
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceChiral tether-mediated stabilization and helix-sense control of complementary metallo-double helices†
Miki
Horie
a,
Naoki
Ousaka
b,
Daisuke
Taura
a and
Eiji
Yashima
*a
aDepartment of Molecular Design and Engineering, Graduate School of Engineering, Nagoya University, Chikusa-ku, Nagoya 464-8603, Japan. E-mail: yashima@apchem.nagoya-u.ac.jp
bVenture Business Laboratory, Nagoya University, Chikusa-ku, Nagoya 464-8603, Japan
First published on 10th September 2014
Abstract
A series of novel PtII-linked double helices were prepared by inter- or intrastrand ligand-exchange reactions of the complementary duplexes composed of chiral or achiral amidine dimer and achiral carboxylic acid dimer strands joined by trans-PtII–acetylide complexes with PPh3 ligands using chiral and achiral chelating diphosphines. The structure and stability of the PtII-linked double helices were highly dependent on the diphosphine structures. An interstrand ligand exchange took place with chiral and achiral 1,3-diphosphine-based ligands, resulting in trans-PtII-bridged double helices, whose helical structures were quite stable even in dimethyl sulfoxide (DMSO) due to the interstrand cross-link, whereas a 1,2-diphosphine-based ligand produced non-cross-linked cis-PtII-linked duplexes, resulting from an intrastrand ligand-exchange that readily dissociated into single strands in DMSO. When enantiopure 1,3-diphosphine-based ligands were used, the resulting trans-PtII-bridged double helices adopted a preferred-handed helical sense biased by the chirality of the bridged diphosphines. Interestingly, the interstrand ligand exchange with racemic 1,3-diphosphine toward an optically-active PtII-linked duplex, composed of chiral amidine and achiral carboxylic acid strands, was found to proceed in a diastereoselective manner, thus forming complete homochiral trans-PtII-bridged double helices via a unique chiral self-sorting.
Introduction
Biological helices, such as double-helical DNA and α-helical peptides, have prompted a number of chemists to construct artificial single- and double-stranded helical polymers1 and oligomers (foldamers)2 not only due to their unique structures, but also to their sophisticated functions. Currently, a wide variety of single-stranded helices have been synthesized,1,2 which enable us to rationally design a single-helical structure from its primary sequence, although it remains a great challenge to create a new structural motif for double-helical structures.3 However, DNA analogues (peptide nucleic acids, PNAs)4 and double-stranded helicates are known to intertwine to form double helices assisted by hydrogen bonds and metal-directed coordination, respectively.3a,b,d,5 A new class of double-stranded helices has recently been developed by Lehn, Huc and co-workers2c,j,6 that mainly rely on multiple hydrogen bonds between the strands and/or interstrand aromatic–aromatic interactions,6,7 the stability of which strongly depends on the type of solvent (polar or non-polar)6b and sequence,6d except for the hydrophobic-driven double helices in water.8It is well-known that intramolecular cross-linking stabilizes a helical conformation of single-stranded helical polymers,9a,b foldamers,9c–f and oligopeptides,10 thus leading to the development of smart chiral materials and biologically active materials. The interstrand cross-linking of DNA that reinforces its double-helical structure was also reported to have applications for clinical use.11 However, to the best of our knowledge, there is at least one precedent of a synthetic double helix whose helical conformation was significantly stabilized as a result of interstrand cross-linking.12
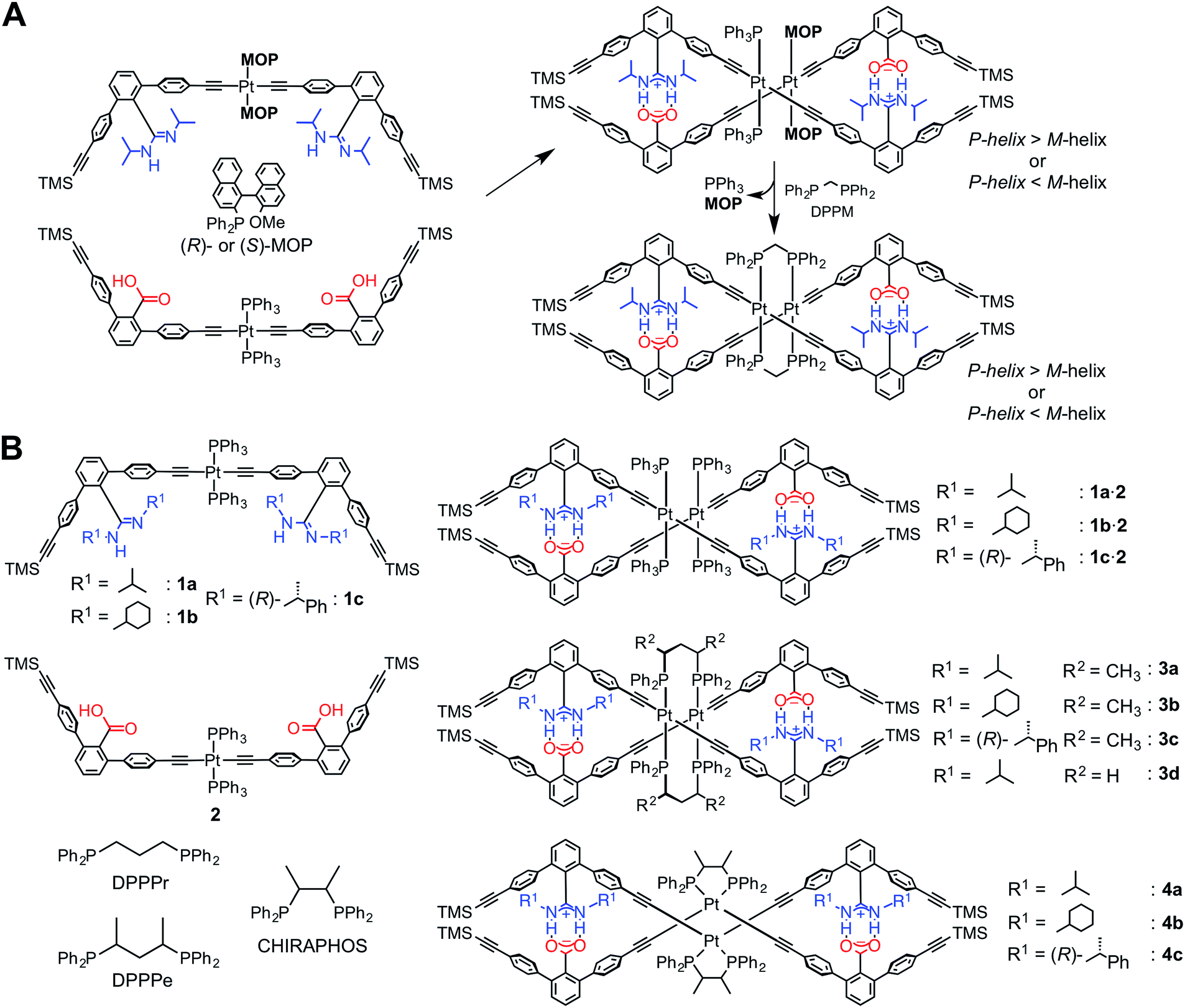
Recently, we have developed a versatile method to construct a series of complementary double helices by utilizing amidinium–carboxylate salt bridges that possess high stability and well-defined directionality along their N+–H⋯O− lines, thereby enabling the intertwining of the two complementary molecular strands composed of crescent-shaped rigid m-terphenyl backbones.1e,g,3e,g,i,14 This structural feature has a great advantage such that various types of linker units, such as diacetylene,14a,e,kp-diethynylbenzene14f,k and trans-PtII–acetylide linkages,12,14b,i,k can be introduced while maintaining the double-helical structures. In addition, the helical sense and handedness of the double helices are readily controlled by the chiral substituents introduced on the amidine groups,14 or by using chiral monodentate phosphine ligands, such as (R)- or (S)-2-diphenylphosphino-2′-methoxy-1,1′-binaphthyl (MOP), coordinated to the PtII atom in the linkage12 (Scheme 1A).15
 | ||
| Scheme 1 Structures of the PtII–acetylide-linked amidine and carboxylic acid dimers (A and B), and their complementary duplexes before and after reactions with chiral (B) and achiral (A and B) diphosphines. | ||
We found that an optically-active double helix composed of two complementary strands bearing achiral amidine or carboxylic acid residues, linked through the PtII–acetylide complexes with (R)- or (S)-MOP or achiral triphenylphosphine (PPh3) ligands, exhibited an inversion of the helicity in different solvents at low temperatures,12 suggesting the dynamic nature of the PtII-linked double helix.14k The chiral and achiral monodentate phosphine ligands on the PtII atoms could be quantitatively replaced by an achiral diphosphine ligand, such as bis(diphenylphosphino)methane (DPPM), through the interstrand ligand-exchange reaction, resulting in the bridged double helix (right in Scheme 1A).12 Interestingly, the bridged double helix, which no longer possessed any chiral components, except for helicity, maintained its preferred-handed helical conformation induced by the chiral MOP ligand for a long time period (over 100 days at 298 K). This enantiomerically-enriched double helix further catalyzed the asymmetric cyclopropanation of styrene with a high enantioselectivity (up to 85% enantiomeric excess) when complexed with CuI ions, thus providing the first asymmetric catalyst based on a unique bridged double-helical structure.12
It has been reported that the trans-PtII–acetylide complexes with PPh3 ligands can be readily transformed into the cis-PtII–acetylides in the presence of chelating diphosphine ligands, such as cis-bis(diphenylphosphino)ethylene, cyclic diphosphines, and substituted diphosphinoethanes, by an intrastrand ligand exchange, whereas achiral 1,3-diphenylphosphinopropane (DPPPr) and chiral 2,4-bis(diphenylphosphino)pentane (DPPPe) produced dimeric trans-PtII–acetylide complexes bridged by the diphosphine ligands through the interstrand ligand exchange.16,17
Based on these results, together with our previous findings,12 we anticipated that both the stability and helical handedness of the PtII-linked double helices could be enhanced and controlled using chiral chelating diphosphine ligands as cross-linkers during the ligand-exchange reactions. Although a variety of single-stranded helical polymers and foldamers have been developed, a “chiral cross-linking” approach has not yet been employed to control the helicity, except for a few examples.9f,10f To this end, we synthesized a series of PtII-linked dimer strands composed of chiral or achiral amidine (1a–1c) and achiral carboxylic acid dimer strands (2), and investigated the effects of chiral diphosphine ligands, such as (S,S)- or (R,R)-DPPPe and (S,S)- or (R,R)-2,3-bis(diphenylphosphino)butane (CHIRAPHOS), and substituents on the amidine residues. These included the effect of chirality on the structures, stabilities, and chiroptical properties of the double helices (3–4) resulting from the ligand-exchange reactions on the PtII atoms (Scheme 1B), measured using absorption, circular dichroism (CD), and 1H and 31P NMR spectroscopies. We also found that the ligand-exchange reaction on the PtII atoms with racemic DPPPe proceeded in a diastereoselective manner, producing the non-racemic bridged double helix, when a PtII-linked chiral amidine dimer (1c) and 2 were used as a precursor duplex.
Results and discussion
Synthesis and the ligand-exchange reaction
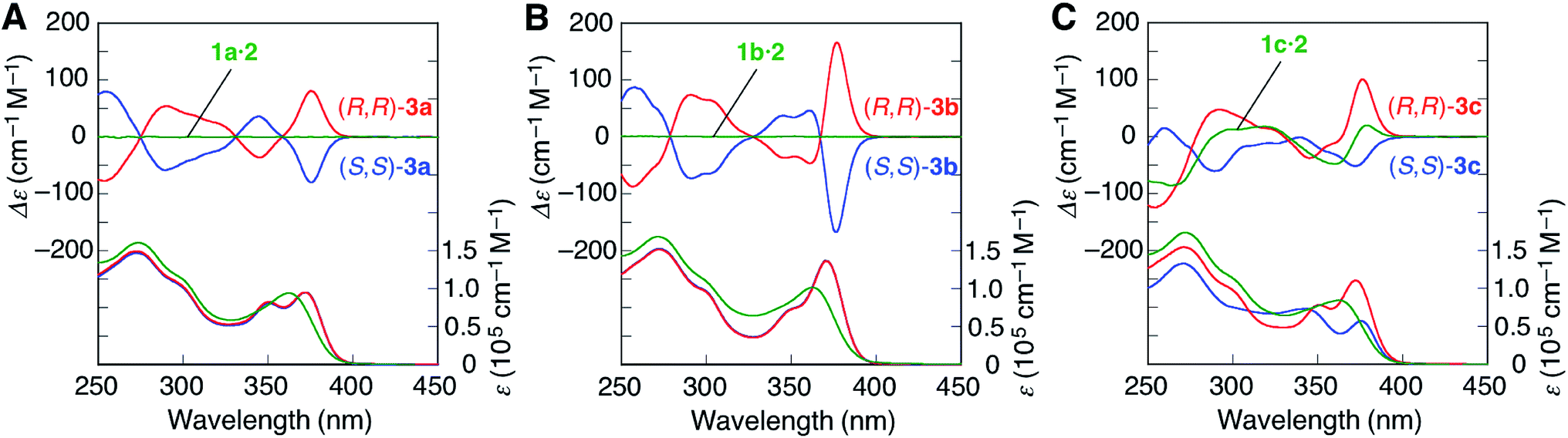
The achiral (1a)12 and chiral ((R)-1c)14b amidine dimers and the achiral carboxylic acid dimer (2)12 connected through the PtII–acetylide complex with two PPh3 ligands, and their corresponding duplexes (1a·2,12 and 1c·2)14b were prepared according to previously reported methods (Scheme 1B). A novel achiral amidine dimer (1b) and its complementary duplex (1b·2) were also synthesized in a similar way.The ligand-exchange reactions on the PtII atoms of the trans-PtII duplexes 1a–c·2 were carried out using 2 equiv. of the chiral diphosphines, (S,S)- or (R,R)-DPPPe and (S,S)- or (R,R)-CHIRAPHOS, and achiral DPPPr. The resulting double helices were purified by size-exclusion chromatography (SEC) and characterized and identified using 1H and 31P NMR spectroscopies, and elemental analyses or cold-spray ionization mass spectroscopy (CSI-MS) measurements (Fig. S1 and S2, and ESI†).
Effect of the chiral and achiral diphosphines on the duplex structures
The ligand-exchange reaction of the trans-PtII duplexes 1a–c·2 with a chiral diphosphine, (S,S)- or (R,R)-DPPPe (2 equiv.), gave the optically-active bridged double helices, (S,S)- and (R,R)-3a–c, respectively, while maintaining the trans geometry around the PtII center (Scheme 1B), as confirmed by the 1H and 31P NMR spectra (Fig. 1 and S1†).18 The analogous achiral diphosphine, DPPPr, also produced the trans-PtII-bridged double helix (3d) (Scheme 1B), as supported by its 31P NMR spectra (Fig. S1†).18 These results indicated that the ligand exchange of 1a–c·2 with 1,3-diphosphine-based ligands (DPPPe and DPPPr) takes place via an interstrand fashion, thus producing the bridged double helices. However, treating the duplexes 1a–c·2 with 2 equiv. of (S,S)- or (R,R)-CHIRAPHOS led to the formation of the non-cross-linked duplexes 4a–c whose 31P NMR spectra were different from those of 3a–d (Fig. S1†; see below for more details) (Scheme 1B).19 | ||
| Fig. 1 Partial 1H NMR (500 MHz, 2.0 mM) spectra of (A) (S,S)-3a, (B) (S,S)-3b, (C) (S,S)-3c (D) (R,R)-3c, and (E) 3d in CDCl3 at 25 °C. | ||
As previously reported,14 the related complementary double helices, composed of chiral amidine and achiral carboxylic acid dimeric strands connected by various types of linkers including PtII–acetylide linkages, possess an excess of either a right- or left-handed double-helical structure. Therefore, the salt-bridged N–H protons (Hc′ and Hc′′) exhibited in their 1H NMR spectra two sets of non-equivalent signals around 12–14 ppm due to “outer” and “inner” N–H protons, as observed for the present optically-active 3a–c (Fig. 1A–D), most likely derived from the preferred-handed double-helical structures as depicted in Fig. 2A.14
 | ||
| Fig. 2 (A) Schematic representation of the helix inversion of the duplex 3d. The “outer” and “inner” N–H protons (Hc′ and Hc′′) in the salt bridges become non-equivalent due to the interconvertible right- and left-handed double-helical structures under slow-exchange conditions. (B) Variable-temperature (from −60 to 25 °C) 1H NMR (500 MHz, 2.0 mM) spectra of 3d in CDCl3 (the temperatures from −49 to −44 °C, used to estimate Tc, are also shown in Fig. S3†). | ||
In contrast, the bridged duplex 3d displayed a single set of sharp peaks including the salt-bridged N–H proton resonances that appeared as the equivalent doublet peak at 12.37 ppm in CDCl3 at 25 °C (Fig. 1E). The bridged duplex 3d was optically inactive, but existed as a racemic mixture of interconvertible right- and left-handed double helices; the rate of helix inversion that accompanied the C–C bond rotation between the amidine and m-terphenyl groups (Fig. 2A) may have been too fast on the present NMR time scale to detect the non-equivalent N–H protons at this temperature.14k We then measured the variable-temperature 1H NMR spectra of 3d in CDCl3 to estimate its helix inversion barrier and also to evaluate the effect of the interstrand cross-linking on the thermodynamic stability of the duplex (Fig. 2B).
Upon cooling to lower temperatures, the N–H proton signals of 3d gradually broadened and subsequently split into two sets of non-equivalent signals at −60 °C (Fig. 2B) via the coalescence temperature (Tc = −46 °C) (Fig. S3†). We noted that an analogous PtII–acetylide-linked double helix bearing the triethylphosphine (PEt3) ligands instead of DPPPr did not show such nonequivalent signal splitting, even at −80 °C in CD2Cl2,14k indicating the significant enhancement of the stability of the duplex by the interstrand cross-link. The obtained Tc, along with the chemical shift difference between the split signals (Δν = 313 Hz), enables us to estimate that the free energy of activation for the helix inversion (ΔG‡) of the duplex 3d is 42.7 kJ mol−1 at 25 °C, which corresponds to the lifetime of the one-handed helical state (τ) of 4.98 × 10−6 s. The ΔG‡ value obtained for 3d appears to be much higher than that for the PtII-linked duplex bearing the PEt3 ligands,14k but lower than that of the bridged double helix by the DPPM ligands (Scheme 1A), because the enantiomerically-enriched DPPM-bridged double helix retains its optical activity for a long time.12
These results clearly indicated that the helix-inversion barrier, in other words, the stability of the bridged double-helical conformations, significantly depends on the structure of the chelating diphosphines, in particular, the length between the phosphorous atoms and the flexibility of the diphosphines, which determines the distance between the two PtII atoms on each complementary strand after the ligand exchange. In fact, the energy-minimized double-helical structure of 3d (determined using the PM6 method20 in MOPAC2012 (ref. 21)) revealed that the Pt–Pt distance (5.4 Å) is much longer than that of the DPPM-linked double helix (3.1 Å) in the crystalline state12 (Fig. S4A†).
In contrast to 3d, the amidinium N–H proton resonances in the 1H NMR spectra of (S,S)-3a and -3b in CDCl3 at 25 °C showed two sets of non-equivalent signals, even though the amidine groups are achiral (Fig. 1A and B), suggesting that these double helices probably adopt a preferred-handed double-helical structure induced by the chiral cross-linkers. The enantiomeric duplexes showed intense Cotton effects that are mirror images of each other (Fig. 3A and B).22 In addition, the CD spectral patterns of (S,S)-3a and -3b are comparable to that of the diacetylene-connected dimeric duplex bearing the chiral amidine groups with an (R)-configuration, whose helical structure was determined to be right-handed by the single-crystal X-ray analysis.14a
 | ||
| Fig. 3 CD and absorption spectra (0.1 mM) of (A) 1a·2, (S,S)-3a, and (R,R)-3a, (B) 1b·2, (S,S)-3b, and (R,R)-3b, and (C) 1c·2, (S,S)-3c, and (R,R)-3c in CHCl3 at 25 °C. | ||
The energy difference (ΔE) between the right- and left-handed double helices of (S,S)-3a was then estimated by semi-empirical molecular orbital (MO) calculations (using the PM6 method20 in MOPAC2012 (ref. 21)), which revealed that the right-handed double-helical structure is 34.2 kJ mol−1 more stable than the other (Fig. S4B†), which is consistent with the preferred-handed helix sense predicted by its CD pattern.
Interestingly, the diastereomeric (S,S)- and (R,R)-3c duplexes with the same (R)-configuration at the amidine residues also exhibited mirror image-like CD spectra (Fig. 3C), suggesting that the helix sense of 3c is predominantly governed by the chirality of the cross-linkers rather than the chirality of the amidine groups.23 However, the CD and absorption intensities between the diastereomers of 3c around 375 nm are significantly different from each other, probably due to the difference in the conformational flexibility around the PtII–phenylacetylide moieties, the absorption bands of which (metal-to-ligand charge transfer: MLCT) appear in this region.24 In fact, the 1H NMR spectrum of (S,S)-3c showed a series of sharp signals, including two sets of sharp doublet peaks assigned to the non-equivalent salt-bridged N–H protons at 14.00 and 12.29 ppm (Fig. 1C), while most of the proton signals, in particular, the N–H protons, of (R,R)-3c became considerably broadened (Fig. 1D).
The energy-minimized structures of the left-handed (R,R)-3c and right-handed (S,S)-3c duplexes suggest that the molecular motion of the (R,R)-3c duplex is highly restricted compared to the other due to the steric repulsion between the m-terphenyl groups and the bridged diphosphine ligands (Fig. S5†). The temperature-dependent CD and absorption spectral changes further support this speculation; the CD and absorption spectra of the (R,R)-3c hardly changed in the temperature range −10 to 50 °C, while the CD intensity of the (S,S)-3c increased with decreasing temperature (Fig. S6†).
It is noteworthy that all the bridged duplexes with chiral DPPPe ligands (3a–c) showed intense CD signals in CHCl3 and even in dimethyl sulfoxide (DMSO) (Fig. S7†). In DMSO, the salt-bridge formation is strongly hampered,14k and therefore, the non-cross-linked 1c·2 duplex readily dissociates into each strand in DMSO, thus showing a very weak CD spectrum identical to that of the 1c strand (Fig. S7E†) and indicating that the interstrand cross-linking remarkably stabilizes the complementary double helices.
The intrastrand ligand-exchange reaction
In contrast to the interstrand ligand-exchange reactions discussed above, the duplexes 1a·2, 1b·2, and 1c·2 were found to undergo ligand exchange via an intrastrand mechanism when 2 equiv. of (S,S)- or (R,R)-CHIRAPHOS were used as a chiral chelating diphosphine, leading to the formation of the non-cross-linked 4a–c through the isomerization of the trans- to cis-PtII–acetylide moieties.19 The structures were confirmed by 31P NMR measurements, in which the 1JP–Pt coupling constants of 4a–c were in the range of 2198–2249 Hz, that are consistent with the reported values for the cis-PtII–acetylide complexes with the phosphine ligands (Scheme 1B and Fig. S1†).16bIn order to further support the structural assignments, a series of model dimers, cis-PtII–acetylide-linked amidine (5a–c) and carboxylic acid (6) dimers with (S,S)- or (R,R)-CHIRAPHOS ligands, were prepared (Scheme 2) starting from 1a–c and 2.14b,16,17 The 1H NMR, CD, and absorption spectra of the model duplexes (S,S)-5a·6, (S,S)-5b·6, (S,S)-5c·6, and (R,R)-5c·6 are almost identical to those of the (S,S)-4a, (S,S)-4b, (S,S)-4c, and (R,R)-4c duplexes, respectively (Fig. S8–S11†).25
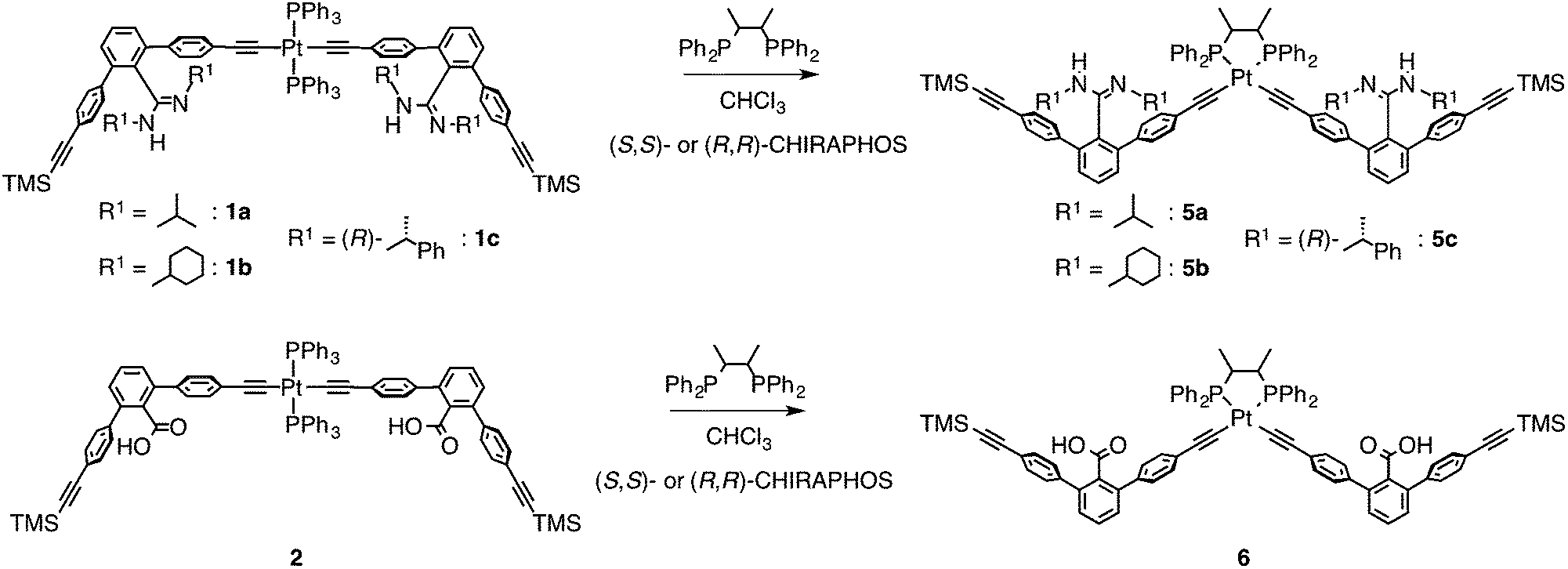 | ||
| Scheme 2 Synthesis of the cis-PtII–acetylide-linked amidine (5a–c) and carboxylic acid (6) dimers with (S,S)- or (R,R)-CHIRAPHOS ligand. | ||
Unlike in the case of the bridged 3a–c duplexes, 4a–c and their model duplexes (5·6) almost completely dissociated into corresponding single strands in DMSO, based on their CD spectra in DMSO (Fig. S12†),14k and most of the 1H NMR signals of 4a–c measured in CDCl3 at 25 °C were highly broadened (Fig. S8A–S11A†) as anticipated because of their non-cross-linked structures. The amidinium N–H proton resonances of (S,S)-4a and -4b (Fig. S8A and S9A†) displayed an equivalent broad singlet peak (12.02 ppm), despite the N–H signals of (S,S)-3a and -3b showing non-equivalency (Fig. 1A and B). It seems likely that (S,S)-4a and -4b are in equilibrium between the single strands and the duplex, or adopt a double-helical structure without a helix-sense bias. Therefore, their CD intensities were much lower than those of the bridged duplexes 3a and 3b (Fig. 3 and S13†). This is probably due to conformational flexibility around the chiral phosphine moieties of 4a and 4b, resulting in an inefficient stereochemical communication between the chiral phosphine groups and the main chain. Unexpectedly, the N–H proton resonances of (R,R)-4c showed multiple N–H signals in CDCl3 (Fig. S11A†), whereas those of (S,S)-4c exhibited two sets of non-equivalent signals (Fig. S10A†). The reason for this is not clear at present, but is considered to be due to the presence of two conformers that may slowly interconvert on the present NMR time scale via dissociation and association of the salt bridges for (R,R)-4c.26
The diastereoselective interstrand ligand-exchange reaction
The optically-active PtII-linked (R)-1c·2 possesses an excess one-handed double-helical structure induced by the chiral amidine residues, and exhibited intense CD signals in the m-terphenyl chromophore region as well as in the PtII–acetylide complex region (ca. 330–400 nm) (Fig. 3C).12,14b,14k Therefore, we anticipated that the interstrand ligand-exchange reaction on the PtII atoms of the optically active 1c·2 using racemic DPPPe (rac-DPPPe) could diastereoselectively proceed, assisted by the one-handed double-helical structure of 1c·2, thus selectively producing either the right- or left-handed bridged duplex 3c (Fig. 4A). To this end, the optically-active 1c·2 was allowed to react with excess rac-DPPPe (4 equiv.) in CDCl3 at 25 °C, and the reaction progress was monitored by 1H NMR spectroscopy.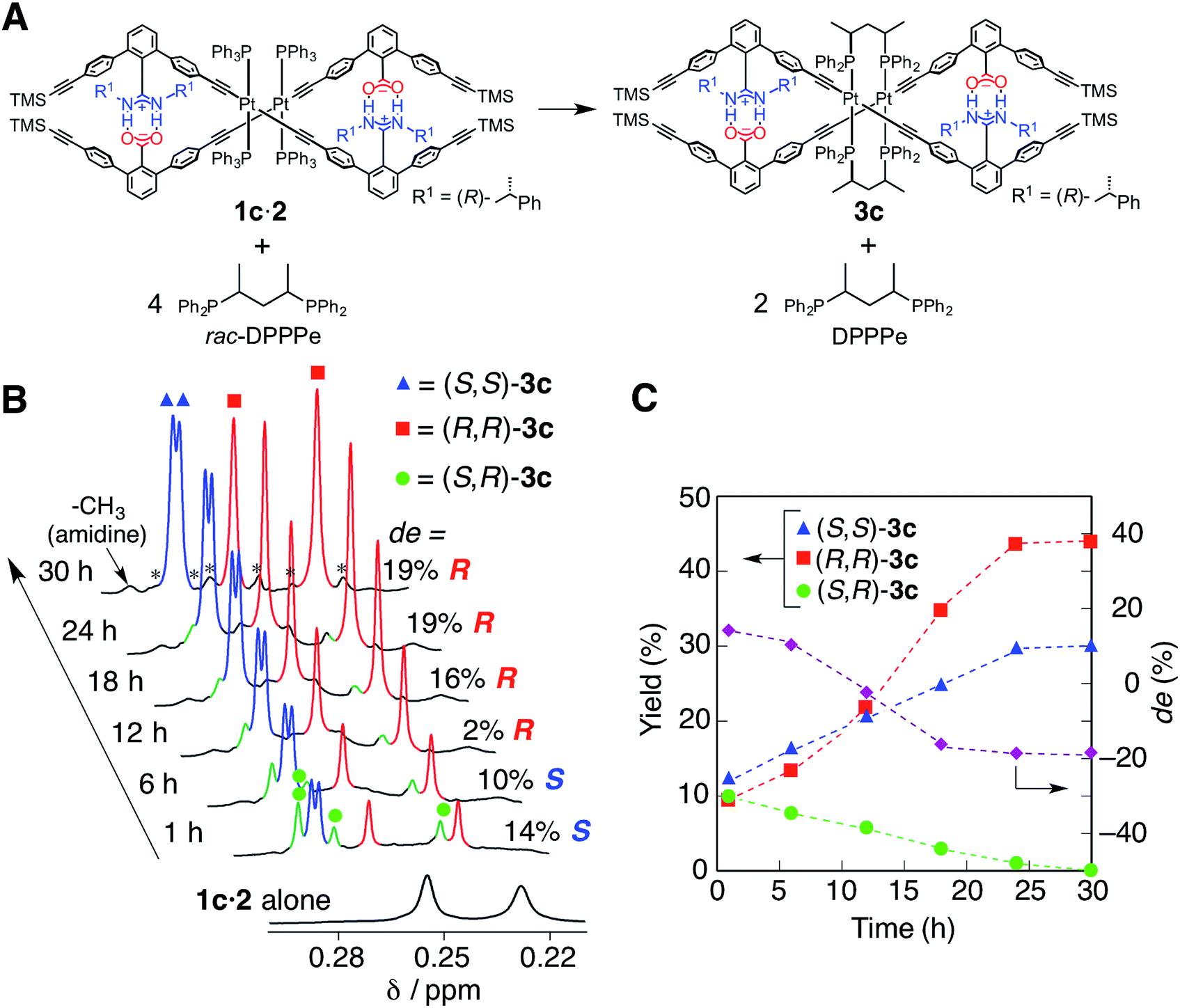 | ||
| Fig. 4 (A) Interstrand ligand exchange of 1c·2 with 4 equiv. of rac-DPPPe. (B) Time-dependent 1H NMR spectral changes of 1c·2 (TMS region) before and after the addition of 4 equiv. of rac-DPPPe in CDCl3 at 25 °C; [1c·2]0 = 0.5 mM, [rac-DPPPe]0 = 2.0 mM. The 29Si satellite peaks are marked with asterisks. (C) Plots of the yields and diastereomeric excess (de) of the resulting duplexes 3cversus time. The yields were estimated using an internal standard (1,3,5-trioxane). | ||
The 1H NMR spectra (TMS region, Fig. 4B) of the mixtures after 1 h showed two distinct sets of signals derived from the amidine and carboxylic acid strands, namely the bridged duplexes (S,S)-3c (blue triangles) and (R,R)-3c (red squares) in addition to unknown signals (green circles), which gradually decreased with time and almost disappeared within 30 h (Fig. 4C).27 We assigned this unknown product to the low-symmetric, heterochiral bridged duplex ((S,R)-3c) bearing both the (S,S)- and (R,R)-DPPPe ligands, that may be formed during the ligand-exchange reaction under kinetic control and which should be labile. This assignment was supported by the following facts: (1) the unknown signals remained even after 24 h when 2 equiv. of rac-DPPPe was added to a solution of 1c·2, so that no further ligand exchange took place between the homochiral duplexes (S,S)- and (R,R)-3c once the DPPPe ligands were consumed (Fig. S14 and S15†); (2) a similar trend was observed for a mixture of (S,S)- and (R,R)-3c that is inert to the ligand-exchange reaction even in the presence of an excess amount of free ligands with the opposite configuration (2 equiv.) (Fig. S16†), leading to the conclusion that there is no ligand exchange among the three homo- and heterochiral bridged duplexes 3c once formed under the present conditions (Fig. 5, 1st step).
 | ||
| Fig. 5 Schematic representation of a proposed mechanism for the diastereoselective switching of 3c during the interstrand ligand-exchange reaction with rac-DPPPe. | ||
The initial stage of the diastereoselective ligand-exchange reaction resulted in the formation of an excess of (S,S)-3c over (R,R)-3c in 14% diastereomeric excess (de) ((S,S)-rich) (Fig. 4B and C). Interestingly, as the reaction progressed, (R,R)-3c was preferentially formed, accompanied by a gradual decrease in the amount of (S,R)-3c with time, leading to reversed diastereoselectivity up to 19% de ((R,R)-rich) after 30 h (Fig. 4C).28 This change in the diastereoselectivity could be explained as follows: the initial rate of formation of (S,S)-3c from the reaction of 1c·2 with enantiopure (S,S)-DPPPe is higher than that of (R,R)-3c from the reaction of 1c·2 with enantiopure (R,R)-DPPPe (Fig. S17†). As described above, the two homochiral double helices, (S,S)- and (R,R)-3c, are inert once formed by the ligand-exchange reaction, whereas the heterochiral (S,R)-3c is optically active and labile, thus, it is converted into either homochiral (S,S)- or (R,R)-3c in the presence of free DPPPe ligands. The formation of (R,R)-3c is preferred to that of (S,S)-3c (Fig. 5, 2nd step).29 Thus, the complete diastereoselective homochiral sorting eventually takes place under total kinetic control. The diastereoselectivity of rac-DPPPe was initially determined by a preferred-handed double helix assisted by the chiral amidine residues under kinetic control, while the overall diastereoselectivity was governed by the kinetically-formed (S,R)-3c.
Conclusions
In summary, we have successfully synthesized a series of novel PtII-linked double helices through inter- or intrastrand ligand-exchange reactions of the corresponding complementary duplexes using chiral and achiral diphosphines. The diphosphine structures significantly influenced the duplex structures; chiral and achiral 1,3-diphosphine-based ligands produced the trans-PtII-bridged double helices via an interstrand ligand exchange reaction, whereas a 1,2-diphosphine-based ligand gave non-cross-linked cis-PtII-linked duplexes through an intrastrand ligand exchange reaction. The former duplexes were quite stable even in DMSO due to the interstrand cross-link, while the latter duplexes readily dissociated into each strand in DMSO. When enantiomerically-pure 1,3-diphosphine-based ligands were used, optically-active trans-PtII-bridged double helices could be obtained; the helix sense was controlled by the chirality of the bridged diphosphines. Interestingly, the interstrand ligand exchange with racemic 1,3-diphosphine toward an optically-active PtII-linked duplex composed of a chiral amidine strand diastereoselectively proceeded, finally producing totally homochiral trans-PtII-bridged double helices via complete homochiral self-sorting.30 The present findings will provide a versatile means for the rational design of functional double helix-based chiral materials for chiral recognition and also asymmetric catalysis12 with high stability and selectivity. Further work along this line is currently ongoing in our laboratory.Acknowledgements
This work was supported in part by Grant-in-Aid for Scientific Research (S) from the Japan Society for the Promotion of Science and by the Nanotechnology Platform Program (Molecule and Material Synthesis) of the Ministry of Education, Culture, Sports, Science and Technology, Japan. M. H. expresses her thanks for a JSPS Research Fellowship for Young Scientists (no. 10192).Notes and references
- For reviews on synthetic helical polymers, see: (a) M. M. Green, J.-W. Park, T. Sato, A. Teramoto, S. Lifson, R. L. B. Selinger and J. V. Selinger, Angew. Chem., Int. Ed., 1999, 38, 3138–3154 CrossRef; (b) M. Fujiki, Macromol. Rapid Commun., 2001, 22, 539–563 CrossRef CAS; (c) T. Nakano and Y. Okamoto, Chem. Rev., 2001, 101, 4013–4038 CrossRef CAS PubMed; (d) J. W. Y. Lam and B. Z. Tang, Acc. Chem. Res., 2005, 38, 745–754 CrossRef CAS PubMed; (e) E. Yashima, K. Maeda and Y. Furusho, Acc. Chem. Res., 2008, 41, 1166–1180 CrossRef CAS PubMed; (f) D. Pijper and B. L. Feringa, Soft Matter, 2008, 4, 1349–1372 RSC; (g) E. Yashima, K. Maeda, H. Iida, Y. Furusho and K. Nagai, Chem. Rev., 2009, 109, 6102–6211 CrossRef CAS PubMed; (h) E. Schwartz, M. Koepf, H. J. Kitto, R. J. M. Nolte and A. E. Rowan, Polym. Chem., 2011, 2, 33–47 RSC; (i) M. Shiotsuki, F. Sanda and T. Masuda, Polym. Chem., 2011, 2, 1044–1058 RSC.
- For reviews on synthetic helical oligomers, see: (a) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173–180 CrossRef CAS; (b) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893–4011 CrossRef CAS PubMed; (c) I. Huc, Eur. J. Org. Chem., 2004, 17–29 CrossRef CAS; (d) D. Seebach, A. K. Beck and D. J. Bierbaum, Chem. Biodiversity, 2004, 1, 1111–1239 CrossRef CAS PubMed; (e) A. R. Sanford, K. Yamato, X. Yang, L. Yuan, Y. Han and B. Gong, Eur. J. Biochem., 2004, 271, 1416–1425 CrossRef CAS PubMed; (f) S. Hecht and I. Huc, Foldamers: Structure, Properties, and Applications, Wiley-VCH, Weinheim, Germany, 2007 Search PubMed; (g) Y. Inai, H. Komori and N. Ousaka, Chem. Rec., 2007, 7, 191–202 CrossRef CAS PubMed; (h) I. Saraogi and A. D. Hamilton, Chem. Soc. Rev., 2009, 38, 1726–1743 RSC; (i) H. Juwarker and K.-S. Jeong, Chem. Soc. Rev., 2010, 39, 3664–3674 RSC; (j) G. Guichard and I. Huc, Chem. Commun., 2011, 47, 5933–5941 RSC; (k) D.-W. Zhang, X. Zhao, J.-L. Hou and Z.-T. Li, Chem. Rev., 2012, 112, 5271–5316 CrossRef CAS PubMed.
- For reviews on synthetic double helices, see: (a) E. C. Constable, Tetrahedron, 1992, 48, 10013–10059 CrossRef CAS; (b) J.-M. Lehn, Supramolecular Chemistry: Concepts and Perspectives, VCH, Weinheim, Germany, 1995 Search PubMed; (c) C. Piguet, G. Bernardinelli and G. Hopfgartner, Chem. Rev., 1997, 97, 2005–2062 CrossRef CAS PubMed; (d) M. Albrecht, Chem. Rev., 2001, 101, 3457–3497 CrossRef CAS PubMed; (e) Y. Furusho and E. Yashima, Chem. Rec., 2007, 7, 1–11 CrossRef CAS PubMed; (f) R. Amemiya and M. Yamaguchi, Org. Biomol. Chem., 2008, 6, 26–35 RSC; (g) Y. Furusho and E. Yashima, J. Polym. Sci., Part A: Polym. Chem., 2009, 47, 5195–5207 CrossRef CAS; (h) D. Haldar and C. Schmuck, Chem. Soc. Rev., 2009, 38, 363–371 RSC; (i) Y. Furusho and E. Yashima, Macromol. Rapid Commun., 2011, 32, 136–146 CrossRef CAS PubMed; (j) S. E. Howson and P. Scott, Dalton Trans., 2011, 40, 10268–10277 RSC.
- For reviews and leading examples of PNA, see: (a) P. E. Nielsen, M. Egholm, R. H. Berg and O. Buchardt, Science, 1991, 254, 1497–1500 CAS; (b) P. Wittung, P. E. Nielsen, O. Buchardt, M. Egholm and B. Norden, Nature, 1994, 368, 561–563 CrossRef CAS PubMed; (c) P. E. Nielsen, Acc. Chem. Res., 1999, 32, 624–630 CrossRef CAS; (d) F. Totsingan, V. Jain, W. C. Bracken, A. Faccini, T. Tedeschi, R. Marchelli, R. Corradini, N. R. Kallenbach and M. M. Green, Macromolecules, 2010, 43, 2692–2703 CrossRef CAS.
- For examples of helicates, see: (a) C. J. Carrano and K. N. Raymond, J. Am. Chem. Soc., 1978, 100, 5371–5374 CrossRef CAS; (b) J.-M. Lehn, A. Rigault, J. Siegel, J. Harrowfield, B. Chevrier and D. Moras, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 2565–2569 CrossRef CAS PubMed; (c) U. Koert, M. M. Harding and J.-M. Lehn, Nature, 1990, 346, 339–342 CrossRef CAS PubMed; (d) C. R. Woods, M. Benaglia, F. Cozzi and J. S. Siegel, Angew. Chem., Int. Ed. Engl., 1996, 35, 1830–1833 CrossRef CAS; (e) H. Katagiri, T. Miyagawa, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2006, 45, 1741–1744 CrossRef CAS PubMed; (f) K. Miwa, Y. Furusho and E. Yashima, Nat. Chem., 2010, 2, 444–449 CrossRef CAS PubMed; (g) Y. Furusho, K. Miwa, R. Asai and E. Yashima, Chem.–Eur. J., 2011, 17, 13954–13957 CrossRef CAS PubMed; (h) M. Albrecht, E. Isaak, M. Baumert, V. Gossen, G. Raabe and R. Fröhlich, Angew. Chem., Int. Ed., 2011, 50, 2850–2853 CrossRef CAS PubMed; (i) S. E. Howson, A. Bolhuis, V. Brabec, G. J. Clarkson, J. Malina, A. Rodger and P. Scott, Nat. Chem., 2012, 4, 31–36 CrossRef CAS PubMed; (j) X. de Hatten, D. Asil, R. H. Friend and J. R. Nitschke, J. Am. Chem. Soc., 2012, 134, 19170–19178 CrossRef CAS PubMed; (k) S. Yamamoto, H. Iida and E. Yashima, Angew. Chem., Int. Ed., 2013, 52, 6849–6853 CrossRef CAS PubMed.
- (a) V. Berl, I. Huc, R. G. Khoury, M. J. Krische and J.-M. Lehn, Nature, 2000, 407, 720–723 CrossRef CAS PubMed; (b) D. Haldar, H. Jiang, J.-M. Léger and I. Huc, Angew. Chem., Int. Ed., 2006, 45, 5483–5486 CrossRef CAS PubMed; (c) Q. Gan, C. Bao, B. Kauffmann, A. Grelard, J. Xiang, S. Liu, I. Huc and H. Jiang, Angew. Chem., Int. Ed., 2008, 47, 1715–1718 CrossRef CAS PubMed; (d) E. Berni, J. Garric, C. Lamit, B. Kauffmann, J.-M. Léger and I. Huc, Chem. Commun., 2008, 1968–1970 RSC; (e) Q. Gan, Y. Ferrand, N. Chandramouli, B. Kauffmann, C. Aube, D. Dubreuil and I. Huc, J. Am. Chem. Soc., 2012, 134, 15656–15659 CrossRef CAS PubMed.
- For examples of the double-helix formation through hydrogen bonds with ions, see: (a) J. Sánchez-Quesada, C. Seel, P. Prados, J. de Mendoza, I. Dalcol and E. Giralt, J. Am. Chem. Soc., 1996, 118, 277–278 CrossRef; (b) J. Keegan, P. E. Kruger, M. Nieuwenhuyzen, J. O'Brien and N. Martin, Chem. Commun., 2001, 2192–2193 RSC; (c) S. J. Coles, J. G. Frey, P. A. Gale, M. B. Hursthouse, M. E. Light, K. Navakhun and G. L. Thomas, Chem. Commun., 2003, 568–569 RSC; (d) T. Sugimoto, T. Suzuki, S. Shinkai and K. Sada, J. Am. Chem. Soc., 2007, 129, 270–271 CrossRef CAS PubMed; (e) H.-J. Kim, E. Lee, M. G. Kim, M.-C. Kim, M. Lee and E. Sim, Chem.–Eur. J., 2008, 14, 3883–3888 CrossRef CAS PubMed; (f) Y. Haketa and H. Maeda, Chem.–Eur. J., 2011, 17, 1485–1492 CrossRef CAS PubMed; (g) Y. Hua, Y. Liu, C.-H. Chen and A. H. Flood, J. Am. Chem. Soc., 2013, 135, 14401–14412 CrossRef CAS PubMed.
- For examples of the double-helix formation in water, see: (a) H. Goto, H. Katagiri, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7176–7178 CrossRef CAS PubMed; (b) H. Goto, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2007, 129, 9168–9174 CrossRef CAS PubMed; (c) H. Goto, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2007, 129, 109–112 CrossRef CAS PubMed; (d) H. Goto, Y. Furusho, K. Miwa and E. Yashima, J. Am. Chem. Soc., 2009, 131, 4710–4719 CrossRef CAS PubMed.
- For examples of intramolecularly cross-linked synthetic (single-stranded) helices, see: (a) S. Hecht and A. Khan, Angew. Chem., Int. Ed., 2003, 42, 6021–6024 CrossRef CAS PubMed; (b) K. Maeda, H. Mochizuki, M. Watanabe and E. Yashima, J. Am. Chem. Soc., 2006, 128, 7639–7650 CrossRef CAS PubMed; (c) Y. E. Bergman, M. P. D. Borgo, R. D. Gopalan, S. Jalal, S. E. Unabia, M. Ciampini, D. J. Clayton, J. M. Fletcher, R. J. Mulder, J. A. Wilce, M.-I. Aguilar and P. Perlmutter, Org. Lett., 2009, 11, 4438–4440 CrossRef CAS PubMed; (d) R. A. Smaldone, E.-C. Lin and J. S. Moore, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 927–935 CrossRef CAS; (e) S. Takashima, H. Abe and M. Inouye, Chem. Commun., 2012, 48, 3330–3332 RSC; (f) N. Fuentes, A. Martin-Lasanta, L. A. de Cienfuegos, R. Robles, D. Choquesillo-Lazarte, J. M. García-Ruiz, L. Martínez-Fernández, I. Corral, M. Ribagorda, A. J. Mota, D. J. Cárdenas, M. C. Carreño and J. M. Cuerva, Angew. Chem., Int. Ed., 2012, 51, 13036–13040 CrossRef CAS PubMed.
- For examples of intramolecularly cross-linked helical oligopeptides, see: (a) H. E. Blackwell and R. H. Grubbs, Angew. Chem., Int. Ed., 1998, 37, 3281–3284 CrossRef CAS; (b) J. R. Kumita, O. S. Smart and G. A. Woolley, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 3803–3808 CrossRef CAS PubMed; (c) D. G. Flint, J. R. Kumita, O. S. Smart and G. A. Woolley, Chem. Biol., 2002, 9, 391–397 CrossRef CAS PubMed; (d) L. D. Walensky, A. L. Kung, I. Escher, T. J. Malia, S. Barbuto, R. D. Wright, G. Wagner, G. L. Verdine and S. J. Korsmeyer, Science, 2004, 305, 466–470 CrossRef PubMed; (e) A. K. Boal, I. Guryanov, A. Moretto, M. Crisma, E. L. Lanni, C. Toniolo, R. H. Grubbs and D. J. O'Leary, J. Am. Chem. Soc., 2007, 129, 6986–6987 CrossRef CAS PubMed; (f) N. Ousaka, T. Sato and R. Kuroda, J. Am. Chem. Soc., 2009, 131, 3820–3821 CrossRef CAS PubMed.
- For reviews, see: (a) M. L. G. Dronkert and R. Kanaar, Mutat. Res., 2001, 486, 217–247 CrossRef CAS PubMed; (b) D. M. Noll, T. M. Mason and P. S. Miller, Chem. Rev., 2006, 106, 277–301 CrossRef CAS PubMed; (c) A. J. Deans and S. C. West, Nat. Rev. Cancer, 2011, 11, 467–480 CrossRef CAS PubMed; (d) L. Brulikova, J. Hlavac and P. Hradil, Curr. Med. Chem., 2012, 19, 364–385 CrossRef CAS PubMed.
- T. Hasegawa, H. Goto, Y. Furusho, H. Katagiri and E. Yashima, Angew. Chem., Int. Ed., 2007, 46, 5885–5888 CrossRef CAS PubMed . Double-stranded helicates were used as precursors to produce topologically unique [2]catenanes and molecular knots after specific interstrand and intrastrand cross-linking at both ends followed by demetallation.13 Obviously, the double helix stability of the stapled helicates was remarkably enhanced.
- For leading examples of stapled double-stranded helicates, see: (a) C. O. Dietrich-Buchecker and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1989, 28, 189–192 CrossRef; (b) C. O. Dietrich-Buchecker, J. Guilhem, C. Pascard and J.-P. Sauvage, Angew. Chem., Int. Ed. Engl., 1990, 29, 1154–1156 CrossRef; (c) Molecular Catenanes, Rotaxanes and Knots, ed. J.-P. Sauvage and C. O. Dietrich-Buchecker, Wiley-VCH, Weinheim, 1999 Search PubMed.
- For examples of complementary double helices based on amidinium–carboxylate salt bridges, see: (a) Y. Tanaka, H. Katagiri, Y. Furusho and E. Yashima, Angew. Chem., Int. Ed., 2005, 44, 3867–3870 CrossRef CAS PubMed; (b) Y. Furusho, Y. Tanaka and E. Yashima, Org. Lett., 2006, 8, 2583–2586 CrossRef CAS PubMed; (c) M. Ikeda, Y. Tanaka, T. Hasegawa, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2006, 128, 6806–6807 CrossRef CAS PubMed; (d) Y. Furusho, Y. Tanaka, T. Maeda, M. Ikeda and E. Yashima, Chem. Commun., 2007, 3174–3176 RSC; (e) H. Ito, Y. Furusho, T. Hasegawa and E. Yashima, J. Am. Chem. Soc., 2008, 130, 14008–14015 CrossRef CAS PubMed; (f) T. Maeda, Y. Furusho, S.-i. Sakurai, J. Kumaki, K. Okoshi and E. Yashima, J. Am. Chem. Soc., 2008, 130, 7938–7945 CrossRef CAS PubMed; (g) H. Iida, M. Shimoyama, Y. Furusho and E. Yashima, J. Org. Chem., 2010, 75, 417–423 CrossRef CAS PubMed; (h) H. Yamada, Y. Furusho, H. Ito and E. Yashima, Chem. Commun., 2010, 46, 3487–3489 RSC; (i) H. Ito, M. Ikeda, T. Hasegawa, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2011, 133, 3419–3432 CrossRef CAS PubMed; (j) H. Yamada, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2012, 134, 7250–7253 CrossRef CAS PubMed; (k) H. Yamada, Z.-Q. Wu, Y. Furusho and E. Yashima, J. Am. Chem. Soc., 2012, 134, 9506–9520 CrossRef CAS PubMed.
- Metal–acetylide complexes, such as PtII–acetylide, have been recognized as useful building blocks to construct well-defined supramolecular architectures because of their well-defined geometry along with their synthetic accessibility, see: (a) P. J. Stang and B. Olenyuk, Acc. Chem. Res., 1997, 30, 502–518 CrossRef CAS; (b) V. W.-W. Yam, Acc. Chem. Res., 2002, 35, 555–563 CrossRef CAS PubMed; (c) K. Onitsuka and S. Takahashi, Top. Curr. Chem., 2003, 228, 39–63 CrossRef CAS PubMed; (d) S. Szafert and J. A. Gladysz, Chem. Rev., 2006, 106, PR1–PR33 CrossRef CAS PubMed; (e) S. Y.-L. Leung, A. Y.-Y. Tam, C.-H. Tao, H. S. Chow and V. W.-W. Yam, J. Am. Chem. Soc., 2012, 134, 1047–1056 CrossRef CAS PubMed.
- (a) G. R. Owen, F. Hampel and J. A. Gladysz, Organometallics, 2004, 23, 5893–5895 CrossRef CAS; (b) A. L. Sadowy, M. J. Ferguson, R. McDonald and R. R. Tykwinski, Organometallics, 2008, 27, 6321–6325 CrossRef CAS.
- For other examples of ligand-exchange reactions at a PtII–acetylide center, see: (a) K. Campbell, R. McDonald, M. J. Ferguson and R. R. Tykwinski, J. Organomet. Chem., 2003, 683, 379–387 CrossRef CAS; (b) K. Campbell, C. A. Johnson II, R. McDonald, M. J. Ferguson, M. M. Haley and R. R. Tykwinski, Angew. Chem., Int. Ed., 2004, 43, 5967–5971 CrossRef CAS PubMed; (c) J. Stahl, W. Mohr, L. de Quadras, T. B. Peters, J. C. Bohling, J. M. Martín-Alvarez, G. R. Owen, F. Hampel and J. A. Gladysz, J. Am. Chem. Soc., 2007, 129, 8282–8295 CrossRef CAS PubMed ; see also ref. 12 and 14b.
- The 1JP–Pt coupling constants of the bridged double helices in the range 2545–2663 Hz are consistent with the reported values for the trans-PtII–acetylide complexes with phosphine ligands.16b.
- Tykwinski and coworkers reported that the trans-PtII–alkenyl complexes with PPh3 ligands could be smoothly transformed into the corresponding cis-form via a ligand-exchange reaction with CHIRAPHOS or cis-bis(diphenylphosphino)ethylene.14b,17a,b.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- J. J. P. Stewart, MOPAC2012, Stewart Computational Chemistry, Colorado Springs, CO, USA, http://openmopac.net/, 2012 Search PubMed.
- The CD and absorption intensities of 3a around 375 nm are smaller than those of 3b, which may be attributed to a conformational flexibility arising from the less bulky amidine substituents of 3a, compared to 3b.
- The right- and left-handed double-helical structures of (S,S)-3c and (R,R)-3c are 53.8 and 18.5 kJ mol−1 more stable than the corresponding opposite-handed duplexes, respectively, estimated by semi-empirical MO calculations (using the PM6 method20 in MOPAC2012 (ref. 21)) (Fig. S4C and D†).
- For discussions on the MLCT band in the PtII–acetylide complexes, see: (a) M. S. Khan, A. K. Kakkar, N. J. Long, J. Lewis, P. Raithby, P. Nguyen, T. B. Marder, F. Wittmann and R. H. Friend, J. Mater. Chem., 1994, 4, 1227–1232 RSC; (b) S. Takahashi, K. Onitsuka and F. Takei, Macromol. Symp., 2000, 156, 69–77 CrossRef CAS; (c) R. D'Amato, A. Furlani, M. Colapietro, G. Portalone, M. Casalboni, M. Falconieri and M. V Russo, J. Organomet. Chem., 2001, 627, 13–22 CrossRef; (d) R. Saha, M. A. Qaium, D. Debnath, M. Younus, N. Chawdhury, N. Sultana, G. Kociok-Köhn, L.-L. Ooi, P. R. Raithby and M. Kijima, Dalton Trans., 2005, 2760–2765 RSC.
- It should be noted that the absorption band around 375 nm observed in 3a–c with the trans-PtII geometry completely disappeared in the non-cross-linked cis-PtII–4a–c and their model duplexes 5a–c·6 (Fig. S8–S11†).14b.
- The rate of chain exchange between the dimeric strands in the related PtII-linked duplexes was reported to be much slower than the NMR time scale.14k.
- The relatively lower yields of 3c during the initial stage of the ligand-exchange reaction may be due to imperfect bridging by DPPPe, resulting in the formation of complicated intermediates, such as duplexes bearing non-bridged DPPPe and PPh3 ligands with different molar ratios, the signals of which would be highly broad. In fact, the TMS signals due to the 1c·2 (0.25 and 0.23 ppm) completely disappeared within 1 h (Fig. 4B).
- A similar diastereoselectivity switching has been observed in the template synthesis of complementary double helices through the formation of dynamic-covalent imine bonds.14j.
- The diastereoselective ligand exchange of heterochiral (S,R)-3c toward (S,S)- and (R,R)-DPPPe was investigated as follows (Fig. S18†); first 1c·2 was allowed to react with 2 equiv. of rac-DPPPe to form unreactive homochiral (S,S)- and (R,R)-3c ((S,S) rich) as well as reactive (S,R)-3c. Upon the further addition of 2 equiv. of rac-DPPPe, the (S,R)-3c preferentially reacted with (R,R)-DPPPe over the antipode (S,S)-DPPPe, thus producing totally homochiral 3c slightly rich in the (R,R)-3c duplex, which clearly revealed the (R,R)-selectivity of the heterochiral (S,R)-3c, although its diastereoselectivity was not high.
- For reviews, see: (a) C. Maeda, T. Kamada, N. Aratani and A. Osuka, Coord. Chem. Rev., 2007, 251, 2743–2752 CrossRef CAS; (b) M. M. Safont-Sempere, G. Fernández and F. Würthner, Chem. Rev., 2011, 111, 5784–5814 CrossRef CAS PubMed; for selected examples of homochiral self-sorting in metallo-supramolecular assemblies, see: (c) M. A. Masood, E. J. Enemark and T. D. P. Stack, Angew. Chem., Int. Ed., 1998, 37, 928–932 CrossRef; (d) S. G. Telfer, T. Sato and R. Kuroda, Angew. Chem., Int. Ed., 2004, 43, 581–584 CrossRef CAS PubMed; (e) M. Hutin, C. J. Cramer, L. Gagliardi, A. R. M. Shahi, G. Bernardinelli, R. Cerny and J. R. Nitschke, J. Am. Chem. Soc., 2007, 129, 8774–8780 CrossRef CAS PubMed; (f) F. Rodler, W. Sicking and C. Schmuck, Chem. Commun., 2011, 47, 7953–7955 RSC; (g) C. Guetz, R. Hovorka, C. Klein, Q.-Q. Jiang, C. Bannwarth, M. Engeser, C. Schmuck, W. Assenmacher, W. Mader, F. Topic, K. Rissanen, S. Grimme and A. Lützen, Angew. Chem., Int. Ed., 2014, 53, 1693–1698 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details and additional spectroscopic data. See DOI: 10.1039/c4sc02275k |
| This journal is © The Royal Society of Chemistry 2015 |