DOI:
10.1039/C4SC02618G
(Edge Article)
Chem. Sci., 2015,
6, 342-348
Semiconductor-driven “turn-off” surface-enhanced Raman scattering spectroscopy: application in selective determination of chromium(VI) in water†
Received
27th August 2014
, Accepted 26th September 2014
First published on 29th September 2014
Abstract
Semiconductor materials have been successfully used as surface-enhanced Raman scattering (SERS)-active substrates, providing SERS technology with a high flexibility for application in a diverse range of fields. Here, we employ a dye-sensitized semiconductor system combined with semiconductor-enhanced Raman spectroscopy to detect metal ions, using an approach based on the “turn-off” SERS strategy that takes advantage of the intrinsic capacity of the semiconductor to catalyze the degradation of a Raman probe. Alizarin red S (ARS)-sensitized colloidal TiO2 nanoparticles (NPs) were selected as an example to show how semiconductor-enhanced Raman spectroscopy enables the determination of Cr(VI) in water. Firstly, we explored the SERS mechanism of ARS–TiO2 complexes and found that the strong electronic coupling between ARS and colloidal TiO2 NPs gives rise to the formation of a ligand-to-metal charge-transfer (LMCT) transition, providing a new electronic transition pathway for the Raman process. Secondly, colloidal TiO2 nanoparticles were used as active sites to induce the self-degradation of the Raman probe adsorbed on their surfaces in the presence of Cr(VI). Our data demonstrate the potential of ARS–TiO2 complexes as a SERS-active sensing platform for Cr(VI) in an aqueous solution. Remarkably, the method proposed in this contribution is relatively simple, without requiring complex pretreatment and complicated instruments, but provides high sensitivity and excellent selectivity in a high-throughput fashion. Finally, the ARS–TiO2 complexes are successfully applied to the detection of Cr(VI) in environmental samples. Thus, the present work provides a facile method for the detection of Cr(VI) in aqueous solutions and a viable application for semiconductor-enhanced Raman spectroscopy based on the chemical enhancement they contribute.
Introduction
In recent years, an increasing interest in the studies of surface-enhanced Raman scattering (SERS) on semiconducting materials (that is semiconductor-enhanced Raman spectroscopy) has emerged owing to its potential application in biological and photoelectronic analyses.1–6 Several semiconductor materials including TiO2, ZnO, graphene, Si, and Ge have been developed as SERS substrates.7–9 However, the application of these materials in SERS-based quantitative measurements is still matter of debate, because of their specific dependence on molecular electronic structure and relatively weak enhancement of SERS signals. Most of the improvement in semiconductor-enhanced Raman spectroscopy is mainly induced by a charge-transfer process, leading to an enhancement of approximately 102 to 104, because the surface plasmon resonance of semiconductor NPs typically lies in the infrared region, which does not usually coincide with the optical laser frequency.2–4,7–13 To date, studies have largely focused on the discovery and interpretation of the SERS phenomena with different semiconductor materials. This represents the most significant bottleneck in the application of such a technique for practical analysis and detection.
The advantage of semiconductor-enhanced Raman spectroscopy is the performance of the semiconductor, which possesses controllable photoelectric properties, good biocompatibility, and environmental stability. In order to exploit these advantages, metal–semiconductor composites were introduced into SERS-based assays with some ingenious designs.14–19 However, the complicated process needed to prepare such composites has strongly limited their application. In addition, semiconductor materials do not respond to the Raman enhancement in these systems.16–19 Considering the photocatalytic properties of semiconductors, quantitative analysis by semiconductor-enhanced Raman spectroscopy would be achieved through the detection of signal degradation of a labeled probe on a semiconductor. Here, we report that semiconductor-enhanced Raman spectroscopy can also be developed into a sensing platform for the detection of metal ions, even without the assistance of a noble metal. In particular, we present a novel “turn-off” SERS strategy and demonstrate its use in a SERS-based assay for the determination of Cr(VI) in water.
Cr has been extensively used in various industrial processes and has become one of the major environmental hazards.20 The toxicological and biological properties of Cr are entirely dependent on its electric charge.21 For instance, Cr(VI) is highly toxic; it generally exists as an oxyanion (CrO42−) in aqueous systems, and is known to be a strong carcinogen.22 In contrast, Cr(III) is relatively non-toxic and is regarded as an essential trace element associated with the metabolism of carbohydrates and lipids.23 Therefore, the reduction of Cr(VI) to Cr(III) is a key process for the detoxification of Cr(VI)-contaminated water and wastewater. In drinking water, the Maximum Contaminant Level (MCL) for Cr(VI) has been identified as 1 μM. However, because no efficient testing method is available for only Cr(VI), the estimated MCL by the World Health Organization (WHO) includes the total amount of Cr.24 Evidently, this definition is not conducive to encouraging the intake of Cr(III) from a daily diet and has pushed up the cost of industrial wastewater treatment. So far, several methods, including atomic spectrometric,25–27 luminescent,28,29 electrometric,30–33 colorimetric,34 and X-ray fluorometric techniques,35 have been developed for the selective determination of Cr(VI). Nevertheless, none of these techniques exhibited the desired sensitivity together with easy manipulation.
