DOI:
10.1039/C4SC02463J
(Edge Article)
Chem. Sci., 2015,
6, 412-417
Metallic behaviour of acid doped highly conductive polymers†
Received
13th August 2014
, Accepted 9th September 2014
First published on 9th September 2014
Abstract
Conductive polymers such as poly(3,4-ethylenedioxythiophene) (PEDOT) are used in a wide range of applications as transparent electrodes, hole injecting layers or thermoelectric materials for room-temperature applications. However, progress is needed to enhance the electrical conductivities of the materials and to provide understanding about their structure–transport relationships. This work presents the synthesis of highly conductive PEDOT-based polymers using iron(III) trifluoromethanesulfonate as oxidant for the first time. The metallic behaviour of the polymer is revealed by conductivity monitoring from 3 to 300 K. The electrical conductivity is further improved (to 2273 S cm−1) using acids, leading to a positive temperature coefficient of resistivity at an unprecedented 45.5% oxidation state. X-ray photoemission spectroscopy (XPS) and time of flight-secondary ion mass spectrometry (ToF-SIMS) analyses demonstrate a complete replacement of the trifluoromethanesulfonate anions by hydrogen sulphate counter ions. This substitution results in an increased concentration of charge carriers (measured in organic electrochemical transistors) along with an enhancement of the mean size of crystalline domains, highlighted by small and wide angle X-ray scattering (SAXS/WAXS), which explains the 80% increase of electrical conductivity.
Introduction
As one of the most easily processable conductive polymers, poly(3,4-ethylenedioxythiophene) (PEDOT) in combination with poly(styrenesulfonate) (PSS) has focused a lot of scientific interest in the past years.1 Its electrical conductivity, σ, has been increased by chemical treatments inducing the partial removal of the excess of PSS, which acts both as dopant and insulating material.2–6 In addition, a successful route to obtain PEDOT has recently been demonstrated through the polymerization of 3,4-ethylenedioxythiophene (EDOT) using iron(III) para-toluenesulfonate (Fe(OTs)3) as oxidative agent and poly(ethylene glycol)-block-poly(propylene glycol)-block-poly(ethylene glycol) (PEG-PPG-PEG) as surfactant.7 The low molecular disorder in such PEDOT layers induces a semi-metallic conduction mechanism according to Crispin et al.8 Taking these reports into account, we considered the possibility to further improve the conductivity of PEDOT materials by using less hindered and poorly coordinating counter anions, which would optimize both the weight fraction and the oxidation level of PEDOT. Accordingly, the present work demonstrates the oxidative chemical polymerization of EDOT using iron(III) trifluoromethanesulfonate (Fe(OTf)3) through a method involving PEG-PPG-PEG. The resulting materials display metallic properties that are additionally strengthened by doping with specific acids. In particular, sulphuric acid enhances σ up to 2273 S cm−1 (1.8 fold increase), and induces a positive temperature coefficient of resistivity (TCR). Though this study is focused on PEDOT polymerized with OTf anion (PEDOT:OTf) due to the novelty of this material, it is noteworthy that the doping method performed is also efficient on PEDOT polymerized with the OTs anion (PEDOT:OTs).
Results and discussion
Electrical conductivity and morphology
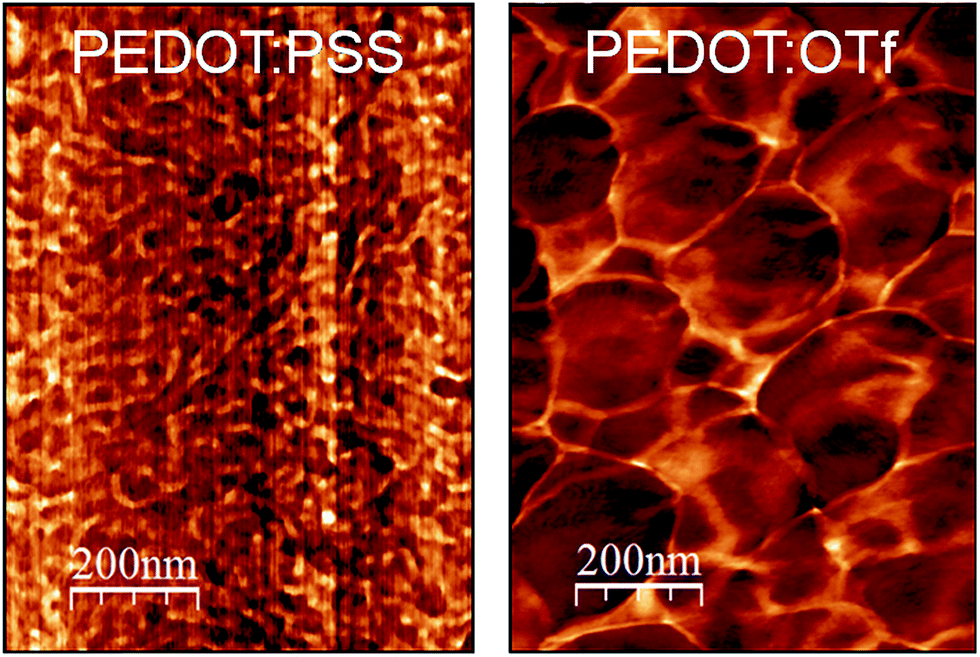
The optimization of the polymerization conditions (Fig. S1†) leads to PEDOT:OTf displaying σ = 1218 S cm−1, whereas the typical electrical conductivity for a spin-coated layer of PEDOT:PSS (used as reference material for morphological and structural discussions in this work) is 1 S cm−1. Atomic force microscopy (AFM) phase contrast images of the surface of PEDOT:OTf and PEDOT:PSS show that PEDOT:OTf has a granular structure with large homogeneous domains, while PEDOT:PSS displays a poorly structured surface, where hard and soft domains are highly entangled (Fig. 1). The amorphous structure of PEDOT:PSS derives from the excess of PSS, resulting in the embedding of PEDOT:PSS grains in a PSS-rich matrix. However, the less sterically hindered OTf ions enable a dense stacking of PEDOT chains resulting in the observed granular structure. The high electrical conductivity of PEDOT:OTf compared with PEDOT:PSS is ascribed to these structural differences, which result in a higher fraction of conductive PEDOT in PEDOT:OTf. The influence of various acid solutions on the electrical conductivity of PEDOT:OTf has been tested. Hydrochloric acid (HCl) and nitric acid (HNO3) induce a downshift of σ to 385 and 740 S cm−1, whereas para-toluenesulfonic (TsOH), trifluoromethanesulfonic (TfOH), and sulphuric (H2SO4) acids increase σ respectively up to 1503, 1613, and 2273 S cm−1. The optimum values of σ are obtained at pH ∼1, whereas when the pH is set below 0.5, the treatment results in the degradation of the polymer layer.
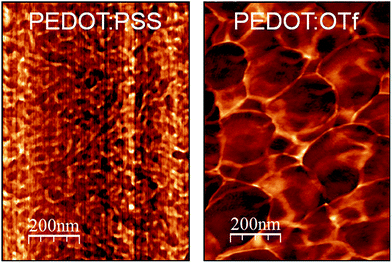 |
| | Fig. 1 AFM phase images of PEDOT:PSS and PEDOT:OTf. | |
Ultraviolet-visible-near infrared spectroscopy
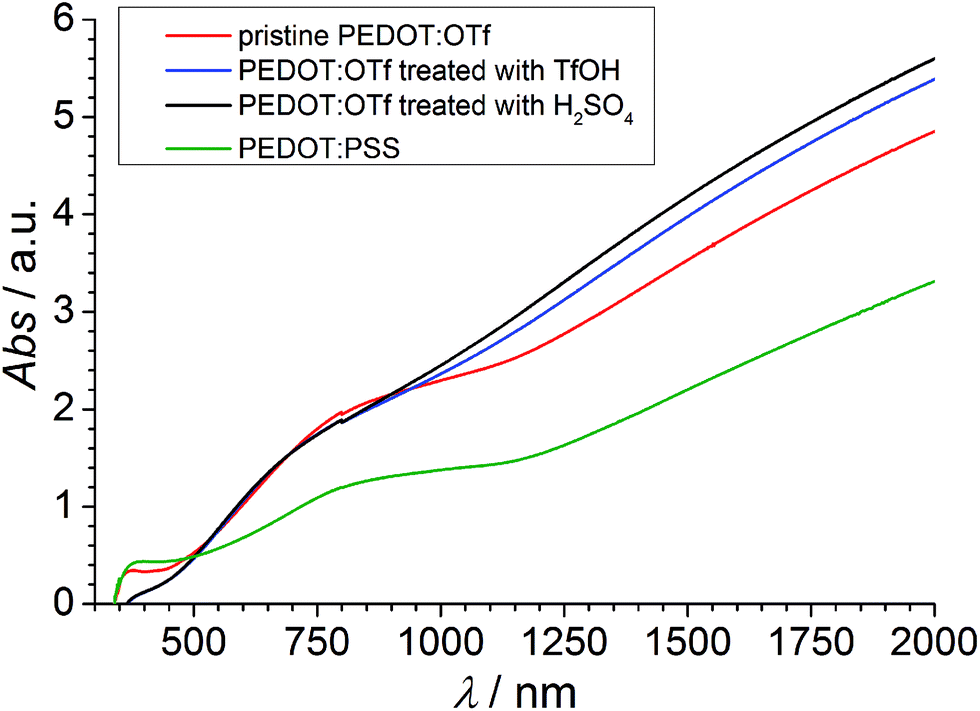
The absorbance spectra of PEDOT:OTf layers in its pristine state, and doped with H2SO4 and TfOH are compared with the absorbance spectrum of a PEDOT:PSS layer (Fig. 2). The NC absorption band (∼590 nm) characterizes the π–π* transition in neutral chains, the RC absorption band (∼750 nm) is distinctive of radical cations and the large DC absorption band (λ > 1250 nm) reveals the presence of dications.9–12 The strong absorbance in RC and DC bands of PEDOT:OTf suggests a higher density of charge carriers compared with PEDOT:PSS, in accordance with phase imaging data. Upon doping with TfOH or H2SO4, the DC band grows wider and its absorbance increases. This observation, along with the increase of σ upon doping, proves the insertion of additional charge carriers in the polymer and an increase of its oxidation level. UV-vis-NIR data showing the influence of other acids are shown in Fig. S2.†
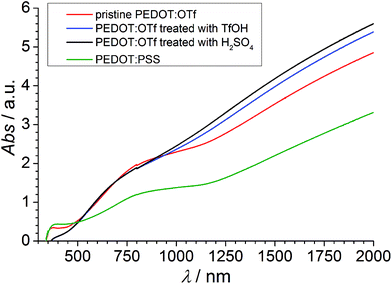 |
| | Fig. 2 UV-vis-NIR spectra of PEDOT:OTf in the pristine state, PEDOT:OTf treated with TfOH and PEDOT:OTf treated with H2SO4, and PEDOT:PSS. | |
X-ray photoelectron spectroscopy
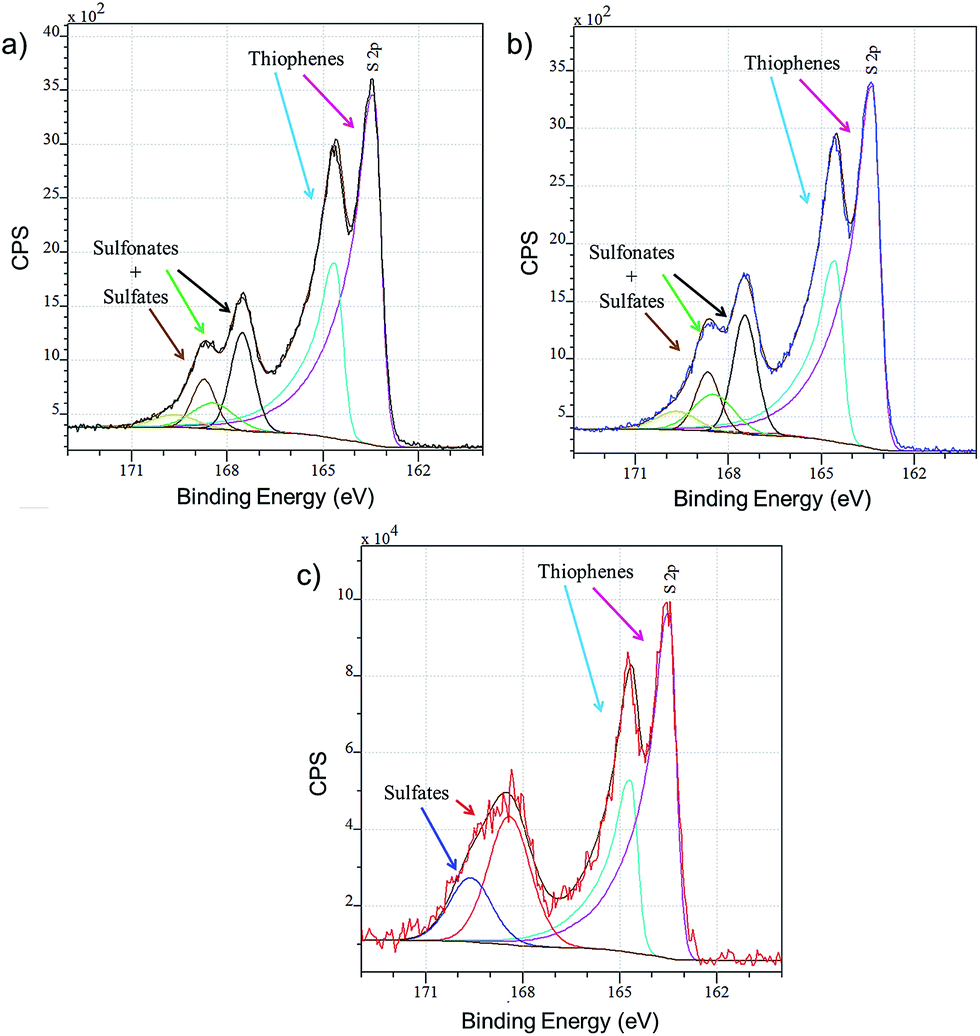
Layers of PEDOT:OTf, PEDOT:OTf doped with TfOH and PEDOT:OTf doped with H2SO4 (called PEDOT:Sulf) are analysed by XPS and the resulting overview spectra are presented in Fig. S3.† The F(1s) signal (688.5 eV), related to fluorinated species, is a good indicator of the presence of OTf ions inside the layer; the intensity of this signal is slightly increased when PEDOT:OTf is doped with TfOH, supporting the hypothesis that new counter ions are inserted in the polymer. In addition, the overview spectra of PEDOT:Sulf has no traces of the F(1s) signal, suggesting that OTf ions have been eliminated from the layer upon doping. The oxidation level of PEDOT layers depends on the ratio of thiophene units to sulphured counter ions; therefore, it can be discussed from the disambiguation of the S(2p) signal related to sulfur.13 For PEDOT:OTf, PEDOT:OTf doped with TfOH, and PEDOT:Sulf (Fig. 3), the S(2p) signal clearly reveals peaks from the thiophene units (163–167 eV), SO3− and HSO4− (167–171 eV). The oxidation level calculated from these contributions is 27.8% for pristine PEDOT:OTf (which is consistent with the oxidation state of pristine PEDOT:OTs reported elsewhere14), and it reaches 35.7% after TfOH doping and 45.5% after H2SO4 doping, which is the highest PEDOT oxidation level ever reported to our knowledge. The increase in the oxidation level supports an increase in the concentration of charge carriers. In the case of H2SO4 doping, HSO4− peaks are the only components of the S(2p) signal in 167–171 eV range. This observation strongly suggests that OTf ions have been replaced by HSO4− upon doping.
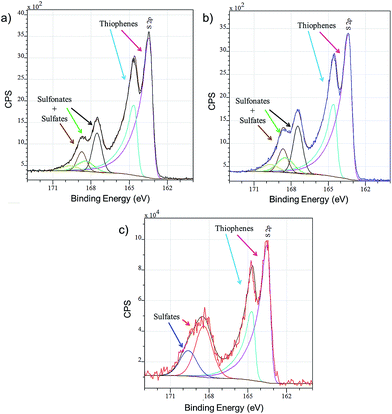 |
| | Fig. 3 XPS S(2p) signals of (a) PEDOT:OTf in the pristine state, (b) PEDOT:OTf treated with TfOH, and (c) PEDOT:Sulf. The disambiguation of the signals is indicated under the experimental curves. | |
Time of flight-secondary ion mass spectroscopy
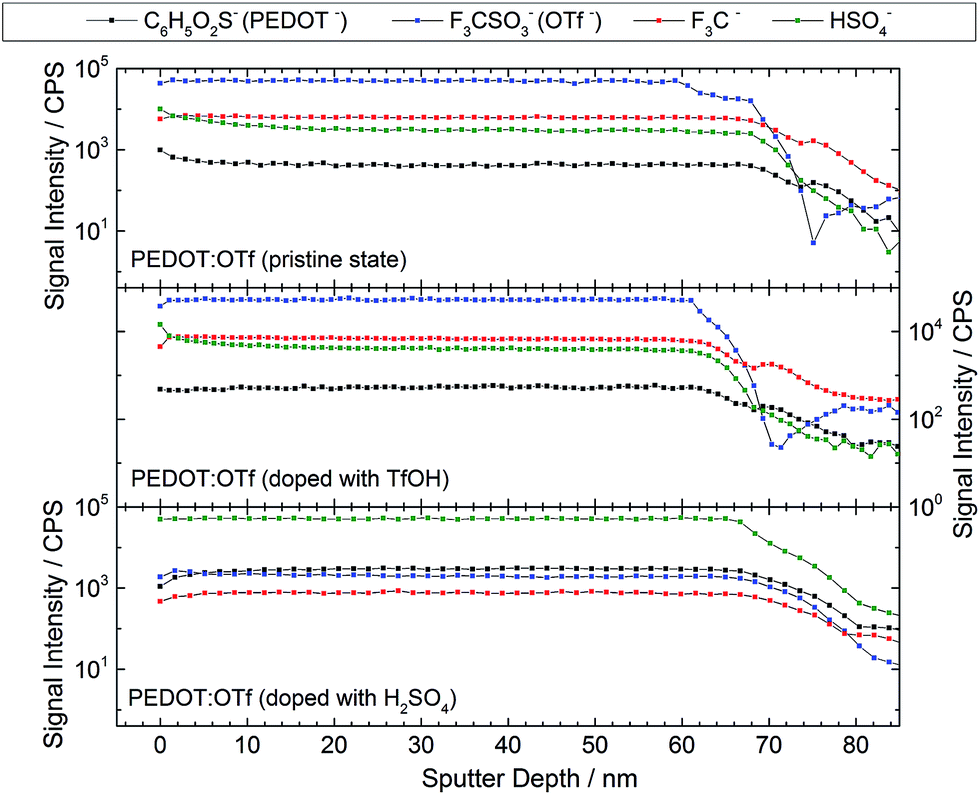
XPS analyses are advantageously combined with ToF-SIMS to probe the homogeneity of the layer at higher thickness ranges. The intensity profiles versus the sputter depth reported in Fig. 4 are those of the most representative ionic species detected during the analysis. The reported species are C6H5O2S− (EDOT−, a fragment of PEDOT macromolecules), OTf, CF3− (an OTf fragment), and HSO4− (either resulting from OTf fragmentation or initially present in the layer). The samples display a homogeneous intensity profile for each ionic species, indicating that the sample composition is uniform with regards to the thickness. Moreover, this confirms that doping reactions take place throughout the polymer layer. All the species signals have a similar intensity when PEDOT:OTf in pristine state was compared with PEDOT:OTf doped with TfOH. In addition, the intensity profiles measured on PEDOT:OTf and PEDOT:Sulf disclose major differences: OTf and CF3− signals are divided by a factor of 154 and 55, respectively, whereas HSO4− signal is multiplied by 23. These observations support the previously stated doping mechanisms, consisting in the replacement of OTf ions with HSO4−, and confirm their occurrence in the entire thickness of the layer.
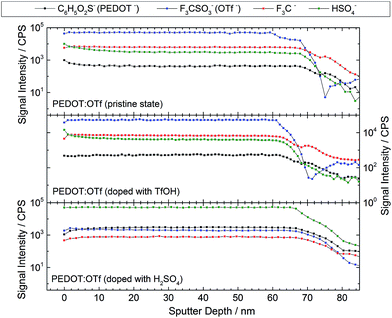 |
| | Fig. 4 ToF-SIMS intensity profiles of different ionic species in ∼65 nm thick layers of PEDOT:OTf in pristine state, PEDOT:OTf treated with TfOH and PEDOT:Sulf. | |
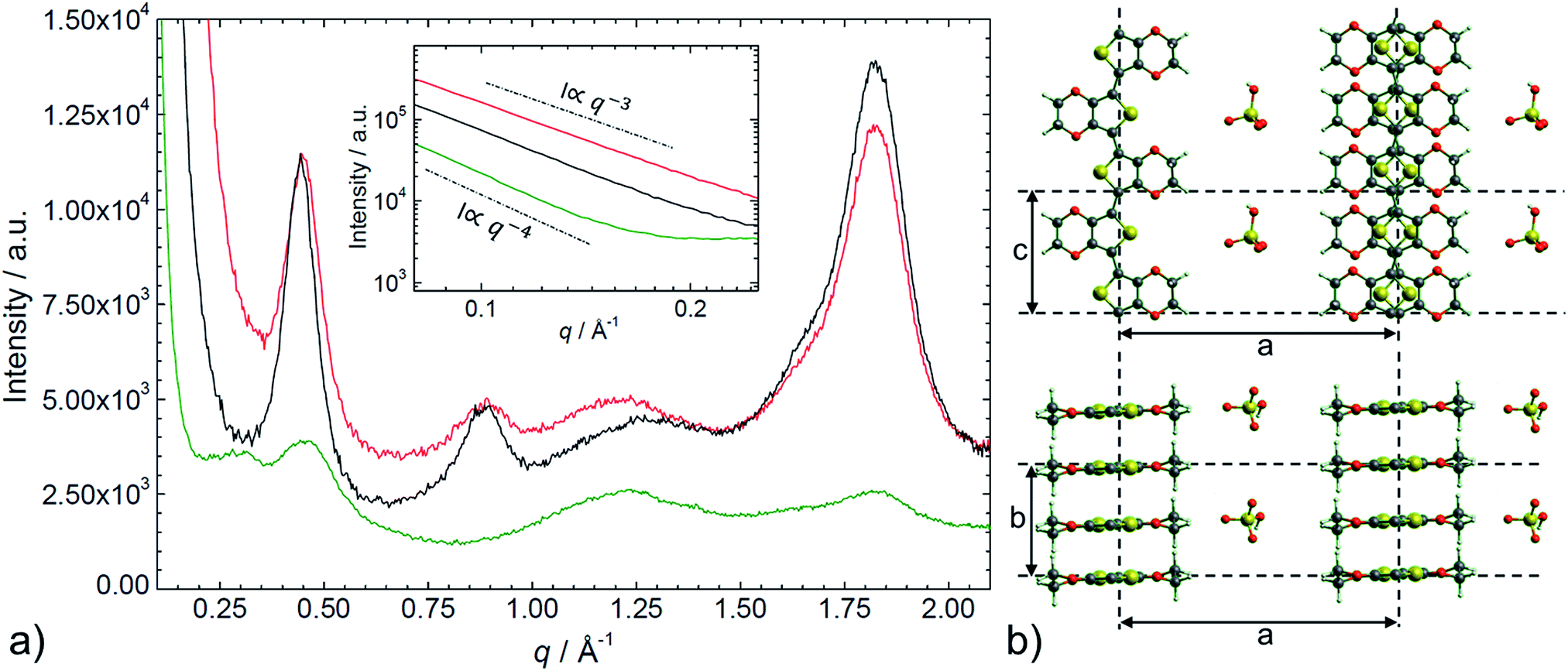
Small and wide angle X-ray scattering
The SAXS/WAXS intensity profiles of thin PEDOT:PSS, PEDOT:OTf, and PEDOT:Sulf films are presented in Fig. 5. To facilitate the reader's understanding, a schematic representation of the crystalline structure of PEDOT:OTf (inspired from previous works on PEDOT:OTs15) is provided in the Fig. 5. PEDOT:PSS intensity profile exhibits two broad humps at q ∼ 0.29 Å−1 (d = 22 Å) and 0.45 Å−1 (d = 14 Å), and two peaks at q = 1.22 Å−1 (d = 5.2 Å) and q = 1.82 Å−1 (d = 3.4 Å). The weak hump centred at 0.29 Å−1 is assigned to the lamellar stacking distance [d(100) = 22 Å] peak occurring from the alternate ordering of the PEDOT and PSS polymer chains. This distance is in agreement with PEDOT and PSS chains widths (7.5 Å and 15.5 Å, respectively).16 The narrow peak at q = 1.82 Å−1 indicates the Bragg reflection from PEDOT oligomers stacking face-to-face along the b-axis with a spacing of 3.4 Å, which is in agreement with the π–π stacking distance [d(010)] of the PEDOT thiophenes.15 From the Scherrer formula17 applied to the 010 diffraction peak, we calculate a crystalline domain size of ∼33 Å along the b-axis. The other peaks at q = 0.46 Å−1 and 1.22 Å−1 are attributed to the interdigitation packing and π–π stacking distance of PSS.15–18 Moreover, for q < 0.2 Å−1, we observed a ∼q−4 power law indicating the contribution of the sharply defined interface separating the PEDOT-rich crystalline grains from the PSS-rich amorphous matrix.19 The intensity profile for PEDOT:OTf and PEDOT:Sulf exhibits several diffraction peaks at q = 0.45 Å−1 (d = 14 Å), 0.89 Å−1 (d = 7.0 Å), and 1.82 Å−1 (d = 3.4 Å) corresponding to the (100), (200), and (020) planes of PEDOT.15 A large shift arises in the lamellar stacking peak position (100) when PEDOT:PSS (q ∼ 0.29 Å−1) is compared to PEDOT:OTf (q ∼ 0.45 Å−1). This corresponds respectively to large [d(100) = 22 Å] and short [d(100) = 14 Å] stacking distances due to the width differences between PSS macromolecules and OTf ions. The low lamellar stacking distance in PEDOT:OTf results in a stronger interaction between PEDOT chains, which is favourable to an increase of the crystalline order degree. All the distinct diffraction peaks in PEDOT:OTf are consistently larger and sharper than in PEDOT:PSS with increase in the thickness and number of crystalline domains. The crystallite size along the b-axis (Lb), derived from the (020) peak is approximately 37 Å in PEDOT:OTf compared to approximately 33 Å in PEDOT:PSS. The crystallite size (La) along the a-axis, derived from the (100) diffraction peak, is approximately 60 Å in the PEDOT:OTf film. These observations are in accordance with the structural changes observed with AFM phase contrast imaging. At small angles (q < 0.2 Å−1), the intensity profile follows a q−3 dependence. This power law in the SAXS domain highlights the absence of discontinuity in the material. Several changes can be noticed when comparing the X-ray patterns of PEDOT:OTf and PEDOT:Sulf. Although the intensities of the diffraction peaks remain essentially unchanged, a sharpening is observed in the (100) and (200) peaks. This is consistent with a constant number of crystalline domains before and after H2SO4 treatment along with an increase of their size along the a-axis La, which is approximately 77 Å in PEDOT:Sulf (calculated from Scherrer formula). Finally, a small peak is observed at q ∼ 1.62 Å−1, corresponding to the distance of ∼3.9 Å, which is comparable to the periodicity of monomer units of PEDOT (∼3.85 Å).15 The effects on the structural order along the a-axis may be attributed to the inserted ions, implying less steric hindrance than OTf anions, and thus inducing more crystalline order at longer distances. This capacity of HSO4− to enable more linear PEDOT structures is supported by the observation of a small peak at q ∼ 1.62 Å−1, matching the periodicity of the monomer unit. Consistent with this interpretation, the tiny peak was not observed on the intensity profile of the pristine PEDOT:OTf sample.
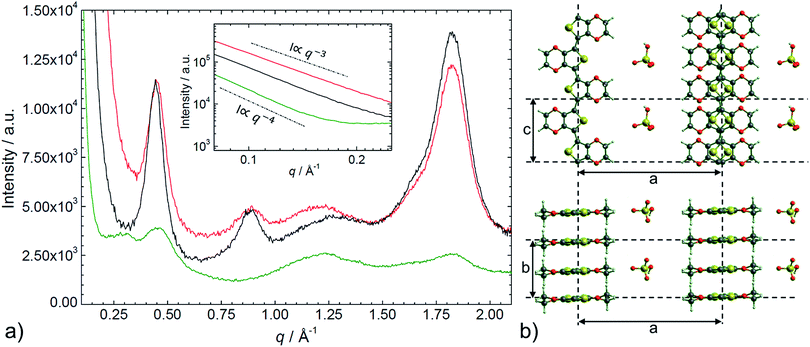 |
| | Fig. 5 a) SAXS/WAXS intensity profile of PEDOT:PSS (green line), PEDOT:OTf in the pristine state (red line), and PEDOT:Sulf (black line). Top inset shows the intensity in the small angles domain. (b) Schematic illustration of the structural model of PEDOT:OTf, in projections along the b-axis (upper) and the c-axis (lower) directions. This illustration is adapted from a ref. 15 and is provided as a support for the discussion. | |
Charge carriers concentration
The mobility of the charge carriers in PEDOT:PSS, PEDOT:OTf, and PEDOT:Sulf is evaluated with an organic electrochemical transistor (OECT) as described by Wei et al. for PEDOT:PSS.20 Apparatus description and output curves are available in Fig. S4 and S5.† The concentration of charge carriers is calculated from the values of the mobility and σ, which are measured on thin polymer films. The electronic mobility in PEDOT:PSS, PEDOT:OTf, and PEDOT:Sulf are 1.3, 0.1, and 0.1 cm2 V−1 s−1, respectively. The high mobility in PEDOT:PSS matches the expectations regarding the quality of polymer chains in commercial formulations. The concentrations of charge carriers of PEDOT:PSS, PEDOT:OTf, and PEDOT:Sulf are 1.1 × 1021, 5.7 × 1022, and 1.6 × 1023 cm−3, respectively. Thus, the value of σ in PEDOT:OTf is linked to the high concentration of charge carriers ascribed to the PEDOT weight fraction. The increase of the concentration of charge carriers in PEDOT:Sulf compared with PEDOT:OTf is consistent with the oxidation levels deduced from the XPS data.
Transport properties
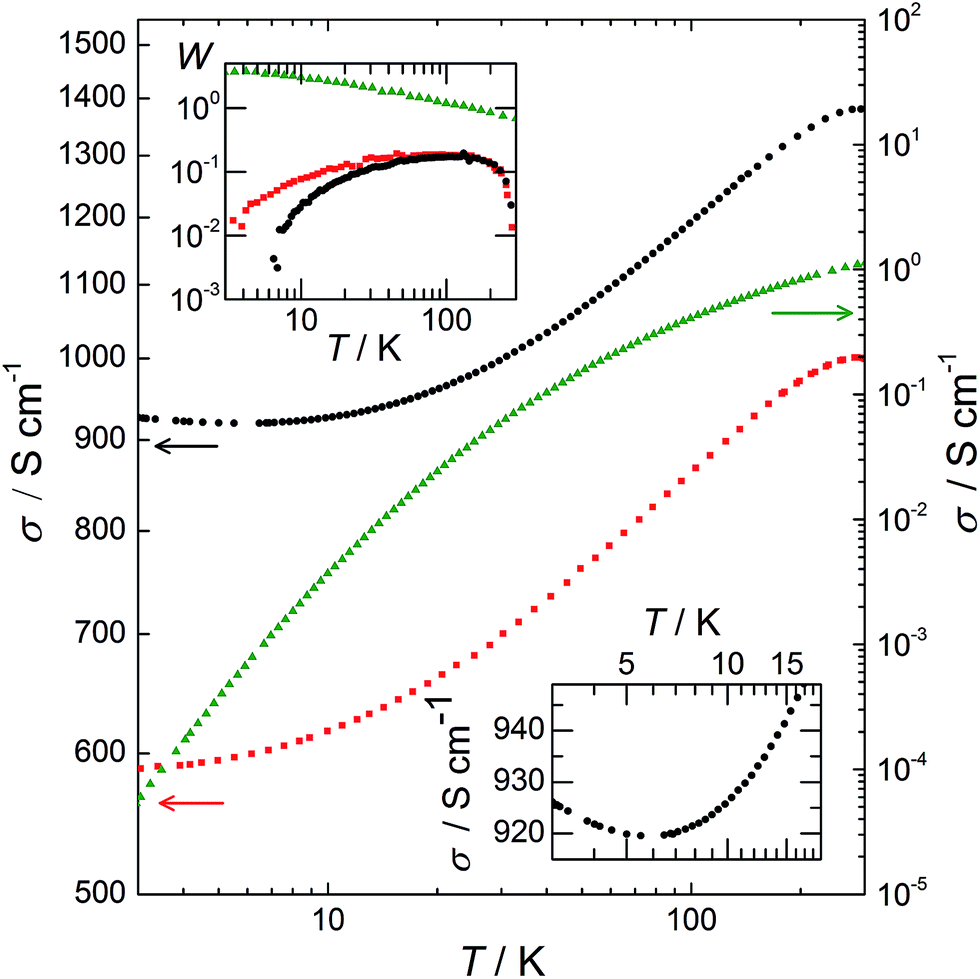
The variation of σ as a function of temperature, T, from 3 to 300 K is plotted in Fig. 6. This variation provides understanding of the transport mechanisms in PEDOT:PSS, PEDOT:OTf, and PEDOT:Sulf. For the PEDOT:PSS sample, σ is ∼1 S cm−1 at room temperature, then it collapses several orders of magnitude at low temperatures, resulting in an extrapolated σ = 0 S cm−1 at T = 0 K (which is typical for semiconductors). The reduced activation energy, defined as W = (d[ln(σ)])/(d[ln(T)])21 is plotted in the top inset of Fig. 6. For PEDOT:PSS, the decrease of W with T and the ratio σ(300 K)/σ(3 K) > 21![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 indicate that the sample is far in the insulating regime. The transport mechanism in PEDOT:PSS fits the VRH model,22 which is consistent with the previous studies,16 and with the embedded structure deduced from our structural observations. For PEDOT:OTf, σ is 1000 S cm−1 at room temperature and the ratio σ(300 K)/σ(3 K) equals 1.7, indicating a highly ordered polymer21 as previously stated (Fig. 1 and 3). From 3 to 45 K, W increases with T; this is typical for conducting polymers on the metallic side of the metal–insulator (M–I) transition.23W has a constant value of 0.19 from 45 to 149 K, and therefore σ ∝ T0.19, which is characteristic for the critical regime.21 The exponent of the power is below 1/3, confirming that this sample is on the metallic side of the M–I transition.23 As a result, the VRH conduction regime does not apply and the transport is governed by a power law, accordingly to the continuity of the material and the density of PEDOT chains stacking. The doping of PEDOT:OTf with H2SO4 results in an increase of σ (about 40% at 300 K for this sample). For T > 5.5 K, W has a positive slope with increasing T, placing PEDOT:Sulf on the metallic side of the M–I transition. The slope of W is steeper than the one of pristine PEDOT:OTf, and the plateau is replaced by a slight slope. Moreover, σ reaches a minimum at 5.5 K, and then it slightly increases from 5.5 K to 3 K (Fig. 6 bottom inset). This means that the TCR is positive in this range. The thermopower values of PEDOT:PSS, PEDOT:OTf, and PEDOT:Sulf 19, 17, and 14 μV K−1, respectively, confirm the stronger metallic behaviour of PEDOT:OTf and PEDOT:Sulf. These values are lower than those observed by Crispin et al. for semi-metallic PEDOT:OTs (55 μV K−1)8 and Pipe et al. for EG-dipped PEDOT:PSS (70 μV K−1).5 Because the mobilities and structures of PEDOT:OTf and PEDOT:Sulf are similar, the strengthened metallic behaviour (i.e. pushing toward the metallic side of the M–I transition) is mainly attributed to the increased concentration of charge carriers (from 5.7 × 1022 to 1.6 × 1023 cm−3) induced by the insertion of HSO4−. The positive TCR observed for PEDOT:Sulf is the characteristic of a “true” metallic behaviour. It is remarkable that after the discovery of conducting polymers in 1977, very few examples of metallic behaviours (i.e. with positive TCR) have been observed in polymer films, such as PF6-doped polypyrroles24 and CSA-doped polyaniline.23,25,26 The origin of such behaviour is still under debate, related discussions are available in previous reports27,28 and their references.
000 indicate that the sample is far in the insulating regime. The transport mechanism in PEDOT:PSS fits the VRH model,22 which is consistent with the previous studies,16 and with the embedded structure deduced from our structural observations. For PEDOT:OTf, σ is 1000 S cm−1 at room temperature and the ratio σ(300 K)/σ(3 K) equals 1.7, indicating a highly ordered polymer21 as previously stated (Fig. 1 and 3). From 3 to 45 K, W increases with T; this is typical for conducting polymers on the metallic side of the metal–insulator (M–I) transition.23W has a constant value of 0.19 from 45 to 149 K, and therefore σ ∝ T0.19, which is characteristic for the critical regime.21 The exponent of the power is below 1/3, confirming that this sample is on the metallic side of the M–I transition.23 As a result, the VRH conduction regime does not apply and the transport is governed by a power law, accordingly to the continuity of the material and the density of PEDOT chains stacking. The doping of PEDOT:OTf with H2SO4 results in an increase of σ (about 40% at 300 K for this sample). For T > 5.5 K, W has a positive slope with increasing T, placing PEDOT:Sulf on the metallic side of the M–I transition. The slope of W is steeper than the one of pristine PEDOT:OTf, and the plateau is replaced by a slight slope. Moreover, σ reaches a minimum at 5.5 K, and then it slightly increases from 5.5 K to 3 K (Fig. 6 bottom inset). This means that the TCR is positive in this range. The thermopower values of PEDOT:PSS, PEDOT:OTf, and PEDOT:Sulf 19, 17, and 14 μV K−1, respectively, confirm the stronger metallic behaviour of PEDOT:OTf and PEDOT:Sulf. These values are lower than those observed by Crispin et al. for semi-metallic PEDOT:OTs (55 μV K−1)8 and Pipe et al. for EG-dipped PEDOT:PSS (70 μV K−1).5 Because the mobilities and structures of PEDOT:OTf and PEDOT:Sulf are similar, the strengthened metallic behaviour (i.e. pushing toward the metallic side of the M–I transition) is mainly attributed to the increased concentration of charge carriers (from 5.7 × 1022 to 1.6 × 1023 cm−3) induced by the insertion of HSO4−. The positive TCR observed for PEDOT:Sulf is the characteristic of a “true” metallic behaviour. It is remarkable that after the discovery of conducting polymers in 1977, very few examples of metallic behaviours (i.e. with positive TCR) have been observed in polymer films, such as PF6-doped polypyrroles24 and CSA-doped polyaniline.23,25,26 The origin of such behaviour is still under debate, related discussions are available in previous reports27,28 and their references.
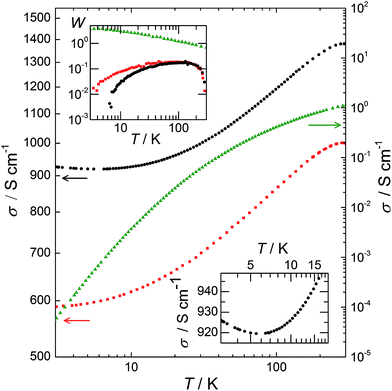 |
| | Fig. 6 log–log plot of σ as a function of temperature from 3 to 300 K. Left Y axis: PEDOT:OTf (red squares) and PEDOT:Sulf (black circles); right Y axis: PEDOT:PSS (green triangles). Top inset: the corresponding activation energy W in a log–log plot. For PEDOT:Sulf, W is negative in the [3 K; 5.5 K] range, as expected for metallic behaviour. Bottom inset: magnified image of the electrical conductivity of PEDOT:Sulf in the [3 K; 20 K] range. | |
Conclusions
In conclusion, Fe(OTf)3 was used as an oxidant for the polymerization of EDOT, resulting in a highly conductive metallic-like PEDOT:OTf polymer that presents an ordered crystalline structure. The treatment of PEDOT:OTf with H2SO4 increases the conductivity up to 2273 S cm−1 and strengthens the metallic behaviour, whereas the crystalline structure remains almost unchanged. The oxidation level of the doped polymer (45.5%) is the highest ever reported for PEDOT, and the dependence of σ on temperature reveals a positive TCR, disclosing this polymer as one of the very few organic materials that demonstrate a metallic behaviour.
Experimental
Chemicals
PEDOT:PSS Clevios PH1000 was purchased from H.C. Starck. P(VDF-HFP), PEG-PPG-PEG (various Mn), acids, EDOT, and ethylene glycol (EG) were purchased from Aldrich and used without further purification.
Polymer films preparation
Glass substrates (Corning Eagle XG, 2.5 cm × 2.5 cm, 1.1 mm thick) were washed with acetone and ethanol. A solution of 20% wt PEG-PPG-PEG (Mn = 5800 g mol−1) and 80% wt ethanol was stirred in an ultrasonic bath for 4 h. Fe(OTf)3 (0.25 mol L−1) was added and the solution was then stirred for 2 h and cooled to 5 °C. EDOT (0.18 mol L−1) was added to the solution. After stirring for 1 min in an ultrasonic bath, the reaction mixture was spin-coated onto the glass substrates and annealed for 10 min at 70 °C. The resulting layers were washed in ethanol and dried. PEDOT:PSS samples, spin-coated from a dilution of PEDOT:PSS Clevios PH1000 in isopropyl alcohol (20% wt), were used as references. Further experimental details are provided in the ESI.†
Doping treatment
All the acids were diluted in water to obtain solutions at pH 0.5. The samples were dipped in the acid solution for 1 h and then dried at 160 °C. Experimental conditions are discussed in the ESI.†
Characterisations
Charge carrier measurements were performed as described by Wei et al. in an OECT,20 using a described ionic gel.29σ was monitored from 3 to 300 K using a He flow cryostat from Oxford Instruments CF 1200 D. The temperature was measured using a RhFe calibrated resistor and σ was obtained after temperature stabilization in the mK range using four-probes in line configuration. X-ray pattern measurements samples were fabricated using thin polymer films according to the aforementioned method. The films were dipped in ethanol, aggregated and dried in order to obtain an ∼0.5 mm thick polymer block. X-ray patterns were measured in transmission geometry using a home-made SAXS/WAXS camera utilizing a point source (source size ∼100 μm × 100 μm) Bruker-Nonius (FR591) rotating anode generator with Cu-Kα radiation (λ = 1.5418 Å) at 45 kV and 26 mA. The samples were freely suspended in the X-ray beam and a multi-wire gas filled large area detector was placed at a distance of 20 cm to record the patterns. Further details are available in the ESI.† The thermopower S was recorded with a ULVAC ZEM-3 system. The thicknesses of the layers and their morphologies were measured by AFM (Bruker Innova). UV-vis-NIR spectra were recorded on a Varian Cary 5000. XPS were performed on a NOVA-KRATOS instrument with an Al Kα monochromatic source. ToF-SIMS was performed with a 5 keV Ar500+2 sputter gun and a 15 keV Bi3+ liquid metal ion gun for the analysis, and the detector was set to the negative ion mode.
Notes and references
- F. Louwet, Synth. Met., 2003, 135–136, 115–117 CrossRef CAS.
- Y. Xia, K. Sun and J. Ouyang, Adv. Mater., 2012, 24, 2436–2440 CrossRef CAS PubMed.
- T.-H. Meen, K.-L. Chen, Y.-H. Chen, W.-R. Chen, D.-W. Chou, W.-H. Lan and C.-J. Huang, Int. J. Photoenergy, 2013, 2013, 1–6 Search PubMed.
- J. Ouyang, Displays, 2013, 34, 423–436 CrossRef CAS PubMed.
- G.-H. Kim, L. Shao, K. Zhang and K. P. Pipe, Nat. Mater., 2013, 12, 719–723 CrossRef CAS PubMed.
- N. Kim, S. Kee, S. H. Lee, B. H. Lee, Y. H. Kahng, Y.-R. Jo, B.-J. Kim and K. Lee, Adv. Mater., 2014, 26, 2268–2272 CrossRef CAS PubMed , 2109.
- M. V. Fabretto, D. R. Evans, M. Mueller, K. Zuber, P. Hojati-Talemi, R. D. Short, G. G. Wallace and P. J. Murphy, Chem. Mater., 2012, 24, 3998–4003 CrossRef CAS.
- O. Bubnova, Z. U. Khan, H. Wang, S. Braun, D. R. Evans, M. Fabretto, P. Hojati-Talemi, D. Dagnelund, J.-B. Arlin, Y. H. Geerts, S. Desbief, D. W. Breiby, J. W. Andreasen, R. Lazzaroni, W. M. Chen, I. Zozoulenko, M. Fahlman, P. J. Murphy, M. Berggren and X. Crispin, Nat. Mater., 2013, 13, 1–5 CrossRef PubMed.
- O. Bubnova and X. Crispin, Energy Environ. Sci., 2012, 5, 9345 CAS.
- S. Garreau, J. L. Duvail and G. Louarn, Synth. Met., 2001, 125, 325–329 CrossRef.
- M. Łapkowski and A. Proń, Synth. Met., 2000, 110, 79–83 CrossRef.
- N. Massonnet, A. Carella, O. Jaudouin, P. Rannou, G. Laval, C. Celle and J.-P. Simonato, J. Mater. Chem. C, 2014, 2, 1278 RSC.
- X. Crispin, S. Marciniak, W. Osikowicz, G. Zotti, A. W. D. van der Gon, F. Louwet, M. Fahlman, L. Groenendaal, F. De Schryver and W. R. Salaneck, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2561–2583 CrossRef CAS.
- M. Fabretto, K. Zuber, C. Hall, P. Murphy and H. J. Griesser, J. Mater. Chem., 2009, 19, 7871 RSC.
- K. E. Aasmundtveit, E. J. Samuelsen, L. A. A. Pettersson, O. Inganäs, T. Johansson and R. Feidenhans'l, Synth. Met., 1999, 101, 561–564 CrossRef CAS.
- N. Kim, B. H. Lee, D. Choi, G. Kim, H. Kim, J.-R. Kim, J. Lee, Y. H. Kahng and K. Lee, Phys. Rev. Lett., 2012, 109, 106405 CrossRef.
-
R. W. James, Optical Principles of the Diffraction of X-Rays, Ox Bow Pre., Woodbridge, 1982 Search PubMed.
- T. H. Lee, K. Do, Y. W. Lee, S. S. Jeon, C. Kim, J. Ko and S. S. Im, J. Mater. Chem., 2012, 22, 21624 RSC.
-
O. Glatter and O. Kratky, Small Angle X-Ray Scattering, Academic P., New York, 1982 Search PubMed.
- Q. Wei, M. Mukaida, Y. Naitoh and T. Ishida, Adv. Mater., 2013, 25, 2831–2836 CrossRef CAS PubMed.
-
R. Menon, in Handbook of Organic Conductive Molecules and Polymers, ed. H. S. Nalwa, Wiley, 1997, vol. 4, p. 3334 Search PubMed.
-
N. F. Mott and E. A. Davis, Electronic Processes in Non-Crystalline Materials, New York, Oxford, 1979 Search PubMed.
- R. Menon, C. Yoon, D. Moses, A. Heeger and Y. Cao, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 17685–17694 CrossRef CAS.
- T. Ishiguro, H. Kaneko, Y. Nogami, H. Ishimoto, H. Nishiyama, J. Tsukamoto, A. Takahashi, M. Yamaura, T. Hagiwara and K. Sato, Phys. Rev. Lett., 1992, 69, 660–663 CrossRef CAS.
- K. Lee, S. Cho, S. H. Park, a. J. Heeger, C.-W. Lee and S.-H. Lee, Nature, 2006, 441, 65–68 CrossRef CAS PubMed.
-
A. J. Heeger, Condens. Coherence Condens. Matter – Proc. Nobel Jubil. Symp., 2003, pp. 30–35 Search PubMed.
- A. B. Kaiser, Rep. Prog. Phys., 2001, 64, 1–49 CrossRef CAS.
-
A. J. Epstein, in Handbook of Conducting Polymers, ed. T. A. Skotheim and J. R. Reynolds, CRC Press, 2007, p. 1680 Search PubMed.
- K. H. Lee, M. S. Kang, S. Zhang, Y. Gu, T. P. Lodge and C. D. Frisbie, Adv. Mater., 2012, 24, 4457–4462 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details for polymer synthesis, SAXS/WAXS, mobility of charge carriers and σ(T) measurements, supplementary UV-vis-NIR spectra, XPS overview spectra. See DOI: 10.1039/c4sc02463j |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence