
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
ITQ-54: a multi-dimensional extra-large pore zeolite with 20 × 14 × 12-ring channels†
Jiuxing
Jiang‡
a,
Yifeng
Yun‡
b,
Xiaodong
Zou
*b,
Jose Luis
Jorda
a and
Avelino
Corma
*a
aInstituto de Tecnología Química (UPV-CSIC), Universidad Politécnica de Valencia – Consejo Superior de Investigaciones Científicas, Av. de los Naranjos s/n, 46022 Valencia, Spain. E-mail: acorma@itq.upv.es
bBerzelii Center EXSELENT on Porous Materials and Inorganic and Structural Chemistry, Department of Materials and Environmental Chemistry, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: xzou@mmk.su.se
First published on 8th October 2014
Abstract
A multi-dimensional extra-large pore silicogermanate zeolite, named ITQ-54, has been synthesised by in situ decomposition of the N,N-dicyclohexylisoindolinium cation into the N-cyclohexylisoindolinium cation. Its structure was solved by 3D rotation electron diffraction (RED) from crystals of ca. 1 μm in size. The structure of ITQ-54 contains straight intersecting 20 × 14 × 12-ring channels along the three crystallographic axes and it is one of the few zeolites with extra-large channels in more than one direction. ITQ-54 has a framework density of 11.1 T atoms per 1000 Å3, which is one of the lowest among the known zeolites. ITQ-54 was obtained together with GeO2 as an impurity. A heavy liquid separation method was developed and successfully applied to remove this impurity from the zeolite. ITQ-54 is stable up to 600 °C and exhibits permanent porosity. The structure was further refined using powder X-ray diffraction (PXRD) data for both as-made and calcined samples.
Introduction
Zeolites have attracted great interest because of their uniformly-sized pore structures in molecular dimensions, and they have wide industrial applications in catalysis, ion exchange, gas adsorption and separation.1–3 Zeolites with extra-large pore sizes (defined by >12 TO4 tetrahedra, T = Si, Al or Ge etc.) are desirable in order for large molecules to be able to diffuse through the channels. In particular, the presence of interconnected large and extra-large channels with different pore sizes and shapes in the same zeolite structure would be interesting, as it would allow both shape selectivity and fast diffusion of large molecules through the channels. Great efforts have been made to synthesize new zeolites,4–6 especially those with extra-large pore openings, multi-dimensional channels and low framework densities. However, extra-large pore zeolites are still very rare,7,8 especially zeolites with extra-large channels in more than one direction. The only examples are found in the gallophosphate cloverite and its aluminophosphate and silicogermanate analogues (-CLO, 20 × 20 × 20-ring),9–11 germanosilicates ITQ-37 (-ITV, gyroidal 30-ring channel),12 and ITQ-40 (16 × 15 × 15-ring).13 In addition, there are only three reported zeolite frameworks that have pore openings ≥20-rings, namely -CLO, -ITV and ITQ-43 (28 × 12 × 12-ring).14Zeolites are often synthesised as polycrystalline powders. Powder X-ray diffraction (PXRD) can be used for structure solution, if the sample is pure and the structure is not too complicated. However, severe peak overlapping due to large unit cell and low symmetry precludes in most cases the structure elucidation by this technique. Electron diffraction (ED) has many advantages compared to X-ray diffraction for structure solution of nano- and micrometer-sized crystals. Crystals considered as powders by X-ray diffraction behave as single crystals by electron diffraction, which makes electron diffraction a promising technique for structural study.15,16 There is no ambiguity for unit cell and space group determination. Although ED data from zone-axis patterns has been used for structure solution of unknown zeolites,17,18 both the data collection and data processing are very demanding and require experts in electron crystallography. In addition, the ED data is incomplete and suffers severe dynamical effects.
Recently two methods for automated collection of three-dimensional electron diffraction data, automated diffraction tomography (ADT)19 and rotation electron diffraction (RED),20–22 have been developed. Both methods allow collection of almost complete 3D ED data sets from a nano- or micrometer-sized crystal starting with an arbitrary orientation. These methods not only make the structure solution from electron diffraction faster and more feasible, but also reduce dynamical effects. Several novel zeolite structures have been solved by the ADT and RED methods; for example a silicoaluminophosphate ITQ-51 with 16-ring channels,23 a germanosilicate PKU-16 with 11 × 11 × 12-ring channels,24 and a germanosilicate ITQ-43 with hierarchical meso-micropores.14 Here we report the silicogermanate ITQ-54 containing three-dimensional intersecting 20 × 14 × 12-ring channels. We show the power of the RED method in structure determination of unknown crystals from multiphasic samples.