Herein, charge-transfer complexes, alizarin red S (ARS)-sensitized colloidal TiO2 NPs, with a facile synthetic route are used to demonstrate how semiconductor-enhanced Raman spectroscopy enables the determination of Cr(VI) in water. We explored the SERS mechanism of ARS–TiO2 complexes and found that the molecular polarizability tensor can be enhanced by a ligand-to-metal charge-transfer (LMCT) transition. Interestingly, the SERS intensities of the ARS–TiO2 complexes have been found to be sensitive to the Cr(VI) concentration due to co-catalysis, indicating their potential for use in the determination of Cr(VI). Several influencing factors such as response time, laser power, pH of the sensing system, and the loading amount of ARS on the colloidal TiO2 NPs were taken into account to optimize the determination conditions. Our experimental results revealed that the ARS–TiO2 complexes exhibit high sensitivity and selectivity toward Cr(VI). The practicality of this proposed method was further validated through the detection of Cr(VI) in real water samples. The method proposed here can be used for the determination of Cr(VI) in aqueous solutions for the accurate assessment of pollution levels. Thus, this work provides a clear proof of concept for extending the applications of semiconductor-enhanced Raman spectroscopy.
Experimental
Materials
Alizarin red S and titanium(IV) butoxide were acquired from Sigma-Aldrich Co. Ltd. and used without further purification. All other chemicals, obtained from Wako Co. Ltd, were analytical grade and employed without further purification. Ultrapure water (18 MΩ cm) was used throughout the study. The tap water and pond water were collected from the Gakuen district of Sanda and a pond near Kwansei Gakuin University, respectively. All the water samples were filtered through 0.2 μm membranes prior to use.
Preparation of colloidal TiO2 nanoparticles
The colloidal TiO2 NPs were synthesized according to a method described in previous reports.36,37 Briefly, a solution of titanium(IV) butoxide (5 mL) dissolved in 2-propanol (95 mL) was added dropwise (1 mL min−1) to an aqueous HNO3 solution (500 mL, pH 1.5) maintained at 1 °C. The solution was continuously stirred for 10–12 hours until a transparent colloid was formed.
SERS measurement
A stock solution of ARS (0.1 M) was prepared in water. ARS solutions with various concentrations were obtained by serial dilution of the stock solution with sodium acetate buffer solution (0.01 M, pH 3.0). The ARS solutions with different concentrations were mixed with the colloidal TiO2 NPs at the same volume and shaken thoroughly. For the detection of metal ions, 10 μL of each sample mixed with 10 μL of ARS–TiO2 was dripped into an aluminum pan (0219-0062, Perkin-Elmer), and the mixture was exposed to a laser beam for 30 s before each SERS measurement. The typical exposure time for each Raman/SERS measurement in this study was 30 s with two accumulations. The error bars represent standard deviations based on three independent measurements.
Instrument
The image of the sample was measured on a Tecnai G2 transmission electron microscope operating at 200 kV. The UV-vis spectra were recorded on a Shimadzu UV-3600 spectrophotometer. A RS-2100 Raman spectrophotometer (Photon Design, Inc.) equipped with a CCD (Princeton Instruments) was used. Radiation with a wavelength of 514.5 nm from an Ar ion laser (Spectra Physics) was employed for the Raman excitation, with a power of 5 mW at the sample. The Raman band of a silicon wafer at 520.7 cm−1 was used to calibrate the spectrometer.
Results and discussion
Synthesis and characterization
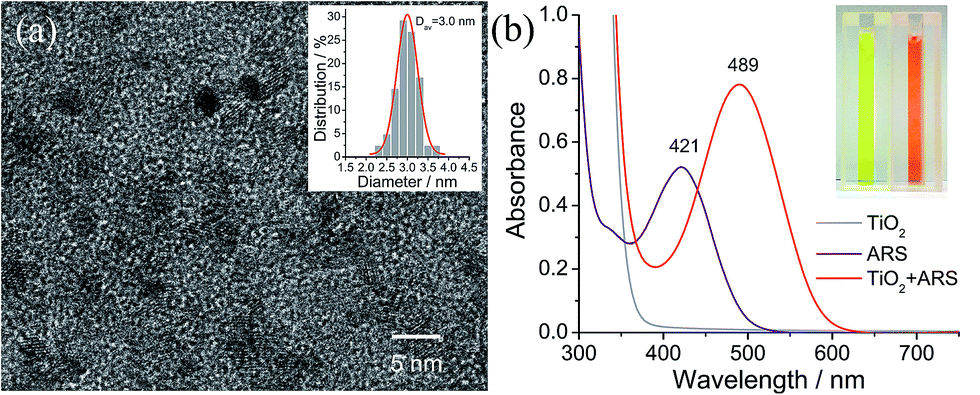
Colloidal TiO2 NPs with an average diameter of 3 nm were prepared by a low-temperature acid hydrolysis route, as described previously (see Fig. 1).36,37 The absorption spectra of the colloidal TiO2 NPs before and after modification with ARS are shown in Fig. 1b, together with that of ARS for the sake of comparison. In contrast to ARS, the ARS–TiO2 complexes exhibit a more intense absorption band in the longer wavelength region with a peak maximum centered at 489 nm. This absorption band has been assigned to the LMCT transition, which arises from the strong electronic coupling between ARS and the colloidal TiO2 NPs.38 Based on the Benesi–Hildebrand analysis for ARS–TiO2 complexes (Fig. S1†), the association constant (Kass) of the complex was determined to be 3.9 × 103 M−1, which indicates the relatively strong binding of ARS on the surface of the TiO2 NPs. An ARS molecule contains many functional groups; the FTIR data of the ARS–TiO2 complexes unambiguously shows that the mode of grafting is bidentate chelation, which involves two hydroxyl groups (Fig. S2†). These observations suggest that the ARS–TiO2 composite material can be used for the development of a semiconductor-supported SERS sensing platform due to the clear charge-transfer transition process.
