Baldo, V. R. Stamenkovic, J. W. Freeland, J. A. Eastman and N. M. Markovic, Nat. Commun., 2014, 5, 4191 Search PubMed.
- C. Iwakura, K. Hirao and H. Tamura, Electrochim. Acta, 1977, 22, 335–340 CrossRef CAS.
- R. Kötz, S. Stucki, D. Scherson and D. M. Kolb, J. Electroanal. Chem., 1984, 172, 211–219 CrossRef.
- H. B. Beer, J. Electrochem. Soc., 1980, 127, 303C CrossRef CAS PubMed.
- S. Trasatti, Electrochim. Acta, 2000, 45, 2377–2385 CrossRef CAS.
- C. Comninellis and G. P. Vercesi, J. Appl. Electrochem., 1991, 21, 136–142 CrossRef CAS.
- C. Comninellis and G. P. Vercesi, J. Appl. Electrochem., 1991, 21, 335–345 CrossRef CAS.
- D. Galizzioli, F. Tantardini and S. Trasatti, J. Appl. Electrochem., 1974, 4, 57–67 CrossRef CAS.
- M. B. Vukmirovic, R. L. Sabatini and R. R. Adzic, Surf. Sci., 2004, 572, 269–276 CrossRef CAS PubMed.
- B. R. Cuenya, S.-H. Baeck, T. F. Jaramillo and E. W. McFarland, J. Am. Chem. Soc., 2003, 125, 12928–12934 CrossRef PubMed.
- F. J. Perez-Alonso, D. N. McCarthy, A. Nierhoff, P. Hernandez-Fernandez, C. Strebel, I. E. L. Stephens, J. H. Nielsen and I. Chorkendorff, Angew. Chem., Int. Ed., 2012, 51, 4641–4643 CrossRef CAS PubMed.
- G. a. Tritsaris, J. Greeley, J. Rossmeisl and J. K. Nørskov, Catal. Lett., 2011, 141, 909–913 CrossRef CAS.
- P. Hernandez-Fernandez, F. Masini, D. N. McCarthy, C. E. Strebel, D. Friebel, D. Deiana, P. Malacrida, A. Nierhoff, A. Bodin, A. M. Wise, J. H. Nielsen, T. W. Hansen, A. Nilsson, I. E. L. Stephens and I. Chorkendorff, Nat. Chem., 2014, 6, 732–738 CAS.
- F. Masini, P. Hernández-Fernández, D. Deiana, C. E. Strebel, D. N. McCarthy, A. Bodin, P. Malacrida, I. Stephens and I. Chorkendorff, Phys. Chem. Chem. Phys., 2014 10.1039/C4CP02144D , accepted.
- H. Haberland, M. Karrais, M. Mall and Y. Thurner, J. Vac. Sci. Technol., A, 1992, 10, 3266 CAS.
- B. Von Issendorff and R. E. Palmer, Rev. Sci. Instrum., 1999, 70, 4497–4501 CrossRef CAS PubMed.
- R. M. Nielsen, S. Murphy, C. Strebel, M. Johansson, I. Chorkendorff and J. H. Nielsen, J. Nanopart. Res., 2009, 12, 1249–1262 CrossRef.
- S. Ardizzone, M. Falciola and S. Trasatti, J. Electrochem. Soc., 1989, 136, 3–8 CrossRef PubMed.
- F. Masini, C. E. Strebel, D. N. McCarthy, A. U. F. Nierhoff, J. Kehres, E. M. Fiordaliso, J. H. Nielsen and I. Chorkendorff, J. Catal., 2013, 308, 282–290 CrossRef CAS PubMed.
- L. Tang, B. Han, K. Persson, C. Friesen and T. He, J. Am. Chem. Soc., 2010, 596–600 CrossRef PubMed.
- X. Yan, P. Lin, X. Qi and L. Yang, Int. J. Mater. Res., 2011, 102, 381–388 CrossRef CAS.
- E. O. Hall and J. Crangle, Acta Crystallogr., 1957, 10, 240–241 CrossRef CAS.
- C. E. Boman, Acta Chem. Scand., 1970, 24, 116–122 CrossRef CAS PubMed.
- J. Greeley, R. J. A. Hellman and J. K. Nørskov, Z. Phys. Chem., 2007, 221, 1209–1220 CrossRef CAS.
- Y. Lee, J. Suntivich, K. J. May, E. E. Perry and Y. Shao-horn, J. Phys. Chem. Lett., 2012, 3, 399–404 CrossRef CAS.
- M. W. Louie and A. T. Bell, J. Am. Chem. Soc., 2013, 135, 12329–12337 CrossRef CAS PubMed.
- H. N. Nong, L. Gan, E. Willinger, D. Teschner and P. Strasser, Chem. Sci., 2014, 5, 2955–2963 RSC.
- R. Frydendal, E. A. Paoli, B. P. Knudsen, B. Wickman, P. Malacrida and E. L. Ifan, ChemElectroChem, 2014 DOI:10.1002/celc.201402262R1 , accepted.
- S. Stucki and A. Menth, Ber. Bunsen-Ges., 1980, 84, 1008–1013 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc02685c |
| This journal is © The Royal Society of Chemistry 2015 |