Results and discussion
The novel zeolite ITQ-54 was obtained using the N,N-dicyclohexylisoindolinium (DCHI) cation as the organic structure directing agent (OSDA). This cation was synthesised from 1,2-bis(bromomethyl)benzene and N,N-dicyclohexylamine (see ESI S2†). The zeolite was obtained after crystallisation at 200 °C for two days from a synthesis gel of composition:0.62 SiO2:0.38 GeO2:0.29 OSDAOH:0.25 NH4F:1.5 H2O.
Elemental analysis shows a significant difference between the experimental C/N ratio measured in the sample (C/N = 14.4) and the theoretical value of the OSDA introduced in the synthesis gel (C/N = 20). This implies that the OSDA decomposed prior to its incorporation to the zeolite. Gas chromatography-mass spectrometry (GC-MS) and solid state 13C MAS-NMR of ITQ-54 show that the organic species occluded in the zeolite is the N-cyclohexylisoindolinium (CHI) cation (see Experimental section). The protonated CHI was generated in situ from DCHI during the synthesis, which seems to be the real OSDA to direct the synthesis of ITQ-54. However, synthesis attempts by using CHI as the sole template or as the co-template only produced even more GeO2 in the final product. When the Ge/Si ratio was decreased, the crystallinity of ITQ-54 also decreased, while the GeO2 content remained almost constant. So far, ITQ-54 can only be synthesised as an impure form by in situ decomposition of DCHI.
The structure of ITQ-54 was determined using the RED method from an as-made sample containing GeO2 as the impurity (see Experimental section). 594 selected area electron diffraction (SAED) frames were collected from an ITQ-54 crystal of ca. 1 μm in size on a JEOL JEM2100 TEM using the software RED-Data Collection,22 with a tilt step of 0.20° and a tilt range of 109.31°. The SAED frames were combined to reconstruct the 3D reciprocal lattice of ITQ-54 using the software RED-Data Processing.22 The RED data shows that ITQ-54 has an I-centered lattice with unit cell parameters a = 26.59 Å, b = 24.93 Å, c = 15.94 Å, α = 89.91°, β = 90.83°, γ = 90.09°. The unit cell parameters were further refined against the PXRD data to be a = 26.9678(13) Å, b = 25.5772(13) Å, c = 16.2577(8) Å, α = β = γ = 90°. The reflection conditions were obtained from the 2D slices h0l, hk0 and 0kl cut from the 3D reciprocal lattice reconstructed from the RED data, as shown in Fig. 1. The possible space groups were deduced from the reflection conditions to be Immm, Imm2, Im2m, I222 and I212121. The structure of ITQ-54 was solved by direct methods using the highest symmetry, Immm. All nine symmetry-independent T-atoms (T = Si/Ge) and 21 symmetry-independent oxygen atoms in the framework were located directly. An additional peak was found in the center of a double 4-ring (D4R) in the difference Fourier map and assigned as fluorine anion.
 | ||
| Fig. 1 (a) 3D reciprocal lattice of ITQ-54 reconstructed from the RED data. The crystal from which the RED data was collected is shown in the insert. The intensity distribution in the h0l (b), hk0 (c) and 0kl (d) planes show 2mm symmetry, indicating that the crystal is orthorhombic. The reflection conditions can be deduced from the RED data as hkl: h + k + l = 2n, hk0: h + k = 2n, h0l: h + l = 2n, 0kl: k + l = 2n, h00: h = 2n, and 00l: l = 2n. The possible space groups are Immm, Imm2, Im2m, I222 and I212121. | ||
All of the nine T atoms are tetrahedrally-coordinated to oxygen; eight are four-connected to other T-atoms and one is three-connected leaving a terminal hydroxyl group. The structure model was refined against the RED data and converged with R1 = 0.317 for 1913 symmetry-independent reflections. The RED data collection, crystal data and structure refinement details are given in Table S1.† The complete crystal structure determination of ITQ-54 by the RED method demonstrates its power for studying sub-micrometer sized crystals and samples containing impurities.
The comparison between 29Si cross polarisation (CP) MAS-NMR and 29Si Bloch decay (BD) MAS-NMR spectra (Fig. S12†) of the as-made sample shows the presence of Q3 species. This confirms the presence of terminal Si–OH groups as determined from the RED data. The 19F MAS-NMR spectrum (Fig. S13†) shows a single resonance band at −11.42 ppm, indicating that fluorine anions are trapped in the D4Rs, which also agrees with the structure model of ITQ-54 determined from the RED data.
The current RED data still suffers dynamical effects which gave a high R1 value in the structure refinement. In order to further confirm the structure model and obtain more accurate atomic positions, the structure needs to be refined against the PXRD data. Despite our efforts of synthesis optimisation, zeolite ITQ-54 was always obtained with a certain amount of GeO2. Thus we developed a separation method based on the different densities of the two compounds, using a heavy liquid to remove the GeO2 impurity as described below. In general, it is not unusual to obtain impurities such as amorphous, dense phases and other zeolitic phases during the synthesis of zeolites.25–27 Although impurities can be eliminated in most cases by optimizing the synthesis conditions, this approach sometimes fails. To the best of our knowledge, there are no reports about simple and general laboratory methods to eliminate impurities in zeolite samples. The only method to obtain a pure sample for property analyses is to pick out the crystals by hand under a microscope, based on the different crystal morphologies. However, this method is only applicable when crystals are both large and have well-defined shapes.25,26 Meanwhile, solid separation methods have already been used for decades in the mineral industry. Different minerals can be separated based on the difference of their densities using a heavy liquid media; phases with densities greater than the liquid will sink down, while those with densities less than the liquid will float to the surface. We found that a 1,1,2,2-tetrabromoethane (TBE)–ethanol solution (TBE, 2.967 g cm−3 Muthmann's solution) was suitable for the complete removal of GeO2 from the ITQ-54 sample, taking advantage of the large difference in the densities of ITQ-54 (2.18 g cm−3) and GeO2 (4.25 g cm−3). A pure ITQ-54 sample was successfully obtained by this approach. Details of the separation method are described in the Experimental section S5.†
Synchrotron PXRD data was collected from the as-made pure ITQ-54 sample (λ = 0.413686 Å). The structure model obtained by RED was refined against the synchrotron PXRD data using the program TOPAS Academic 4.1.28 The final refinement converged with Rwp = 0.0678, Rexp = 0.0535 and RB = 0.0216, see Fig. S4 and Table S2.† All of the bond distances and angles in the refined structure of ITQ-54 are chemically reasonable. More details about the Rietveld refinement are described in the ESI.†
The OSDA could be completely removed by a slow stepwise calcination procedure while retaining the ITQ-54 framework, as described in the Experimental section S5.† Finally a calcined pure ITQ-54 sample was obtained, and its PXRD pattern was collected. Rietveld refinement was performed using the program FULLPROF,29 which converged with Rwp = 0.095, Rexp = 0.035, RB = 0.038 and RF = 0.041. The chemical formula of the calcined ITQ-54 is Si0.51Ge0.49O1.97(OH)0.06. The Rietveld plots are shown in Fig. 2 and crystallographic data is given in Table S3.†
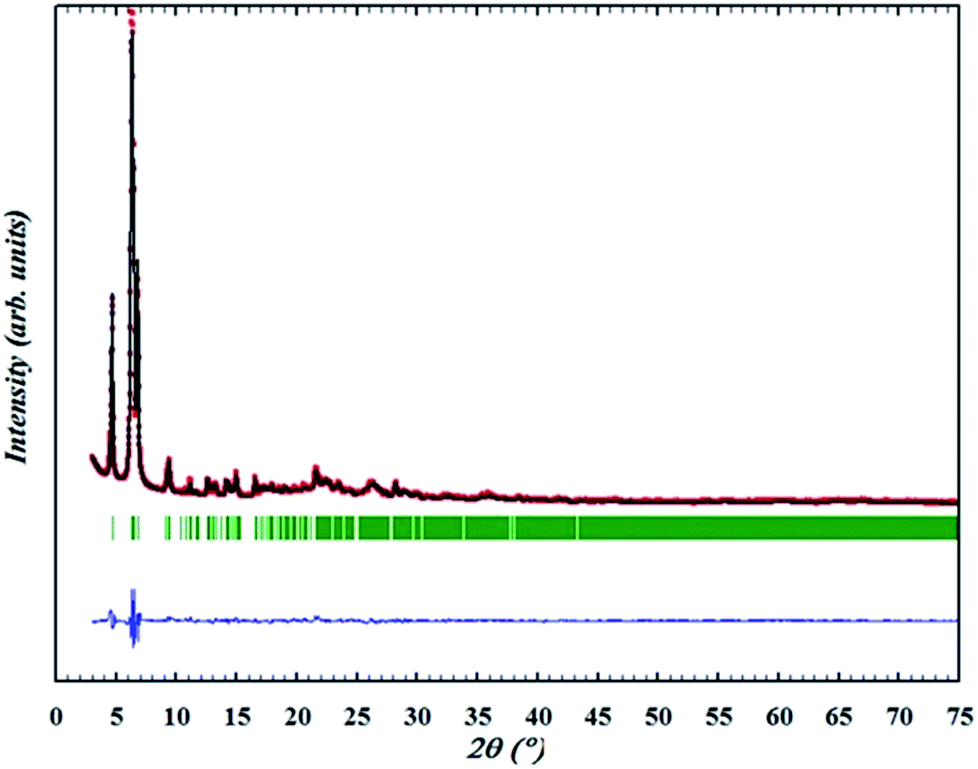 | ||
| Fig. 2 Rietveld refinement of the PXRD pattern of ITQ-54 upon calcination at 873 K (data collected at 303 K, Cu Kα radiation, λ = 1.5418 Å). Data points show the observed PXRD pattern; the line along these points is the calculated pattern, with the difference profile at the bottom. The vertical tick marks below the pattern give the positions of the Bragg reflections. | ||
The framework structure of ITQ-54 is built from a novel [435661] composite building unit (CBU) consisting of 16 TO4 tetrahedra (Fig. 3a). One of the TO4 tetrahedra in the [435661] CBU is three-connected, leaving a terminal –OH group. Each [435661] CBU connects to three neighbouring [435661] CBUs via their 4-rings to form two D4Rs and one 4–2 unit (Fig. 3b), defining a layer containing 12-rings in the bc-plane (Fig. 3c). Each layer is further connected to its closest neighbouring layers via additional D4Rs to form a 3D framework with tri-directional 20 × 14 × 12-ring channels (Fig. 3d and e). The free diameters are 10.3 Å × 5.8 Å for the 20-ring channels along the c-axis, 9.8 Å × 6.6 Å for the 14-ring channels along the b-axis and 7.2 Å × 6.8 Å for the 12-ring channels along the a-axis, as shown in Fig. S6.† ITQ-54 has a framework density of 11.1 T-atoms per 1000 Å3, which is one of the lowest among zeolites.9,10,12,13,30–32
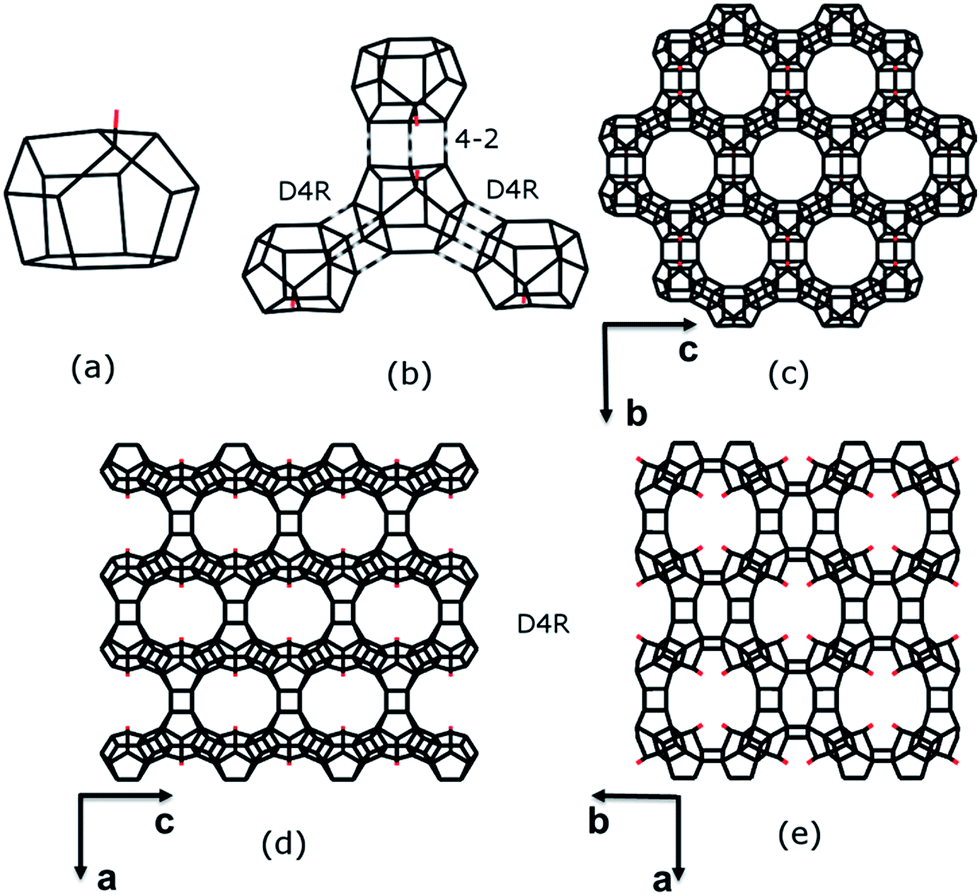 | ||
| Fig. 3 Construction of the framework structure of ITQ-54. (a) The new [435661] CBU. (b) Connectivity of the new CBU to the other three CBUs via D4R, D4R and 4–2 units, respectively, to form (c) a layer in the bc-plane with 12-ring pores. (d and e) The 12-ring layers connect to each other via D4Rs to form a 3D framework. 14-ring channels are created along the b-axis (d) and 20-ring channels are created along the c-axis (e). Only T–T connections and the terminal O atoms (in red) are shown. | ||
ITQ-54 has a new zeolite topology, with intersecting extra-large channels (20- and 14-ring channels). The coordination sequences and vertex symbols are given in Table S5.† The channel system and cavities are better illustrated by natural tiling, where the intersection of the 20- and 14-ring channels as well as the 20-ring and 12-ring channels can be clearly seen by tracing the colours of the tiles (Fig. 4 and S7†). The framework can be described by a four-coordinated net crb with each node occupied by a [435661] CBU (Fig. S8†).
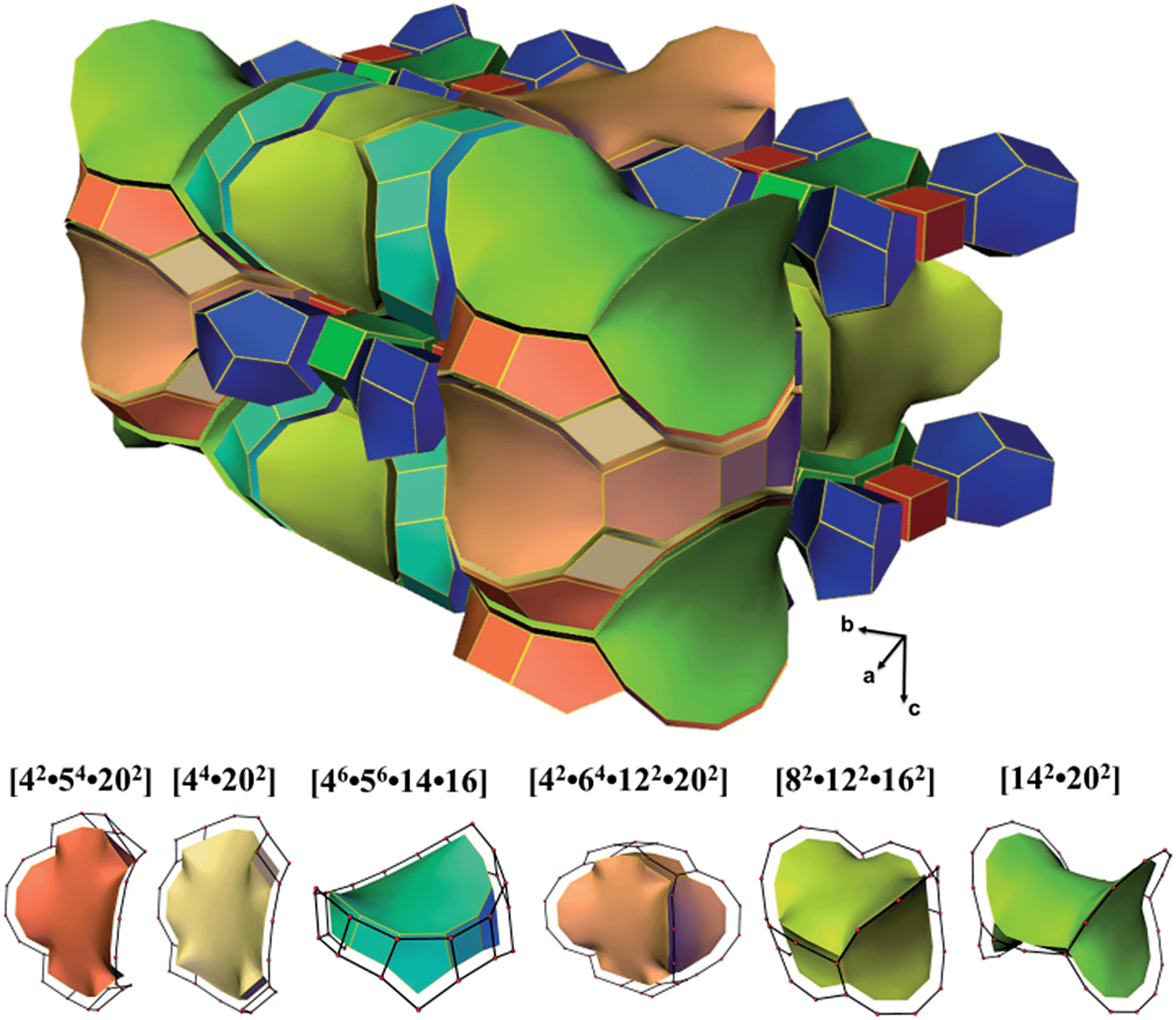 | ||
| Fig. 4 Illustration of the channel system and cavities in ITQ-54 by natural tiling. More descriptions are given in the ESI.† | ||
ITQ-54 is stable up to 600 °C and exhibits permanent porosity. Like most other zeolites with ≥20-rings, ITQ-54 contains germanium and the calcined sample is not stable in the presence of moisture. For practical applications, it is necessary to substitute Ge by Si and/or Al through direct or post-synthesis methods. These have been demonstrated in several germanosilicates.33–35 Similar procedures are promising to improve the thermal stability of ITQ-54, which is helpful for its use as a catalyst to process bulk molecules.
Conclusions
In conclusion, a new zeolite ITQ-54 with extra-large pores has been synthesised. The zeolite synthesis was directed by in situ decomposition of the OSDA. Its structure was solved using the rotation electron diffraction (RED) method from a multiphasic sample. The structure of ITQ-54 contains straight intersecting 20 × 14 × 12-ring channels along the three crystallographic axes. It is one of the few zeolites with extra-large channels in more than one direction. The framework density of ITQ-54 also has one of the lowest framework densities among the known zeolites. The multi-dimensional extra-large channels in ITQ-54 can facilitate the diffusion of large molecules through the channels, which is especially interesting for applications in separation and catalysis. The hydroxyl groups pointing towards the channels can be used for further functionalisation of the material. We have successfully applied a separation method based on the use of a heavy liquid to remove GeO2 and obtain pure ITQ-54 samples. We show the power of the RED method in the structure solution of submicrometer-sized crystals, and in discovering new structures in multi-phasic samples. This is important for the targeting of interesting materials so that the synthesis conditions can be optimised according to the structures.Experimental sections
Synthesis
In a typical synthesis of zeolite ITQ-54, Ludox (SiO2, AS-40, 40% Sigma-Aldrich), OSDA (8–16% in water solution), GeO2 (99.998%, Sigma-Aldrich) and NH4F (99.99% Aldrich, 10 wt% in water solution), were introduced into 3 ml Teflon vials and stirred for ∼3 h under infrared lamp heating. The air temperature around the synthesis gel was controlled to be within 80–90 °C using a thermometer, and the intensity of infrared light was adjusted to ensure a proper decomposition of the OSDA and evaporation of water until the desired water content was achieved. Then, the Teflon vials were inserted into the 3 × 5 multiautoclave and the crystallisation was carried out at 200 °C for two days in static conditions. Crystalline products were washed with abundant quantities of water and acetone prior to drying at 100 °C. The samples were characterised by powder X-ray diffraction; all of the as-made samples contained some GeO2 impurity which could not be avoided regardless of synthesis optimisation. The initial sample used for structure elucidation by electron crystallography was synthesised in a gel composition of 0.62SiO2:0.38GeO2:0.29OSDAOH:0.25NH4F:1.5H2O. The synthesis was optimised into a gel composition of 0.68SiO2:0.32GeO2:0.25OSDAOH:0.23NH4F:1.1H2O and the resulting sample was used for the study of GeO2 removal and further characterisations.Compared with previous silicogermanate zeolite syntheses, the synthesis of ITQ-54 needs a relative high temperature (80–90 °C) during the gel preparation to induce the proper decomposition of the OSDA. If the heating temperature is not high enough, the product either contains BEC impurity or transforms completely to the BEC zeolite. If the heating temperature is too high, the product becomes trigonal GeO2.
Analysis of occluded organic species in ITQ-54 by GC-MS and 13C NMR
In order to know which organic species are occluded, 2 g of as-made ITQ-54 were dissolved with 37% HCl, and the organic species was extracted with 100 ml of chloroform. The organic layer was first washed by saturated K2CO3 and anhydrous MgSO4 solutions, and then analysed by gas chromatography-mass spectrometry (GC-MS). One major peak (73.9% estimated by area integration) and several minor peaks (total 26.1%) were observed in the gas chromatography spectra. The main peaks can be recognised as N,N-cyclohexylisoindolinium (CHI) by comparing the GC-MS spectra with synthetic CHI (Fig. S9†). The solid state 13C MAS-NMR of ITQ-54 and the liquid 13C NMR of the protonated CHI are in good agreement (Fig. S10†), as well as the C/N ratio (experimental = 14.4, theoretical = 14).Structure determination by RED
Powders of the as-made ITQ-54 were crushed, dispersed in absolute ethanol and treated by sonication for 2 minutes. Then, a droplet of the suspension was transferred onto a copper grid. The TEM sample was observed on a JEOL JEM-2100 microscope operated at 200 kV using a single-tilt tomography sample holder. The RED data sets were collected using the software package RED-data collection.22 594 SAED frames were collected covering a tilt range of 109.31°. Energy-dispersive spectroscopy (EDS) was carried out on the same crystals after the RED data collection to determine the Si/Ge ratio. The SAED frames were combined to reconstruct the 3D reciprocal lattice using the software RED-data processing,22 which performed shift correction, peak search, unit cell determination, indexation of the reflections and intensity extraction. A list of hkl intensity was extracted from the RED data and used for the structure solution by direct methods, followed by the structure refinement. The intensities I(hkl) obtained from RED were directly used as |F(hkl)|2 for structure determination, i.e. using kinematical approximation. Final structure refinement was performed using the SHELXL-97 program by minimising the sum of squared deviations of F2 using a full-matrix technique. All of the T (T = Si/Ge), O and F atoms were refined isotropically.Separation of ITQ-54 and GeO2
The separation was performed by the heavy liquid method based on the different densities of the zeolite and the GeO2 impurity. A TBE–ethanol solution (density 2.21 g cm−3) was chosen as a working medium. Before the separation, the sample was dispersed in ethanol to break the aggregation of both species (see ESI†). After a first separation in a 2.21 g cm−3 (87.1%) TBE–ethanol solution, the top layer (2.21 g) and bottom layer (0.21 g) were collected separately. The GeO2 content in the top layer decreased from 8.7% to 1.5%, based on density contribution. The separation procedure was then repeated twice, with the GeO2 content decreased to 0.97% and <0.5%, respectively. In fact, in the PXRD pattern of the last sample, no diffraction peaks from GeO2 could be observed.Powder X-ray diffraction
Synchrotron PXRD data was collected from the as-made pure ITQ-54 sample on the beamline 11-BM at the Advanced Photon Source, Argonne National Laboratory (λ = 0.413686 Å). PXRD data of the calcined pure ITQ-54 was collected on a PANalytical X'Pert PRO diffractometer from a sample obtained through direct in situ calcination of the as-made pure ITQ-54 sample in an Anton Parr XRK-900 chamber, at a heating rate of 3 °C min−1. The sample was kept at constant temperatures for 2 hours at 100, 150, 200, 250 and 300 °C, and for 4 hours at 350, 400, 450, 500, 550 and 600 °C. The PXRD pattern was collected after the sample being cooled down to room temperature.Topology analysis
The nets of ITQ-54 were computed using TOPOS.36,37 The visualisation of nets and tiling was generated using 3dt.38Acknowledgements
This work was supported by the Spanish Government (MAT2012-38567-C02-01, Consolider Ingenio 2010-Multicat CSD-2009-00050 and Severo Ochoa SEV-2012-0267), Generalitat Valenciana (Project Prometeo), the Swedish Research Council (VR), the Swedish Governmental Agency for Innovation Systems (VINNOVA) and the Knut & Alice Wallenberg Foundation through a grant for purchasing the TEMs and the project grant 3DEM-NATUR. Y. F. Yun thanks the China Scholarship Council (CSC). The authors acknowledge ESRF, Grenoble for the use of the beamline ID31 and the mail-in system of 11-BM at the Advanced Photon Source, Argonne National Laboratory, USA.Notes and references
- M. E. Davis, Nature, 2002, 417, 813–821 CrossRef CAS PubMed.
- P. Atienzar, M. J. Díaz-Cabañas, M. Moliner, E. Peris, A. Corma and H. García, Chem.–Eur. J., 2007, 13, 8733–8738 CrossRef CAS PubMed.
- A. Corma, J. Catal., 2003, 216, 298–312 CrossRef CAS.
- Z. Wang, J. Yu and R. Xu, Chem. Soc. Rev., 2012, 41, 1729–1741 RSC.
- Y. Li, J. Yu and R. Xu, Angew. Chem., Int. Ed., 2013, 52, 1673–1677 CrossRef CAS PubMed.
- X. Meng and F.-S. Xiao, Chem. Rev., 2014, 114, 1521–1543 CrossRef CAS PubMed.
- C. Baerlocher and L. McCusker, 2014, Database of Zeolite Structures:http://www.iza-structure.org/databases/, (accessed August 20, 2014).
- J. Jiang, J. Yu and A. Corma, Angew. Chem., Int. Ed., 2010, 49, 3120–3145 CrossRef CAS PubMed.
- M. Estermann, L. B. McCusker, C. Baerlocher, A. Merrouche and H. Kessler, Nature, 1991, 352, 320–323 CrossRef CAS.
- J. Su, Y. Wang, J. Lin, J. Liang, J. Sun and X. Zou, Dalton Trans., 2013, 1360–1363 RSC.
- Y. Wei, Z. Tian, H. Gies, R. Xu, H. Ma, R. Pei, W. Zhang, Y. Xu, L. Wang, K. Li, B. Wang, G. Wen and L. Lin, Angew. Chem., Int. Ed., 2010, 49, 5367–5370 CrossRef CAS PubMed.
- J. Sun, C. Bonneau, Á. Cantín, A. Corma, M. J. Díaz-Cabañas, M. Moliner, D. Zhang, M. Li and X. Zou, Nature, 2009, 458, 1154–1157 CrossRef CAS PubMed.
- A. Corma, M. J. Díaz-Cabañas, J. Jiang, M. Afeworki, D. L. Dorset, S. L. Soled and K. G. Strohmaier, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 13997–14002 CrossRef CAS PubMed.
- J. Jiang, J. L. Jorda, J. Yu, L. A. Baumes, E. Mugnaioli, M. J. Diaz-Cabanas, U. Kolb and A. Corma, Science, 2011, 333, 1131–1134 CrossRef CAS PubMed.
- L. McCusker and C. Baerlocher, Z. Kristallogr., 2013, 228, 1–10 CAS.
- T. Willhammar, Y. Yun and X. Zou, Adv. Funct. Mater., 2014, 24, 182–199 CrossRef CAS.
- D. L. Dorset, Z. Kristallogr., 2003, 218, 525–530 CAS.
- P. Wagner, O. Terasaki, S. Ritsch, J. G. Nery, S. I. Zones, M. E. Davis and K. Hiraga, J. Phys. Chem. B, 1999, 103, 8245–8250 CrossRef CAS.
- U. Kolb, T. Gorelik, C. Kübel, M. T. Otten and D. Hubert, Ultramicroscopy, 2007, 107, 507–513 CrossRef CAS PubMed.
- D. Zhang, P. Oleynikov, S. Hovmöller and X. Zou, Z. Kristallogr., 2010, 225, 94–102 CrossRef CAS.
- X. Zou, S. Hovmoller and P. Oleynikov, Electron Crystallography: Electron Microscopy and Electron Diffraction, Oxford University Press, 2011 Search PubMed.
- W. Wan, J. Sun, J. Su, S. Hovmoller and X. Zou, J. Appl. Crystallogr., 2013, 46, 1863–1873 CAS.
- R. Martinez-Franco, M. Moliner, Y. Yun, J. Sun, W. Wan, X. Zou and A. Corma, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3749–3754 CrossRef CAS PubMed.
- W. Hua, H. Chen, Z.-B. Yu, X. Zou, J. Lin and J. Sun, Angew. Chem., Int. Ed., 2014, 53, 5868–5871 CrossRef CAS PubMed.
- T. Conradsson, M. S. Dadachov and X. D. Zou, Microporous Mesoporous Mater., 2000, 41, 183–191 CrossRef CAS.
- Z. Liu, X. Song, J. Li, Y. Li, J. Yu and R. Xu, Inorg. Chem., 2012, 51, 1969–1974 CrossRef CAS PubMed.
- L. Tang, L. Shi, C. Bonneau, J. Sun, H. Yue, A. Ojuva, B.-L. Lee, M. Kritikos, R. G. Bell, Z. Bacsik, J. Mink and X. Zou, Nat. Mater., 2008, 7, 381–385 CrossRef CAS PubMed.
- R. A. Young, The Rietveld Method, Oxford University Press, 1995 Search PubMed.
- J. Rodríguez-Carvajal, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 192, 55–69 CrossRef.
- N. Zheng, X. Bu, B. Wang and P. Feng, Science, 2002, 298, 2366–2369 CrossRef CAS PubMed.
- Y. Xu, Y. Li, Y. Han, X. Song and J. Yu, Angew. Chem., Int. Ed., 2013, 52, 5501–5503 CrossRef CAS PubMed.
- Y. Han, Y. Li, J. Yu and R. Xu, Angew. Chem., Int. Ed., 2011, 50, 3003–3005 CrossRef CAS PubMed.
- F. Gao, M. Jaber, K. Bozhilov, A. Vicente, C. Fernandez and V. Valtchev, J. Am. Chem. Soc., 2009, 131, 16580–16586 CrossRef CAS PubMed.
- H. Xu, J. Jiang, B. Yang, L. Zhang, M. He and P. Wu, Angew. Chem., Int. Ed., 2014, 53, 1355–1359 CrossRef CAS PubMed.
- L. Burel, N. Kasian and A. Tuel, Angew. Chem., Int. Ed., 2014, 53, 1360–1363 CrossRef CAS PubMed.
- V. A. Blatov, A. P. Shevchenko and D. M. Proserpio, Cryst. Growth Des., 2014, 14, 3576–3586 CAS.
- V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS.
- O. Delgado-Friedrichs, Theor. Comput. Sci., 2003, 303, 431–445 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis, structure determination by RED, separation using the heavy liquid method, Rietveld refinement against PXRD of as-made and calcined samples, topology analysis, textural properties, 29Si and 19F solid state NMR, TG analysis and CIFs. CCDC 1021735. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc02577f |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2015 |