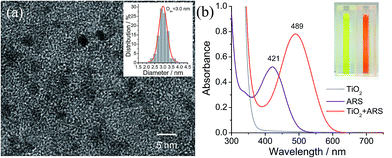 |
| | Fig. 1 (a) High-resolution transmission electron microscopy (TEM) image of the colloidal TiO2 nanoparticles. Inset is the size distribution of the colloidal TiO2 nanoparticles (Dav is the average particle size). (b) Optical absorption spectra of the colloidal TiO2 nanoparticles (30 mM), ARS (0.1 mM), and ARS–TiO2 complexes at pH 1.5. The inset shows the corresponding sample photographs. | |
Mechanism for SERS of the ARS–TiO2 system
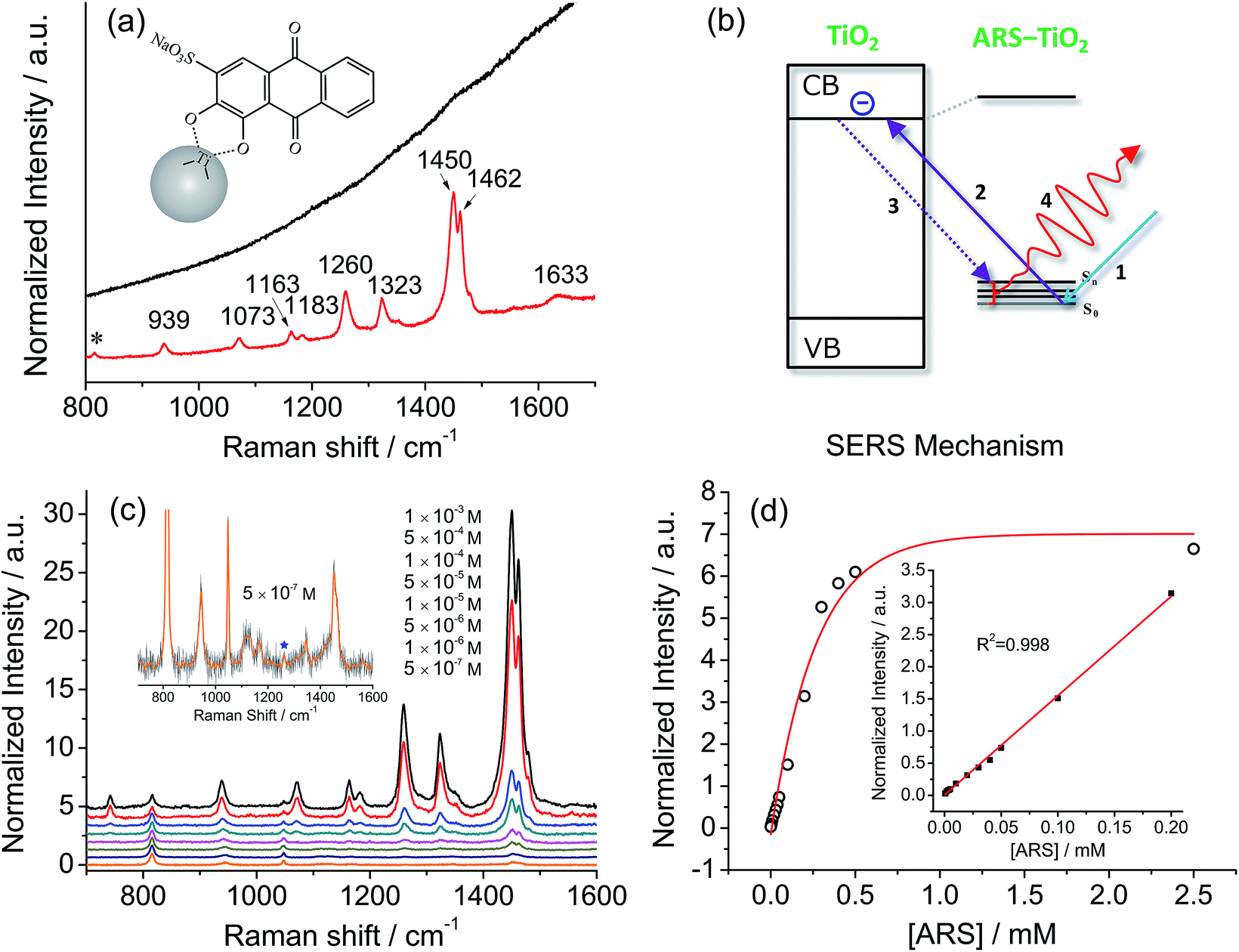
Fig. 2a compares a Raman spectrum of 0.1 M ARS in aqueous solution and a SERS spectrum of ARS–TiO2 complexes with 514.5 nm excitation. The vibrational mode assignments listed in Table S1† are based primarily on earlier IR and Raman studies of related alizarin dyes.39–42 The SERS spectrum is characterized by a significant enhancement in the 1200–1500 cm−1 region, where the bands are typically assigned to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C–O–R stretching modes. This observation confirms the presence of strong coupling between the electronic transitions and the CC/C–O–R bond stretching modes in ARS ligands, which is consistent with the conclusions obtained from the absorption and IR spectra of the ARS–TiO2 complexes. Such coupling has also been observed on other semiconductor NPs in colloidal suspensions such as CeO2, Fe2O3, ZrO2, etc.43 Furthermore, the concentration-dependent SERS experiments displayed in Fig. 2c clearly demonstrate that a concentration as low as 5 × 10−7 M ARS can be detected.44 The intensities of the Raman signals can be fitted with the BET model well and represent a saturation effect (Fig. 2d). In addition, a linear correlation was found between the intensity at 1260 cm−1 and the ARS concentration in the range of 5 × 10−7 to 2 × 10−4 M.
C and C–O–R stretching modes. This observation confirms the presence of strong coupling between the electronic transitions and the CC/C–O–R bond stretching modes in ARS ligands, which is consistent with the conclusions obtained from the absorption and IR spectra of the ARS–TiO2 complexes. Such coupling has also been observed on other semiconductor NPs in colloidal suspensions such as CeO2, Fe2O3, ZrO2, etc.43 Furthermore, the concentration-dependent SERS experiments displayed in Fig. 2c clearly demonstrate that a concentration as low as 5 × 10−7 M ARS can be detected.44 The intensities of the Raman signals can be fitted with the BET model well and represent a saturation effect (Fig. 2d). In addition, a linear correlation was found between the intensity at 1260 cm−1 and the ARS concentration in the range of 5 × 10−7 to 2 × 10−4 M.
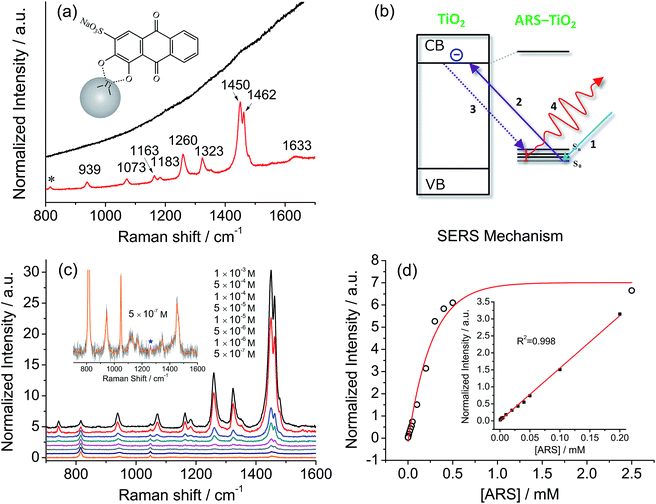 |
| | Fig. 2 (a) A Raman spectrum of 0.1 M ARS in aqueous solution (top) and a SERS spectrum of 10−3 M ARS adsorbed on colloidal TiO2 NPs (bottom). (b) Suggested mechanism for the photoexcitation charge-transfer process of ARS–TiO2 complexes with 514.5 nm excitation. (c) SERS spectra of ARS with variable concentrations adsorbed on colloidal TiO2 NPs. Inset: the magnified SERS spectrum of ARS–TiO2 at 5 × 10−7 M. (d) Normalized Raman intensity at 1260 cm−1versus the concentration of ARS. The inset shows the low concentration range. All the intensities of the bands are normalized to the intensity of the signal due to 2-propanol at 816 cm−1. | |
Of note is that the enhancement arises from the strong coupling interaction between the dye molecules and the colloidal TiO2 NPs and, more importantly, the formation of charge-transfer complexes opens up a new electronic transition pathway for the Raman process.45,46 In this case, the ground-state electrons of the ARS–TiO2 complexes are initially excited from the highest occupied molecular orbital (HOMO) level to the conduction band (CB) of the TiO2 NPs by the incident light (Fig. 2b). Then, the excited electrons immediately transfer back to the vibrational energy level of the ARS molecule and subsequently release a Raman photon with the ARS molecule at some vibrational state. The molecular polarizability tensor can be enhanced by such a charge-transfer process due to the vibronic coupling of the conduction band states of the semiconductor with the excited states of the probe molecule through a Herzberg–Teller coupling term.47 Therefore, unlike in resonance Raman spectroscopy, where the molecule itself should reach a resonant state with excitation by the incident light, the enhancement can be considered in this case as a SERS phenomenon, which arises from the chemical enhancement mechanism via the Herzberg–Teller contribution.2,45,48
Mechanism for responding to Cr(VI)
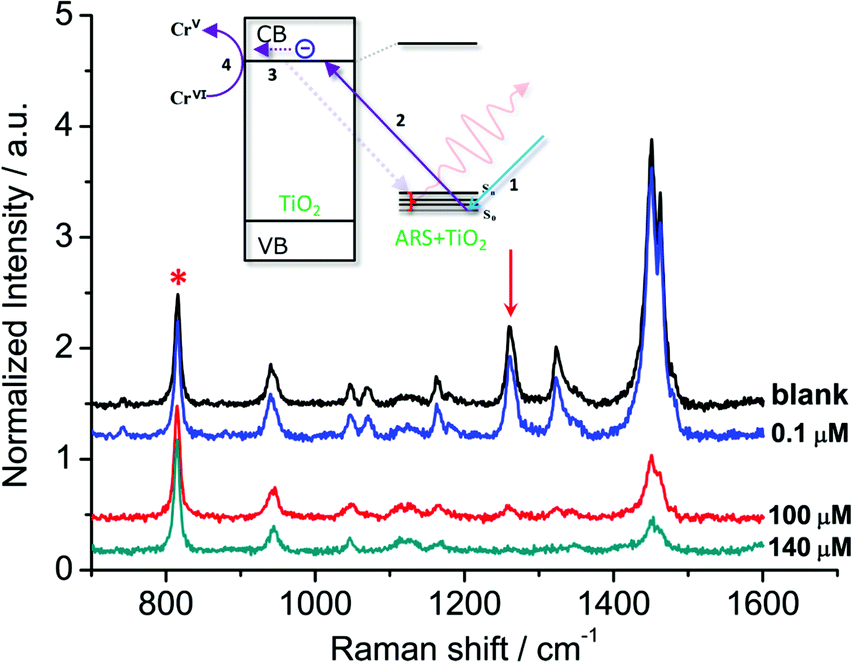
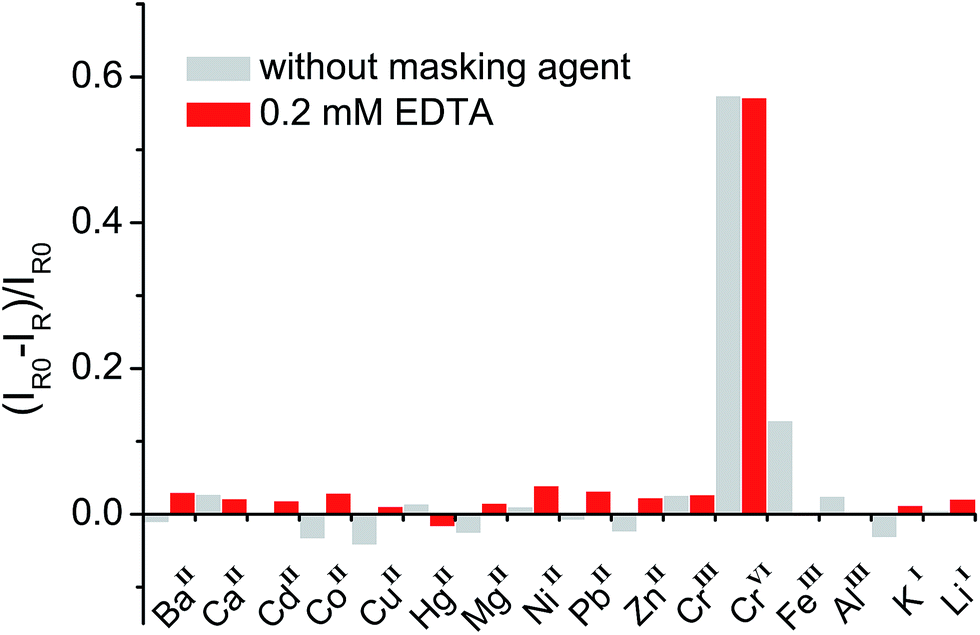
In general, organic ligands are susceptible to decomposition on bulk TiO2, owing to their adsorption through physisorption or weak chemisorption. Compared with bulk TiO2, colloidal TiO2 NPs possess abundant under-coordinated Ti defect sites, which provide plenty of coordination sites for ARS to bind via bidentate chelation. This chelation mode is favorable to the recombination between ARS+ and an electron, thereby stabilizing the ARS molecule. Consequently, ARS adsorbed on the surface of colloidal TiO2 NPs cannot be easily oxidized by visible light irradiation and preserves the SERS properties even after exposure to high laser power (Fig. S4†). However, the SERS intensities are decreased with the addition of Cr(VI), indicating the decomposition of ARS adsorbed on the colloidal TiO2 NP surfaces (Fig. 3 and S5†). As shown in Fig. 4 and S6,† the ARS–TiO2 complexes exhibit a remarkably high selectivity and lower interference in the determination of Cr(VI), particularly in the presence of Cr(III). This specificity originates from the favorable redox potential of the couple Cr(VI)/Cr(V) (+0.55 V) for the reduction promoted by those electrons trapped in inter-band-gap states, together with the strong interaction between Cr(VI) and Ti(IV) atoms with unfilled valence orbitals at the TiO2 surface. This reduction can result in the formation of Cr(V) and decomposition of ARS–TiO2 complexes (Fig. S5†), thus leading to a decrease in the SERS intensities. The redox potential of the Fe(III)/Fe(II) couple (+0.77) is close to that of the Cr(VI)/Cr(V) couple. However, the relatively weak interaction between Fe(III) and the positively charged colloidal TiO2 NPs only causes minor disturbance. To minimize the interference from Fe(III), 0.2 mM EDTA was added to the sodium acetate buffer as a masking agent. As expected, the interference from Fe(III) was found to be negligible in the presence of EDTA. Based on these results, it was inferred that the SERS intensities of ARS are sensitive to the Cr(VI) concentration, indicating the possibility of Cr(VI) detection using ARS-sensitized colloidal TiO2 NPs.
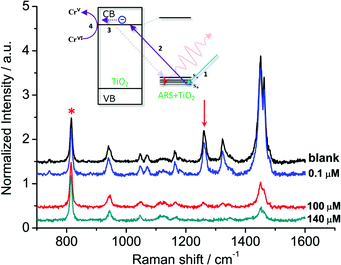 |
| | Fig. 3 SERS spectra obtained from the ARS–TiO2 (50 μM ARS) mixtures with the various Cr(VI) concentrations. The intensities of the bands are normalized to that of the signal from 2-propanol at 816 cm−1. Inset: suggested mechanism for the photoexcitation catalysis process of ARS–TiO2 complexes in the presence of Cr(VI). | |
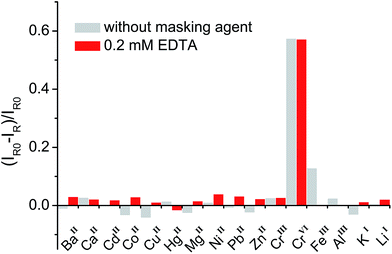 |
| | Fig. 4 Relative Raman intensity [(IR0 − IR)/IR0] of ARS–TiO2 complexes in the presence of metal ions with and without the addition of a masking reagent. The concentration of Cr(VI) was 20 μM and each of the other metal ions was 100 μM. IR and IR0 represent the Raman intensities of ARS–TiO2 at 1260 cm−1 in the presence and absence of metal ions, respectively. | |
Optimization of the sensing system
Prior to the application of such a SERS sensing platform to the detection of Cr(VI), several influencing factors, such as response time, laser power, pH of the sensing system, and the loading amount of ARS on the colloidal TiO2 NPs, must be considered. Firstly, the Cr(VI) induced SERS decrease was found to be fast and reached an equilibrium within 30 s (Fig. S7†), and thus the mixture was exposed to a laser beam for 30 s before each SERS measurement. This fast sample preparation is beneficial for high-throughput Cr(VI) assays. Secondly, we compared the affinity of Cr(VI) for the colloidal TiO2 NP surfaces at different pH values, because acidity affects the surface charge of the colloidal TiO2 NPs and the existing species of Cr(VI). As shown in Fig. S8,† no changes in the SERS intensities were observed with a pH ranging from 2.0 to 5.0. Within this range, HCrO4− is the major Cr(VI) species in the sensing system. Meanwhile, the colloidal TiO2 surface is positively charged, which is favorable to the adsorption of negatively charged ions, such as HCrO4−, CrO42−, and Cr2O72−. Moreover, an aggregation–sedimentation phenomenon was observed at a pH larger than 5.0, providing us with a simple method to recycle the TiO2 NPs from the analyte. Finally, the response mechanism was based on a co-catalysis scheme, in which both Cr(VI) and ARS are activated by the available Ti coordination sites on the surface of the colloidal TiO2 NPs. Thus, the catalytic efficiency with different loading amounts of ARS on the colloidal TiO2 NPs was also investigated to determine the optimum ARS-sensitized concentration. It was clearly found that increasing the loading amount of ARS results in an increase in the SERS intensity, but the best performance was obtained at 50 μM (Fig. S9†).
Application
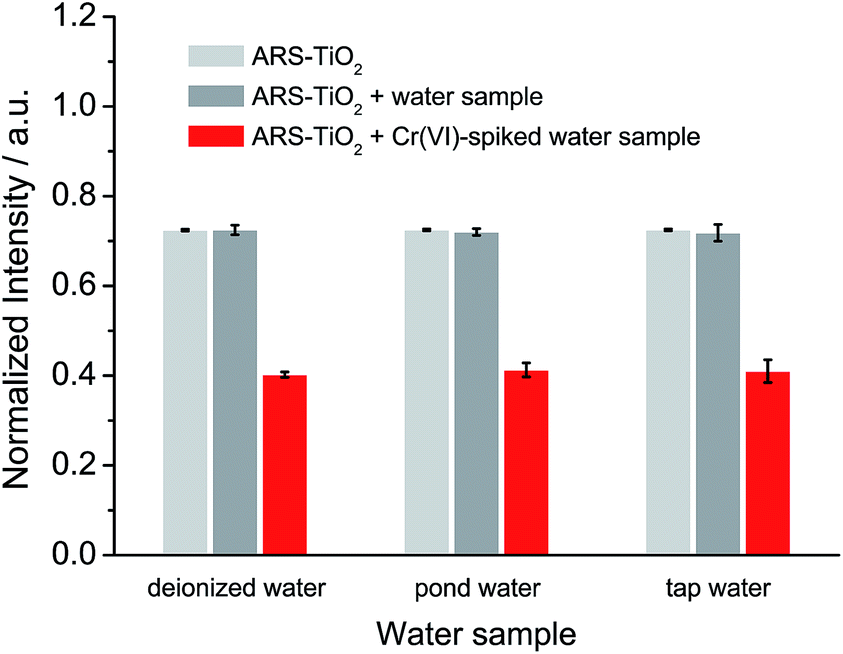
Based on the optimized conditions, the sensitivity and linearity of this sensing system were evaluated with different concentrations of Cr(VI) (Fig. 5). A good inverse proportionality was observed between the SERS intensity and the amount of Cr(VI) in the concentration range of 0.6–10 μM. The lowest concentration at which Cr(VI) could be detected is 0.6 μM. This concentration is lower than the maximum level of Cr(VI) in drinking water allowed by the WHO. In addition, the water samples spiked with different concentrations of Cr(VI) were also measured by employing our sensing system (Fig. 6). The measurements, which accurately reported the concentrations of the added standard Cr(VI) with good recoveries (Table S2†), confirmed that the sensing system proposed in this work has great potential for the quantitative analysis of Cr(VI) in environmental samples.
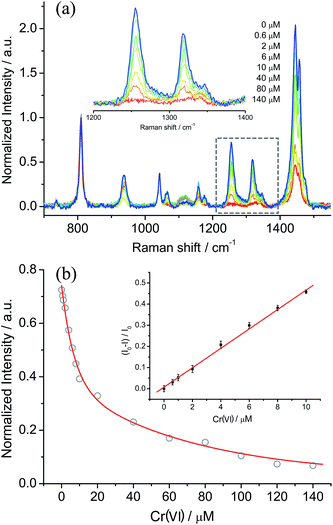 |
| | Fig. 5 (a) SERS spectra of ARS–TiO2 complexes (50 μM ARS) in the absence and presence of Cr(VI) with different concentrations. The intensities of the bands are normalized to that of the signal from 2-propanol at 816 cm−1. (b) The normalized Raman intensity at 1260 cm−1versus the Cr(VI) concentration. The inset shows the relative Raman intensity [(IR0 − IR)/IR0] in the low concentration range. | |
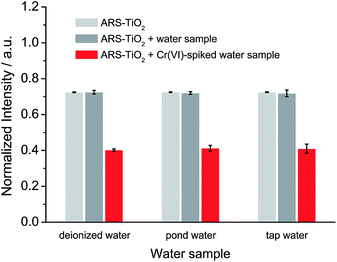 |
| | Fig. 6 SERS response of ARS–TiO2 to different water samples and water samples spiked with 10 μM Cr(VI) at pH 3. | |
Conclusions
In this work, we showed that semiconductor-enhanced Raman spectroscopy can be used as a sensing platform for the detection of metal ions. Firstly, the possibility of utilizing the dye-sensitized TiO2 system to promote SERS is discussed. It is found that the strong coupling interaction between the dye molecules and the colloidal TiO2 NPs leads to the formation of charge-transfer complexes and thus opens up a new electronic transition pathway for the charge-transfer process. The molecular polarizability tensor can be enhanced by such a charge-transfer process due to the vibronic coupling of the conduction band states of the semiconductor with the excited states of the probe molecule through a Herzberg–Teller coupling term. Secondly, a novel “turn-off” SERS strategy has been proposed and its use in a SERS-based assay for Cr(VI) has been demonstrated. Colloidal TiO2 NPs can be employed not only as an effective substrate to elicit the SERS signals of an ARS molecule, but also as a catalytic center to induce the self-degradation of the ARS response to Cr(VI). The “turn off” SERS signal upon laser irradiation allows the development of a facile assay to measure Cr(VI). Of note is that this method does not require complex pretreatment and complicated instruments, but provides a high sensitivity in a high-throughput fashion and excellent selectivity toward Cr(VI) over other common anions. Furthermore, the experiments using solutions spiked with Cr(VI) revealed that our method is effective in monitoring the Cr(VI) in real water samples. Based on this “turn-off” SERS strategy, other metal ions can also be detected by utilizing different semiconductor enhancement systems in which the energy level of the semiconductor is matched with the redox potential of the determined metal ion. Thus, we believe that the data described in this contribution clearly demonstrate that semiconductor-enhanced Raman spectroscopy integrated with the catalysis of semiconductor materials can be used as a reliable detection method for metal ions in practical applications.
Acknowledgements
This work was supported by a Support Project to Assist Private Universities in Developing Bases for Research (Research Centre for Single Molecule Vibrational Spectroscopy) from the Ministry of Education, Culture, Sports, Science and Technology of Japan. We are grateful to Dr Tamitake Itoh (National Institute of Advanced Industrial Science and Technology, Japan) for helpful discussions. W. J. acknowledges the support from the Japanese Society for Promotion of Science (JSPS, P13332).
Notes and references
- L. G. Quagliano, J. Am. Chem. Soc., 2004, 126, 7393–7398 CrossRef CAS PubMed.
- A. Musumeci, D. Gosztola, T. Schiller, N. M. Dimitrijevic, V. Mujica, D. Martin and T. Rajh, J. Am. Chem. Soc., 2009, 131, 6040–6041 CrossRef CAS PubMed.
- X. Ling, L. M. Xie, Y. Fang, H. Xu, H. L. Zhang, J. Kong, M. S. Dresselhaus, J. Zhang and Z. F. Liu, Nano Lett., 2010, 10, 553–561 CrossRef CAS PubMed.
- X. Wang, W. Shi, G. She and L. Mu, J. Am. Chem. Soc., 2011, 133, 16518–16523 CrossRef CAS PubMed.
- C. Qiu, L. Zhang, H. Wang and C. Jiang, J. Phys. Chem. Lett., 2012, 3, 651–657 CrossRef CAS.
- I. Alessandri, J. Am. Chem. Soc., 2013, 135, 5541–5544 CrossRef CAS PubMed.
- L. B. Yang, X. Jiang, W. D. Ruan, B. Zhao, W. Q. Xu and J. R. Lombardi, J. Phys. Chem. C, 2008, 112, 20095–20098 CAS.
- Y. F. Wang, W. D. Ruan, J. H. Zhang, B. Yang, W. Q. Xu, B. Zhao and J. R. Lombardi, J. Raman Spectrosc., 2009, 40, 1072–1077 CrossRef CAS.
- X. Wang, W. Shi, G. She and L. Mu, Phys. Chem. Chem. Phys., 2012, 14, 5891–5901 RSC.
- D. Finkelstein-Shapiro, P. Tarakeshwar, T. Rajh and V. Mujica, J. Phys. Chem. B, 2010, 114, 14642–14645 CrossRef CAS PubMed.
- S. Ma, R. Livingstone, B. Zhao and J. R. Lombardi, J. Phys. Chem. Lett., 2011, 2, 671–674 CrossRef CAS.
- X. X. Xue, W. Ji, Z. Mao, H. J. Mao, Y. Wang, X. Wang, W. D. Ruan, B. Zhao and J. R. Lombardi, J. Phys. Chem. C, 2012, 116, 8792–8797 CAS.
- J. R. Lombardi and R. L. Birke, J. Phys. Chem. C, 2014, 118, 11120–11130 CAS.
- W. Ji, X. X. Xue, W. D. Ruan, C. X. Wang, N. Ji, L. Chen, Z. S. Li, W. Song, B. Zhao and J. R. Lombardi, Chem. Commun., 2011, 47, 2426–2428 RSC.
- W. Ji, Y. Kitahama, X. X. Han, X. X. Xue, Y. Ozaki and B. Zhao, J. Phys. Chem. C, 2012, 116, 24829–24836 CAS.
- X. H. Li, G. Y. Chen, L. B. Yang, Z. Jin and J. H. Liu, Adv. Funct. Mater., 2010, 20, 2815–2824 CrossRef CAS.
- W. Xu, X. Ling, J. Xiao, M. S. Dresselhaus, J. Kong, H. Xu, Z. Liu and J. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9281–9286 CrossRef CAS PubMed.
- C. Wen, F. Liao, S. Liu, Y. Zhao, Z. Kang, X. Zhang and M. Shao, Chem. Commun., 2013, 49, 3049–3051 RSC.
- X. X. Han, L. Chen, U. Kuhlmann, C. Schulz, I. M. Weidinger and P. Hildebrandt, Angew. Chem., Int. Ed., 2014, 53, 2481–2484 CrossRef CAS PubMed.
- S. M. Booker and C. Pellerin, Environ. Health Perspect., 2000, 108, 402–407 CrossRef PubMed.
- S. A. Katz and H. Salem, J. Appl. Toxicol., 1993, 13, 217–224 CrossRef CAS.
- R. Saha, R. Nandi and B. Saha, J. Coord. Chem., 2011, 64, 1782–1806 CrossRef CAS.
- J. B. Vincent, Acc. Chem. Res., 2000, 33, 503–510 CrossRef CAS PubMed.
-
Guidelines for Drinking-Water Quality, World Health Organization, 4th edn, 2011, also available at http://www.who.int/water_sanitation_health/publications/2011/dwq_guidelines/en/, accessed March 2014 Search PubMed.
- M. Sperling, S. Xu and B. Welz, Anal. Chem., 1992, 64, 3101–3108 CrossRef CAS.
- X. Zhang and J. A. Koropchak, Anal. Chem., 1999, 71, 3046–3053 CrossRef CAS PubMed.
- M. Gardner and S. Comber, Analyst, 2002, 127, 153–156 RSC.
- D. F. Marino and J. D. Ingle, Anal. Chem., 1981, 53, 294–298 CrossRef CAS.
- H. Q. Chen and J. C. Ren, Talanta, 2012, 99, 404–408 CrossRef CAS PubMed.
- L. Yong, K. C. Armstrong, R. N. Dansby-Sparks, N. A. Carrington, J. Q. Chambers and Z.-L. Xue, Anal. Chem., 2006, 78, 7582–7587 CrossRef CAS PubMed.
- G. Liu, Y.-Y. Lin, H. Wu and Y. Lin, Environ. Sci. Technol., 2007, 41, 8129–8134 CrossRef CAS.
- W. Jin, G. Wu and A. Chen, Analyst, 2014, 139, 235–241 RSC.
- B. K. Jena and C. R. Raj, Talanta, 2008, 76, 161–165 CrossRef CAS PubMed.
- X. Y. Wu, Y. B. Xu, Y. J. Dong, X. Jiang and N. N. Zhu, Anal. Methods, 2013, 5, 560–565 RSC.
- I. Tsuyumoto and Y. Maruyama, Anal. Chem., 2011, 83, 7566–7569 CrossRef CAS PubMed.
- D. Bahnemann, A. Henglein, J. Lilie and L. Spanhel, J. Phys. Chem., 1984, 88, 709–711 CrossRef CAS.
- H. N. Ghosh, J. Phys. Chem. B, 1999, 103, 10382–10387 CrossRef CAS.
- Y. D. Iorio, E. S. Román, M. I. Litter and M. A. A. Grela, J. Phys. Chem. C, 2008, 112, 16532–16538 Search PubMed.
- L. C. T. Shoute and G. R. Loppnow, J. Chem. Phys., 2002, 117, 842–850 CrossRef CAS PubMed.
- D. Pan, D. Hu and H. P. Lu, J. Phys. Chem. B, 2005, 109, 16390–16395 CrossRef CAS PubMed.
- P. M. Jayaweera and T. A. U. Jayarathne, Surf. Sci., 2006, 600, L297–L300 CrossRef CAS PubMed.
- M. L. de Souza and P. Corio, Vib. Spectrosc., 2010, 54, 137–141 CrossRef CAS PubMed.
- S. J. Hurst, H. C. Fry, D. J. Gosztola and T. Rajh, J. Phys. Chem. C, 2011, 115, 620–630 CAS.
- The lowest quantifiable concentration of ARS could be reduced through tuning the size of the colloidal TiO2 NPs. In general, decreasing the particle size increases the amount of under-coordinated Ti defect sites and subsequently increases the number of probe molecules adsorbed on a particle’s surface. Thus, the enhancement effects will increase with decreasing particle size. However, when the particle size reaches its Bohr radius region (which is 1.5 nm for TiO2), the enhancement effects may plummet due to the quantization effects. The quantization effects will result in the use of only discrete allowed levels from the conduction and valence band continuum, thus decreasing the chemical enhancement borrowed from the allowed transitions within continuum states through Herzberg–Teller vibronic coupling.
- P. C. Redfern, P. Zapol, L. A. Curtiss, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 11419–11427 CrossRef CAS.
- R. Huber, S. Spörlein, J. E. Moser, M. Grätzel and J. Wachtveitl, J. Phys. Chem. B, 2000, 104, 8995–9003 CrossRef CAS.
- The evaluation of vibronic coupling usually involves a complex mathematical treatment. Actually, the direct calculation of vibronic couplings has been very limited so far. For SERS theory, a matrix element (Herzberg–Teller coupling term) is introduced into the coupling system, which represents the vibronic mixing of semiconductor states with molecular states.
- J. R. Lombardi and R. L. Birke, Acc. Chem. Res., 2009, 42, 734–742 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed Benesi–Hildebrand plot, IR spectra, Raman assignments, and experiment optimization. See DOI: 10.1039/c4sc02618g |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence