
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Carbon–hydrogen (C–H) bond activation at PdIV: a Frontier in C–H functionalization catalysis
Joseph J.
Topczewski
and
Melanie S.
Sanford
*
Department of Chemistry, University of Michigan, Ann Arbor, MI 48108, USA. E-mail: mssanfor@umich.edu; Fax: +1 734 647 4865; Tel: +1 734 615 0451
First published on 29th September 2014
Abstract
The direct functionalization of carbon–hydrogen (C–H) bonds has emerged as a versatile strategy for the synthesis and derivatization of organic molecules. Among the methods for C–H bond activation, catalytic processes that utilize a PdII/PdIV redox cycle are increasingly common. The C–H activation step in most of these catalytic cycles is thought to occur at a PdII centre. However, a number of recent reports have suggested the feasibility of C–H cleavage occurring at PdIV complexes. Importantly, these latter processes often result in complementary reactivity and selectivity relative to analogous transformations at PdII. This mini review highlights proposed examples of C–H activation at PdIV centres. Applications of this transformation in catalysis as well as mechanistic details obtained from stoichiometric model studies are discussed. Furthermore, challenges and future perspectives for the field are reviewed.
 Joseph J. Topczewski | Joseph Topczewski earned a B.S. with honours from the University of Wisconsin-Parkside in 2007 and conducted research with Professor Lori Allen. He received his Ph.D. in 2011 from the University of Iowa and was named an ACS Medicinal Chemistry Predoctoral Fellow while conducting research in the lab of David F. Wiemer. At the University of Iowa, he conducted postdoctoral studies with Professors Hien Nguyen and Daniel Quinn before joining the Sanford Lab in 2013. He is currently and NIH NRSA postdoctoral fellow studying the application of high valent transition metal catalysis. |
 Melanie S. Sanford | Melanie Sanford obtained her B.S. and M.S. degrees from Yale University and conducted research with Professor Robert Crabtree. She completed her Ph.D. at the California Institute of Technology with Professor Robert Grubbs and conducted postdoctoral research with Professor John Groves at Princeton University before joining he faculty at the University of Michigan in 2003. After receiving numerous awards, she was promoted in 2007 and 2010 and is currently the Moses Gomberg Collegiate Professor of Chemistry. The Sanford lab is engaged in unveiling the reactivity of high valent transition metals and elucidating the mechanism of this reactivity. |
Introduction
Over the past 15 years, catalytic C–H bond functionalisation has emerged as a rich and highly active field of research.1 C–H functionalisation reactions proceeding via PdII/IV catalytic cycles are particularly prevalent due to their operational simplicity, wide scope, excellent functional group tolerance, and opportunities to access both C–C and C-heteroatom bond construction.2 PdII/IV-catalysed C–H functionalization reactions are generally proposed to proceed via catalytic cycles exemplified by that shown in red in Fig. 1. This involves three elementary steps: C–H activation at PdII, 2e− oxidation to PdIV (or a PdIII dimer)3 with an appropriate stoichiometric oxidant (oxidant-X), and finally C–X bond-forming reductive elimination from the high valent palladium centre to release the product.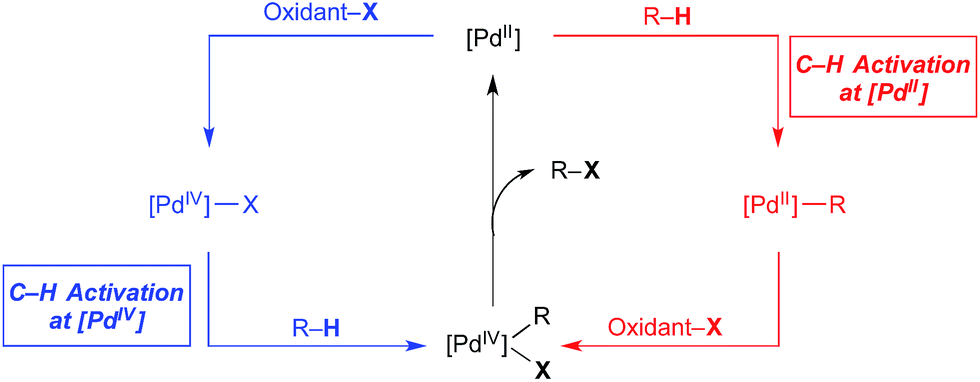 | ||
| Fig. 1 Examples of catalytic cycles involving C–H activation at PdII (red) versus PdIV (blue). | ||
Extensive research has established that the steps occur in this order for the vast majority of PdII/IV-catalysed C–H functionalisations.2 However, several recent reports have suggested that C–H cleavage can also occur at PdIV centres (Fig. 1, blue) and, further, that this process may be governed by different selectivity and reactivity principles than analogous transformations at PdII. This offers the exciting possibility for alternative catalytic cycles for Pd-catalysed C–H functionalisation, involving, for example, oxidation of PdII to PdIV, C–H bond activation at PdIV, and reductive elimination to release the product and regenerate the PdII catalyst. Notably, in both cycles in Fig. 1, additional and/or alternative steps are possible; however, the key distinguishing feature of the blue cycle, discussed herein, is that at least one C–H activation event occurs at PdIV. This mini review summarizes examples where arene C–H activation at a PdIV centre is proposed in both catalytic transformations and in stoichiometric model systems.4 For some of these systems, clear experimental evidence demonstrates C–H activation at PdIV while for others, the role of C–H activation at PdIV is strongly suspected. Both synthetically useful catalytic cycles and mechanistic details are presented and discussed.5
C–H activation at PdIV
To the best of our knowledge, the first report implicating a C–H activation reaction at PdIV involved the dimerization of 2-aryl pyridines.6 In this system, Pd(OAc)2 catalyses the C–H/C–H oxidative coupling of a variety of substituted 2-aryl pyridines at room temperature using Oxone as the terminal oxidant. A representative example is the conversion of 2 equiv. of 1 into 2 (Fig. 2).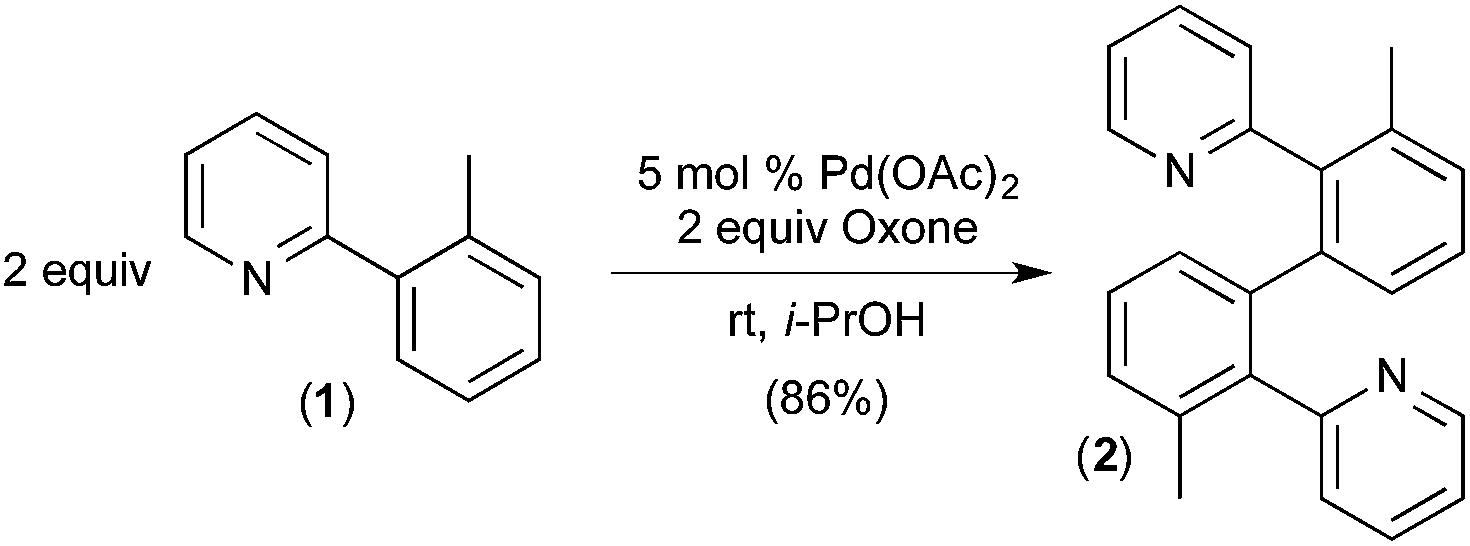 | ||
| Fig. 2 Oxidative coupling of 2-arylpyridine derivatives via proposed C–H activation at PdIV. | ||
Several experiments were conducted that suggest that this transformation involves two discrete C–H activation steps that have very different selectivities. For example, the unsymmetrically-substituted substrate 3 undergoes stoichiometric cyclometalation with PdII(OAc)2 to afford a single isomeric product 4via selective cleavage of C–HA (Fig. 3a). When this complex is subjected to Oxone and substrate 1 under the standard conditions, a single isomer of the coupled product is formed (6a, Fig. 3c). In contrast, when the sequence is reversed (i.e., 1 is first cyclometalated at PdII to form 5 (Fig. 3b), and this intermediate is subjected to analogous conditions with substrate 3), a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of the isomeric products 6a and 6b is produced (Fig. 3d). These results implicate two different C–H activation steps with different selectivities: (i) the initial cyclometalation of 3 at PdII(OAc)2 (>99:1 selectivity for activation of HA) and (ii) a subsequent C–H activation of 3 (5:1 selectivity for HAversus HB).
:1 mixture of the isomeric products 6a and 6b is produced (Fig. 3d). These results implicate two different C–H activation steps with different selectivities: (i) the initial cyclometalation of 3 at PdII(OAc)2 (>99:1 selectivity for activation of HA) and (ii) a subsequent C–H activation of 3 (5:1 selectivity for HAversus HB).
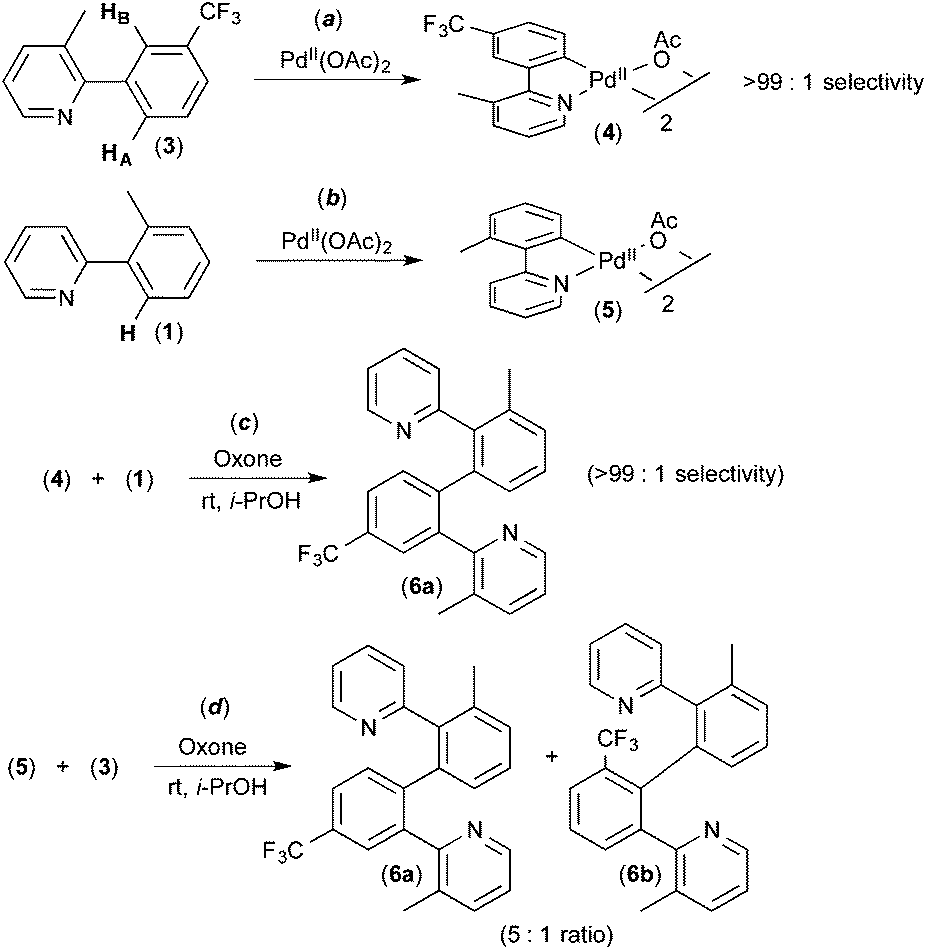 | ||
| Fig. 3 Experiments implicating two different C–H activation steps with different selectivities in activation of substrate 3. | ||
A variety of additional experiments, including cross-over studies and reactivity studies of possible intermediates, implicated the mechanism shown in Fig. 4. Here, an initial C–H activation at PdII (step (i)), is followed by oxidation of the resulting palladacycle intermediate A with Oxone to yield PdIV species B (step (ii)). The second C–H activation then occurs at this PdIV intermediate to yield C (step (iii)), which undergoes C–C bond-forming reductive elimination to complete the catalytic cycle (step (iv)).
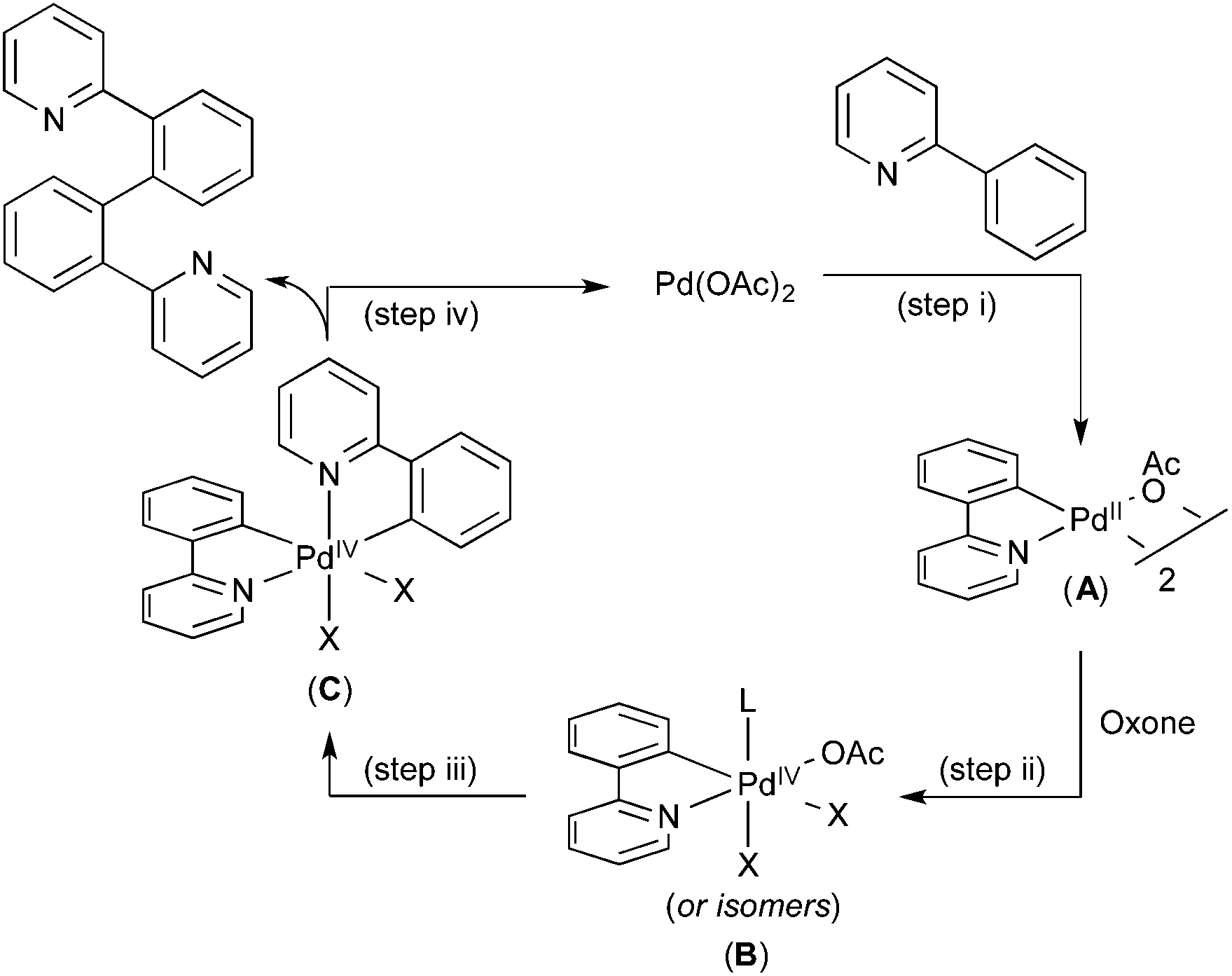 | ||
| Fig. 4 Proposed mechanism for Pd-catalysed oxidative coupling of 2-aryl pyridines. | ||
Synthetic applications of C–H activation at PdIV
Subsequent work has taken advantage of proposed arene C–H activation reactions at PdIV to achieve synthetically useful catalytic transformations. In one elegant example, Michael demonstrated the PdII/IV-catalysed aminoarylation of terminal olefins with NFSI as the oxidant (Fig. 5).7 This reaction was discovered during an investigation of the Pd-catalysed diamination of 7 (Fig. 5a). When the solvent for this transformation was changed from EtOAc to toluene, the aminoarylation product 8 was formed via toluene C–H activation. A variety of substituted arenes can also be used in this transformation, with substituents including Br, CH3, and CH3O. Furthermore, mono-substituted arenes react with extremely high selectivity at the para position (c.f., products 9–12 of Fig. 5). This high selectivity is in marked contrast to most other Pd-catalysed functionalisations of mono-substituted arenes, which typically form mixtures of isomeric products.8 Additionally, Michael's aminoarylations proceed efficiently at room temperature, which is significantly milder than most PdII-catalysed C–H functionalizations of simple arenes. The authors propose that arene C–H activation occurs at a PdIV centre and that this feature is responsible for the unusually high selectivity and reactivity.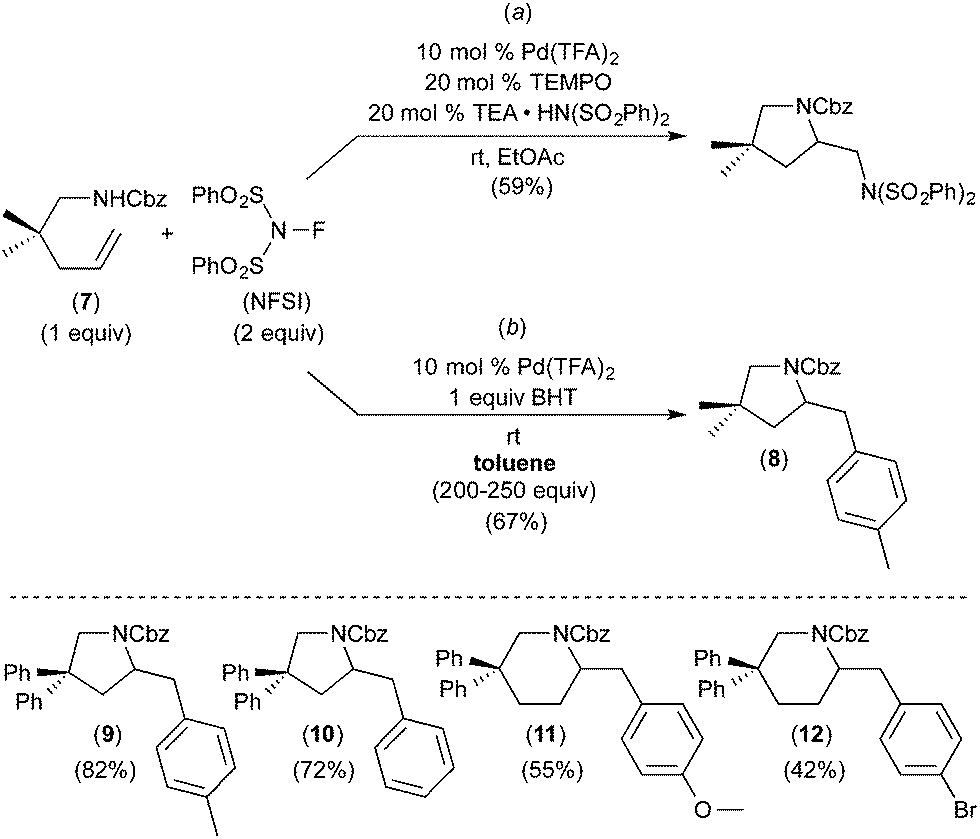 | ||
| Fig. 5 Alkene aminoarylation via proposed C–H activation at PdIV. | ||
A number of experiments were conducted to gain further insights into the mechanism of this process. First, the electronic requirements of the C–H activation step were investigated using competition experiments between benzene and other arenes. These studies showed that benzene reacts faster (by a factor of ∼2.5) than both anisole and bromobenzene.
A competition between toluene and toluene-d8 showed an intermolecular H/D competition isotope effect of 1.1 (14-d0/14-d7 = 1.1, Fig. 6a). In contrast, the use of 1,3,5-trideuterobenzene as the substrate resulted in a much larger intramolecular H/D competition isotope effect of 4 (15-d3/15-d2 = 4, Fig. 6b). In combination, these results implicate a 2-step C–H activation process, in which the two different steps occur with distinct selectivities. As shown in Fig. 7, the authors propose that the two steps are π-coordination of the arene to the PdIV centre (which determines the intermolecular isotope effect, step (iii) in Fig. 7) followed by C–H cleavage of the π-coordinated substrate (which dictates the intramolecular isotope effect, step (iv) in Fig. 7). Notably, C–H activation reactions at PdII centres generally show much higher intermolecular competition isotope effects (typically ranging from 2 to 6).9
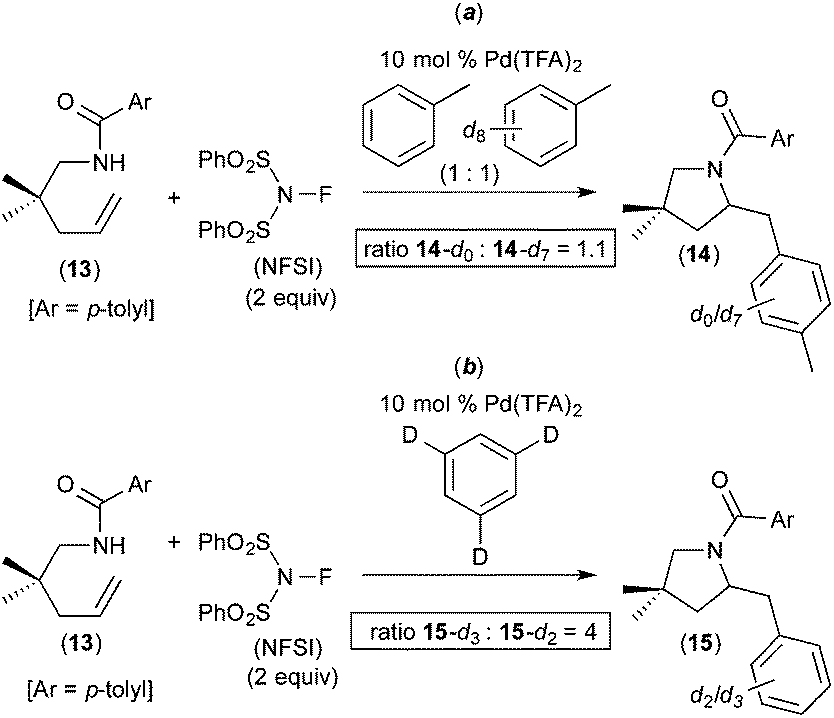 | ||
| Fig. 6 Kinetic isotope effect studies of alkene aminoarylation reaction. | ||
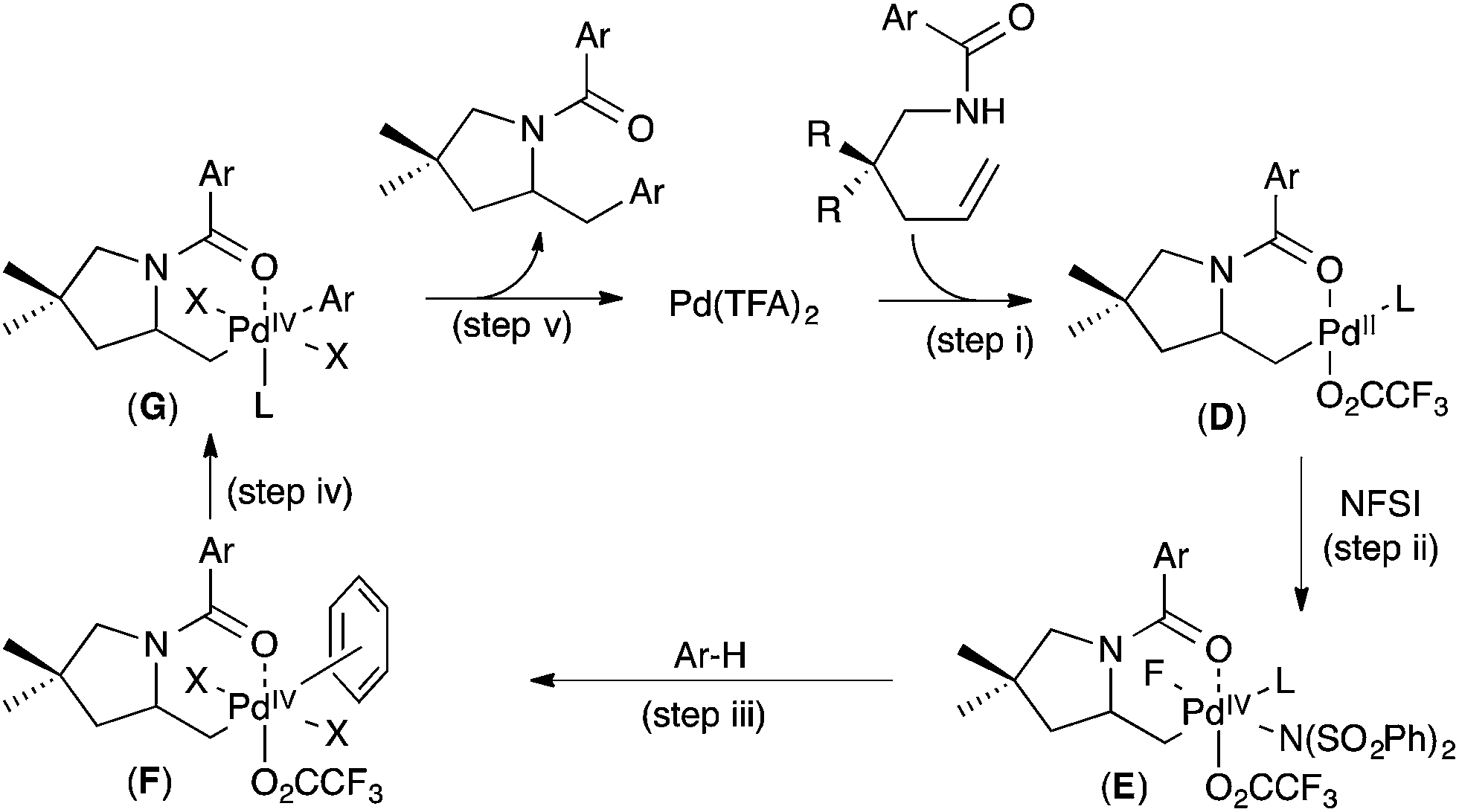 | ||
| Fig. 7 Proposed mechanism for aminoarylation. | ||
On the basis of these (and additional) studies, a full catalytic cycle was proposed. As shown in Fig. 7, the cycle begins with intramolecular anti-aminopalladation to produce alkyl PdII intermediate D (step (i)). D then undergoes oxidation with NFSI to produce PdIV intermediate E (step (ii)) π-coordination of the arene substrate to E to generate F (step (iii)) is followed by C–H cleavage (step (iv)) to afford aryl alkyl PdIV complex G. Finally, C–C bond-forming reductive elimination (step (v)) closes the catalytic cycle.
The Yu group reported a related PdII/IV-catalysed C–H functionalization reaction involving the oxidative coupling of perfluorobenzamides with simple arenes using NFSI as the oxidant (Fig. 8).10 Similar to Michael's work, this transformation proceeds with very high para selectivity, ranging from 12:1 with a bromo- or ethyl-substituent to “para only,” with methoxy- or fluoro-substituents. The authors rationalize this unusually high selectivity based on a mechanism involving two sequential C–H activation events: ligand-directed C–H activation at PdII followed by arene C–H activation at PdIV. They propose that the weakly coordinating perfluorobenzamide directs an initial C–H activation at PdII. The NFSI then oxidizes this palladacyclic intermediate to a PdIV fluoride complex, which promotes para-selective arene C–H activation.11
 | ||
| Fig. 8 Arene C–H oxidative coupling involving proposed C–H activation at PdIV. | ||
Several studies were conducted to shed further light on the mechanism of this transformation. While substantial yields of oxidative coupled products were obtained with a number of different oxidants (e.g., Selectfluor, N-fluoropyridinium K2S2O8), only F+ oxidants afforded high levels of para-selectivity. This led the authors to propose that the presence of a fluoride ligand on the PdIV center is crucial for achieving para-selective C–H activation.
As shown in Fig. 9, an isotope effect study revealed that the initial reaction rate is identical with toluene and toluene-d8 as the arene substrate (kH/kD = 1). This result suggests that the C–H activation at PdIV is not the slow step of the catalytic cycle. Unlike the Michael system, no competition or intramolecular isotope effect studies were reported in this system, so the possible role of π-coordination cannot be assessed from this report.10
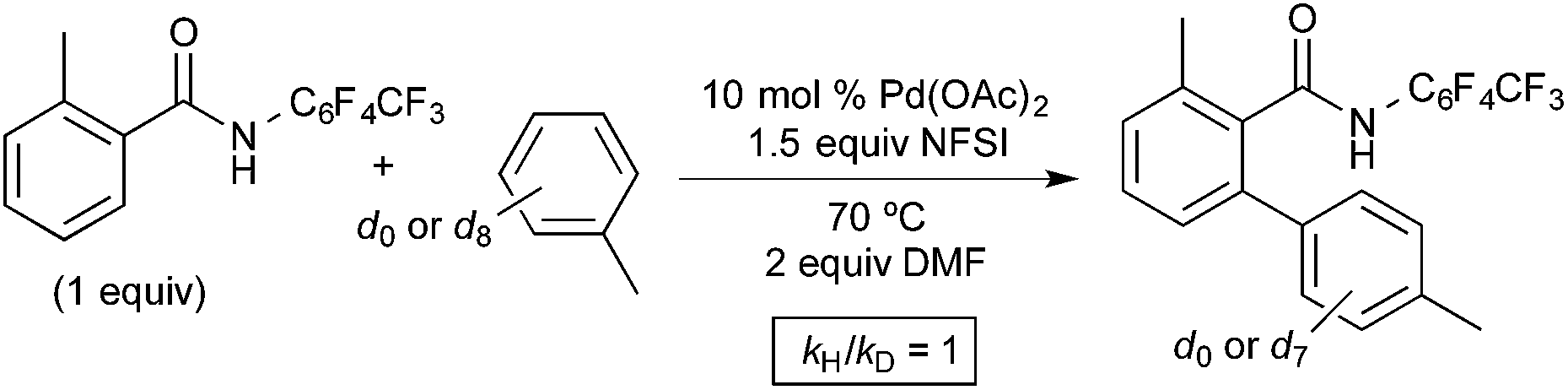 | ||
| Fig. 9 Isotope effect study for arene C–H oxidative coupling. | ||
A number of groups have used naphthalene as a substrate in C–H arylation reactions that are believed to proceed via C–H activation at PdIV. For example, in 2008, Inoue and coworkers demonstrated the PdCl2-catalysed C–H arylation of naphthalene with aryl stannanes.12 This reaction was selective for arylation at the α-position of naphthalene (α:β ratio = 3.5:1, Fig. 10a) and afforded modest 40% yield. A variety of other substrates were evaluated and phenanthrene was found to afford the best yield (80%) as well as high selectivity for the 9-position (Fig. 10b). A mechanism involving naphthalene or phenanthrene C–H activation at PdIV was proposed; however, minimal evidence is provided to support this pathway.
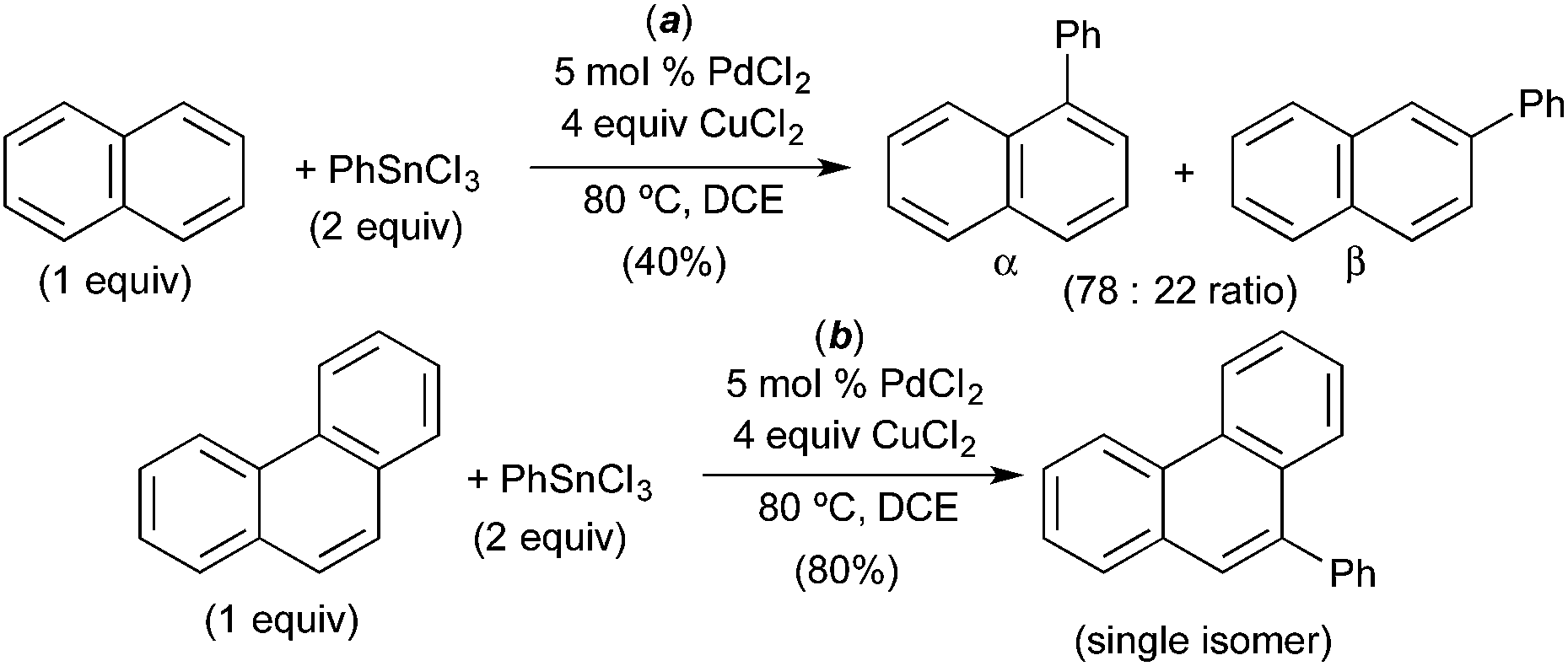 | ||
| Fig. 10 C–H arylation of arenes with PhSnCl3via proposed C–H activation at PdIV. | ||
More recently, our group demonstrated the C–H arylation of naphthalene using diaryliodonium salts as both the oxidant and aryl source (Fig. 11).13 In this system, the selectivity of C–H cleavage could be tuned through the appropriate selection of supporting ligand. Simple Pd salts, such as Pd(OAc)2 and PdCl2, afforded modest yields and selectivities for the C–C coupled products (yields ranging from 12–24% and α:β selectivities from 5:1 to 13:1). The yield and selectivity could be enhanced dramatically through the use of N–N chelating L type ligands, and the optimal diimine Pd catalyst (16 in Fig. 11) afforded 70% yield and >70:1 selectivity for the α-arylated product. Notably, since this work, complementary β selectivity has been achieved in the same transformation by employing a platinum catalyst.4c
 | ||
| Fig. 11 C–H arylation of naphthalene with [Ph2I]BF4via proposed C–H activation at PdIV. | ||
Rate studies of the Pd-catalyzed naphthalene arylation showed 1st order kinetics in [Ar2I]+ and zero order dependence on [naphthalene]. Isotope effect studies were conducted using naphthalene and naphthalene-d8. The initial rate of the C–H arylation reaction was essentially identical with each of these two substrates (kH/kD = 1, Fig. 12b). Furthermore, a competition between naphthalene and naphthalene-d8 afforded a product ratio corresponding to an H/D competition isotope effect of 1.08 (17-d0/17-d7 = 1.08, Fig. 12a). This is very similar to the results obtained by Michael in analogous competition experiments (Fig. 6). Naphthalene was found to be the best substrate for this reaction, and arenes without an extended π-system (e.g., anisole, benzene, chlorobenzene, veratrole) afforded low yields and selectivities. On the basis of these investigations, the oxidation of the PdII catalyst by the aryliodonium salt was proposed to be the rate-determining step, and the C–H activation of naphthalene was proposed to occur at the resulting PdIV centre. Additionally, a two-step C–H activation mechanism analogous to that put forth by Michael (Fig. 7) was proposed in this system. The first step is proposed to involve π-coordination of the substrate to PdIV (a step that should be facilitated by the extended π-system of naphthalene) followed by subsequent C–H cleavage at the PdIV centre.
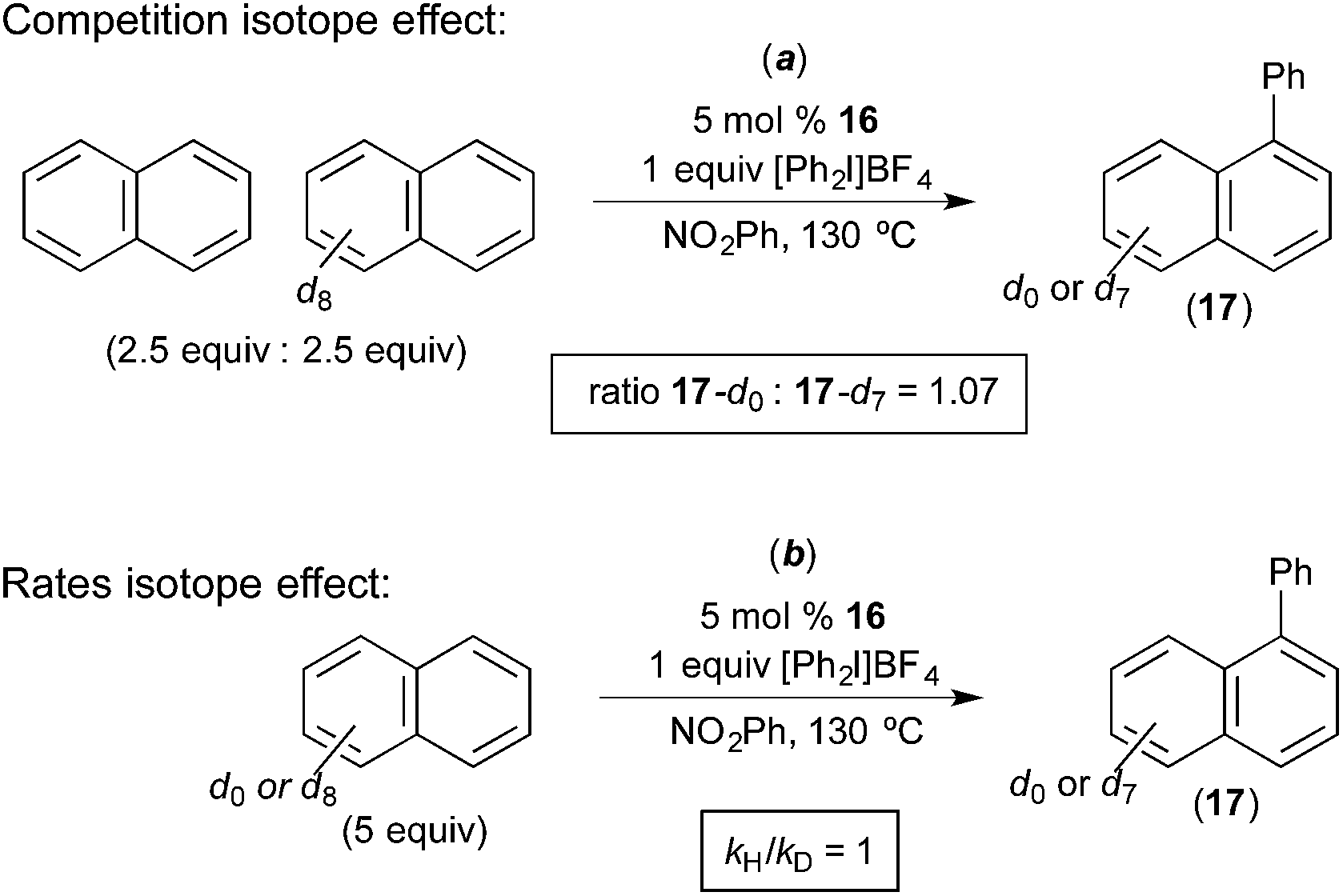 | ||
| Fig. 12 Isotope effect studies of naphthalene C–H arylation. | ||
Seayad recently described the selective C–H/C–H oxidative coupling of furans with arenes (Fig. 13).14 In this system, the site selectivity of furan C–H activation could be modulated based appropriate selection of the terminal oxidant. Using AgCO3 as oxidant, the authors observed poorly selective activation of the furan (C-4/C-5 arylated products were formed in an ∼1:1 ratio, Fig. 13a). Notably, Ag2CO3 is unlikely to promote oxidation of PdII to PdIV. In contrast, the use of N-fluoropyridinium triflate (NFTP), an “F+” oxidant that is well known to promote the oxidation of PdII to PdIV,15 afforded >20:1 C-5 selectivity in most cases. In the NFTP system, large isotope effect (ratio of products 22-d0/22-d5 = 4.8) was observed when the reaction was run as a competition between benzene and benzene-d6 (Fig. 14b). In contrast, the competition between furan 18 and deuterated furan 19-d resulted in a relatively small quasi isotope effect of 1.7 (the substrates are slightly different, so this is not a true isotope effect; however, the authors state that the rate of arylation is similar for the two substrates) (Fig. 14a). The authors propose a mechanism initiated by initial oxidation of ligated PdII to PdIV by NFPT and subsequent C–H activation of the two substrates. An alternative possibility involving benzene activation at PdII, oxidation with NFPT and subsequent furan activation at PdIV is also possible, and perhaps more likely based on the related reactions described above.
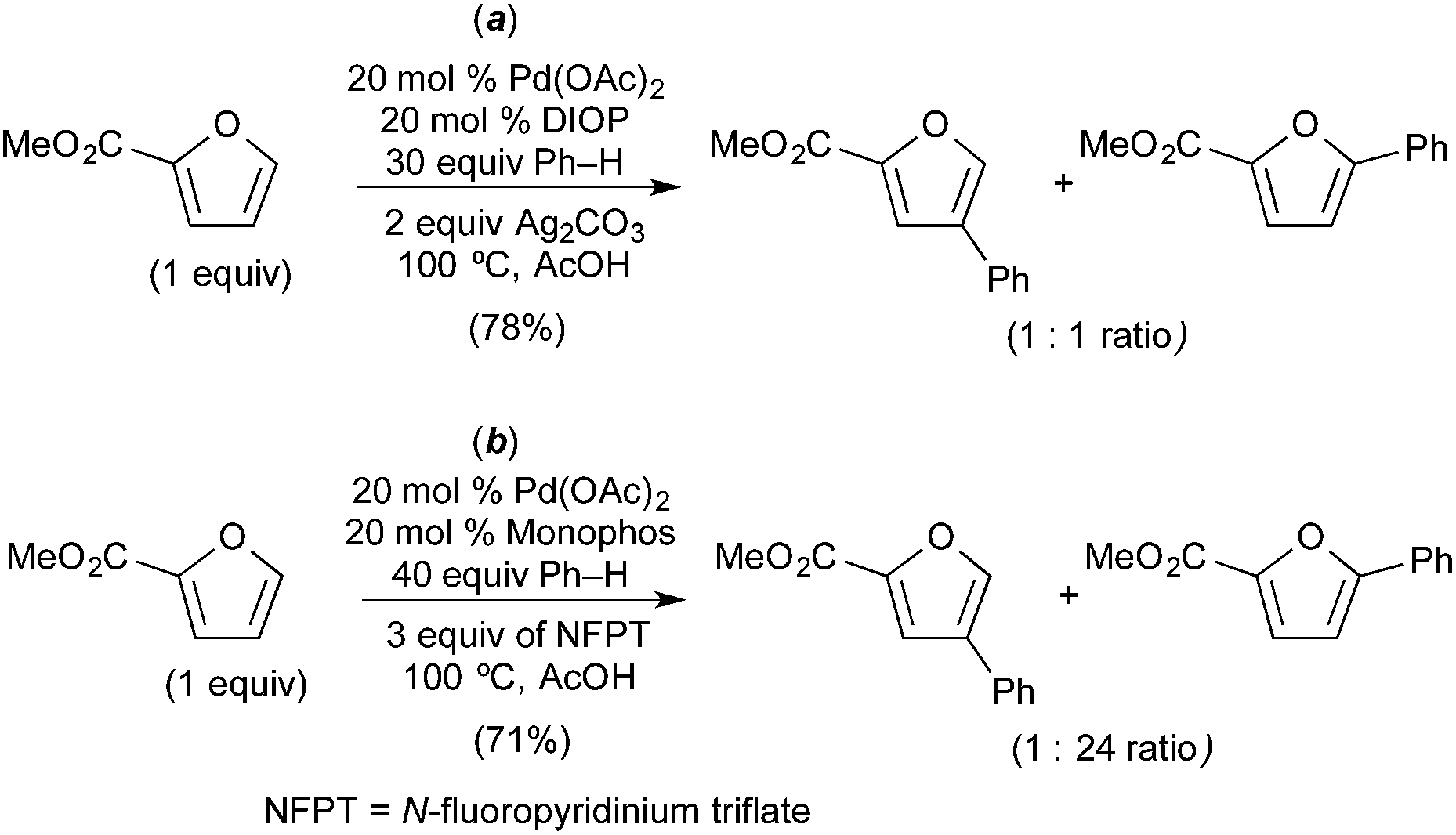 | ||
| Fig. 13 Oxidative coupling of furans and benzene: proposed furan C–H activation at PdII (a) and PdIV (b) depending on oxidant. | ||
 | ||
| Fig. 14 Isotope effect studies in benzene/furan oxidative coupling with NFPT as oxidant. | ||
The examples described above summarize the current state of the art in catalytic transformations proposed to proceed via C–H activation at PdIV. These examples show encouraging selectivity trends and demonstrate that valuable synthetic methods can be achieved with C–H activation at PdIV as a likely step. At this stage, most of these transformations have been discovered serendipitously rather than through reaction design. However, moving forward it would be important to rationally design catalytic sequences involving C–H activation at a PdIV centre. A uniting feature of the transformations discussed above is the use of strong oxidants, with “F+” reagents, hypervalent iodine reagents, and inorganic peroxides being particularly common choices. In addition, many these transformations are believed to involve the generation of a PdII–C bond prior to oxidation of PdII to PdIV. This likely serves to accelerate the oxidation event. Finally, most of the catalysts and intermediates in these transformations possess oxidatively stable ligands that are unlikely to participate in competing reductive elimination. All of these features should serve as key design considerations as new reactions are developed.
Direct observation of C–H activation at Pd(IV)
Our group has developed organometallic model systems in order to directly observe and study this fundamental reaction. Such studies should ultimately assist in the rational design of new catalytic processes that incorporate this elementary step. The PdIV model complexes were carefully designed to accelerate C–H activation while slowing competing reductive elimination processes from PdIV. These complexes were designed so that the C–H activation would be intramolecular. For example, in complex 24 (Fig. 15), studied by Racowski et al.,16 the biphenyl ligand was incorporated to enable intramolecular C–H activation, which is typically more facile than the corresponding intermolecular reactions. In addition, the CF3 ligand was included because aryl-CF3 reductive elimination is known to be relatively slow from PdIV, particularly at low temperatures.17 Complex 24 was generated in situ by the oxidation of (bpy)PdII(2-biphenyl) (CF3) complex 23 with PhICl2 at −30 °C. Warming complex 24 to room temperature resulted in intramolecular activation of the 2-aryl substituent to form the cyclometalated PdIV product 25. To our knowledge, this was the first direct observation of C–H activation at a PdIV centre.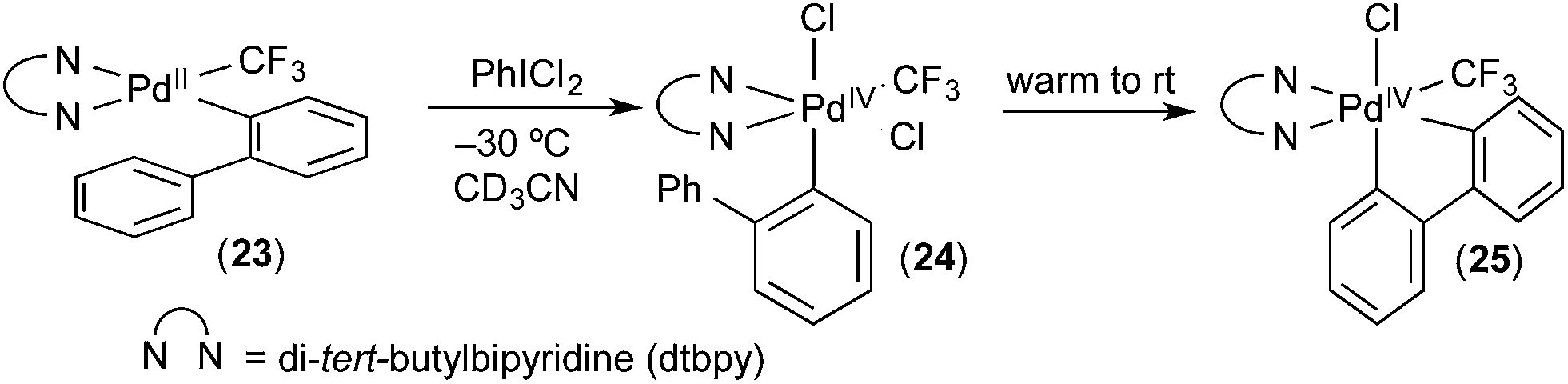 | ||
| Fig. 15 First direct observation of C–H activation at PdIV. | ||
In a follow up study, the related complex 26 (Fig. 16) was synthesized via oxidation of a PdII precursor with PhICl2.18 The tridentate tris(2-pyridyl)-methane ligand (Py3CH) was a key design feature in this study. This strongly coordinating tridentate ligand is well-known to stabilize octahedral PdIV species relative to analogues with bidentate nitrogen donors like bipyridine.19 Thus, it was anticipated that the Py3CH ligand would slow C–H activation and enable more detailed mechanistic investigations of this process. Indeed, the PdIV aryl complex 26 proved stable at room temperature and could be fully characterized by 1D and 2D NMR, HRMS, and X-ray crystallography. Complex 26 did not undergo C–H activation, even upon heating to 90 °C in CDCl3. Instead, C–Cl bond-forming reductive elimination was observed under these conditions. However, when one of the chloride ligands in 26 was exchanged for an acetate, the resulting intermediate 27 underwent clean cyclometalation at room temperature to yield 28. This result suggests that C–H activation at PdIV in this system likely occurs via a concerted metalation-deprotonation mechanism, analogous to C–H activation at PdII.20
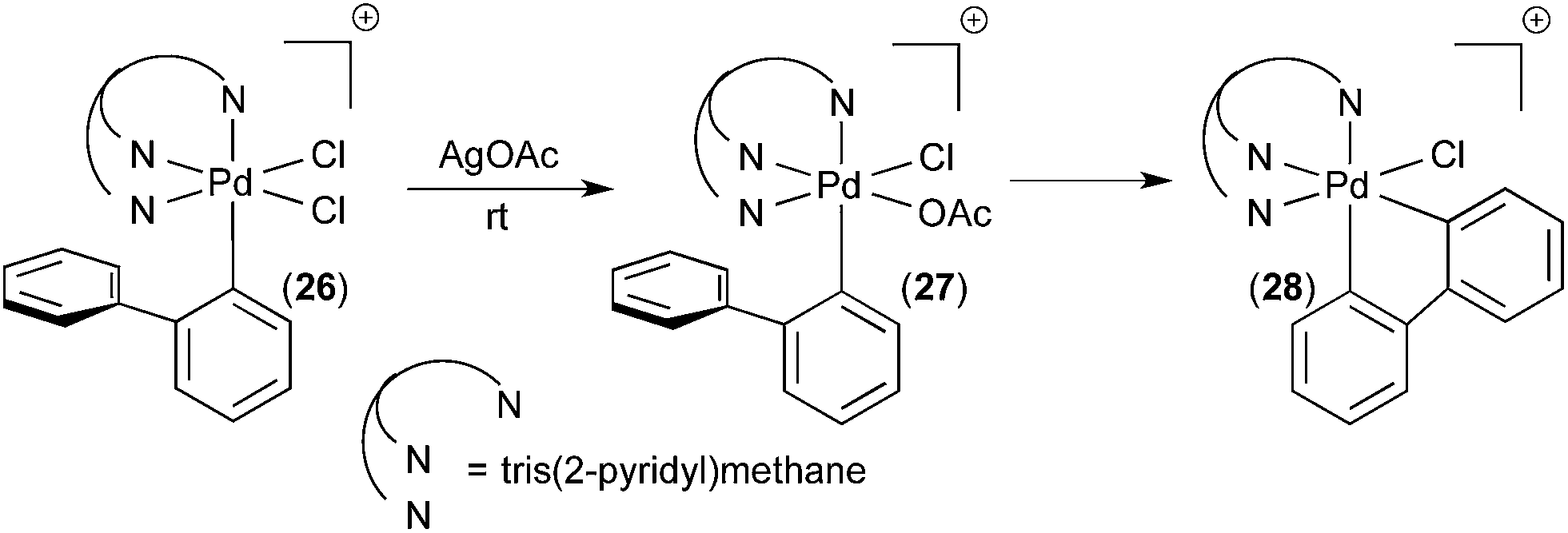 | ||
| Fig. 16 C–H activation at PdIV complex 27. | ||
When complex 29 (Fig. 17) was treated with acetate, a product ratio consistent with an H/D competition isotope effect of 7 was obtained. In contrast, the initial rate of C–H activation at 30versus30-d2 was essentially identical (kH/kD = 1). These results in combination with a variety of studies of the dynamic behaviour of complex 26 indicate that C–H cleavage is not the rate determining step in the C–H activation process in this system. The similarity between the KIE observed on this system to those observed in catalytic systems further supports the mechanisms proposed for the catalytic reactions described above.
 | ||
| Fig. 17 Isotope effects in C–H activation at PdIV. | ||
These two examples chronicle C–H activation at discrete and, in one case, isolable PdIV complexes. It is also noteworthy that C–H activation at the PdIV centre can be facile at room temperature or below with bidentate ligands or in the presence of acetate ion. Future work on isolated palladium complexes may explore an intermolecular C–H activation event at PdIV.
Conclusions
The catalytic functionalization of C–H bonds via high valent palladium is a powerful manifold for developing synthetically useful transformations. This mini-review has summarized experiments supporting the viability of C–H bond activation at PdIV and has described mechanistic studies of the C–H bond cleavage event. Synthetically useful catalytic cycles that utilize C–H activation at PdIV remain limited; however, there are great opportunities in this area due to the potential for unique selectivity in these transformations. Although the presently described work is mostly limited to catalytic C–H arylation sequences, as this method is more fully understood, one can anticipate its application to more diverse scaffolds and functionalizations.Acknowledgements
We thank the NIH (GM073836) and NSF (CHE-1111563 and CHE-1361542) for support of this research. In addition, JJT gratefully acknowledges the NIH (F32 GM109479) for a post-doctoral fellowship.Notes and references
- For reviews on C–H activation: (a) A. D. Ryabov, Chem. Rev., 1990, 90, 403 CrossRef CAS; (b) A. E. Shilov and G. B. Shul'pin, Chem. Rev., 1997, 97, 2879 CrossRef CAS PubMed; (c) V. Ritleng, C. Sirlin and M. Pfeffer, Chem. Rev., 2002, 102, 1731 CrossRef CAS PubMed; (d) A. R. Dick and M. S. Sanford, Tetrahedron, 2006, 62, 2439 CrossRef CAS PubMed; (e) I. A. I. Mkhalid, J. H. Barnard, T. B. Marder, J. M. Murphy and J. F. Hartwig, Chem. Rev., 2010, 110, 890 CrossRef CAS PubMed; (f) M. P. Doyle, R. Duffy, M. Ratnikov and L. Zhou, Chem. Rev., 2010, 110, 704 CrossRef CAS PubMed; (g) H. M. Davies, J. Du Bois and J. Q. Yu, Chem. Soc. Rev., 2011, 40, 1855 RSC; (h) D. A. Colby, R. G. Bergman and J. A. Ellman, Chem. Rev., 2009, 110, 624 CrossRef PubMed.
- For reviews on C–H activation at PdII: (a) X. Chen, K. M. Engle, D. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2009, 48, 5094 CrossRef CAS PubMed; (b) T. W. Lyons and M. S. Sanford, Chem. Rev., 2010, 110, 1147 CrossRef CAS PubMed; (c) L. Ackermann, Chem. Rev., 2011, 111, 1315 CrossRef CAS PubMed; (d) S. R. Neufeldt and M. S. Sanford, Acc. Chem. Res., 2012, 45, 936 CrossRef CAS PubMed.
- (a) D. C. Powers, E. Lee, A. Ariaford, M. S. Sanford, B. F. Yates, A. J. Canty and T. Ritter, J. Am. Chem. Soc., 2012, 134, 12002 CrossRef CAS PubMed; (b) D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840 CrossRef CAS PubMed.
- For reports proposing allylic and/or vinylic C–H activation at PdIV: (a) L. T. Pilarski, N. Selander, D. Bose and K. J. Szabo, Org. Lett., 2009, 11, 5518 CrossRef CAS PubMed; (b) N. Selander, B. Willy and K. J. Szabo, Angew. Chem., Int. Ed., 2010, 49, 4051 CrossRef CAS PubMed; (c) R. Alam, L. T. Pilarski, E. Pershagen and K. J. Szabo, J. Am. Chem. Soc., 2012, 134, 8778 CrossRef CAS PubMed.
- For examples of C–H activation at PtIV see: (a) G. B. Shul'pin, G. V. Nizova and A. T. Nikitaev, J. Organomet. Chem., 1984, 276, 115 CrossRef; (b) M. Lersch and M. Tilset, Chem. Rev., 2005, 105, 2471 CrossRef CAS PubMed; (c) A. M. Wagner, A. J. Hickman and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 15110 CrossRef PubMed.
- K. L. Hull, E. L. Lanni and M. S. Sanford, J. Am. Chem. Soc., 2006, 128, 14047 CrossRef CAS PubMed.
- (a) C. F. Rosewall, P. A. Sibbald, D. V. Liskin and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 9488 CrossRef CAS PubMed; (b) P. A. Sibbald, C. F. Rosewall, R. D. Swartz and F. E. Michael, J. Am. Chem. Soc., 2009, 131, 15945 CrossRef CAS PubMed.
- For examples of modest selectivity for C–H activation at PdII, see ref. 1b and 2d as well as: (a) Y. Fujiwara, R. Asano, I. Moritani and S. Teranishi, J. Org. Chem., 1976, 41, 1681 CrossRef CAS; (b) L. J. Ackerman, J. P. Sadighi, D. M. Kurtz, J. A. Labinger and J. E. Bercaw, Organometallics, 2003, 22, 3884 CrossRef CAS.
- Examples of isotope effects for C–H activation at PdII: (a) H. A. Chiong, Q. Pham and O. J. Daugulis, J. Am. Chem. Soc., 2007, 129, 9879 CrossRef CAS PubMed; (b) Z. J. Shi, B. Li, X. Wan, J. Cheng, Z. Fang, B. Cao, C. Qin and Y. Wang, Angew. Chem., Int. Ed., 2007, 46, 5554 CrossRef CAS PubMed; (c) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS PubMed.
- X. Wang, D. Leow and J. Q. Yu, J. Am. Chem. Soc., 2011, 133, 13864 CrossRef CAS PubMed.
- (a) K. Engle, T. S. Mei, X. Wang and J. Q. Yu, Angew. Chem., Int. Ed., 2011, 50, 1478 CrossRef CAS PubMed; (b) A. J. Hickman and M. S. Sanford, Nature, 2012, 484, 177 CrossRef CAS PubMed.
- H. Kawai, Y. Kobayashi, S. Oi and Y. Inoue, Chem. Commun., 2008, 1464 RSC.
- A. J. Hickman and M. S. Sanford, ACS Catal., 2011, 1, 170 CrossRef CAS.
- N. Asyikin, B. Juwaini, J. K. P. Ng and J. Seayad, ACS Catal., 2012, 2, 1787 CrossRef.
- (a) N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2009, 131, 3796 CrossRef CAS PubMed; (b) N. B. Ball, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2010, 132, 2878 CrossRef CAS PubMed; (c) J. M. Racowski, J. B. Gary and M. S. Sanford, Angew. Chem., Int. Ed., 2012, 51, 3414 CrossRef CAS PubMed.
- J. M. Racowski, N. D. Ball and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 18022 CrossRef CAS PubMed.
- N. D. Ball, J. B. Gary, Y. Ye, J. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2011, 133, 7577 CrossRef CAS PubMed.
- A. Maleckis, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 6618 CrossRef CAS PubMed.
- (a) P. K. Byers, A. J. Canty, B. W. Skelton and A. H. White, J. Chem. Soc., Chem. Commun., 1987, 1093 RSC; (b) P. K. Byers, A. J. Canty, B. W. Skelton and A. H. White, Organometallics, 1990, 9, 826 CrossRef CAS.
- (a) D. L. Davies, S. M. A. Donald and S. A. Macgregor, J. Am. Chem. Soc., 2005, 127, 13754 CrossRef CAS PubMed; (b) M. Lafrance and K. Fagnou, J. Am. Chem. Soc., 2006, 128, 16496 CrossRef CAS PubMed; (c) S. I. Gorelsky, D. Lapointe and K. Fagnou, J. Am. Chem. Soc., 2008, 130, 10848 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |